
Abstract
A right aortic arch (RAA) with a left arterial duct (LAD) together encircle the trachea and have the potential to cause tracheobronchial compression and published guidelines recommend bronchoscopy in symptomatic patients. The aim of the study was to describe the incidence of tracheal compression in a cohort of prenatally diagnosed RAA and LAD. Retrospective review of clinical course and imaging of prenatal cases of RAA and LAD assessed with flexible bronchoscopy over an 11-year period. 34 cases of prenatally diagnosed RAA with LAD underwent bronchoscopy at median age of 9 months (range 0.4–123) of whom 11 had respiratory symptoms and 23 were asymptomatic. In the neonatal period, three cases demonstrated respiratory symptoms. An aberrant left subclavian artery (ALSA) was identified in 29 cases. Pulsatile tracheal compression was identified in 32/34 (94%) cases and two cases showed normal tracheal appearances. Significant tracheal compression (> 70% occlusion) was present in 25/34 (74%) cases of which 16 were asymptomatic. Significant carinal compression (> 70% occlusion) was identified in 14/34 (42%) cases, an ALSA was observed in 13/14. Surgical relief of a vascular ring has been performed in 27 (79%) cases at a median age of 15 months (range 0.6–128 months). At surgery, a fibrous remnant of an atretic left aortic arch was identified in 11/27 (41%) cases. Significant tracheal compression may be present in infants even without symptoms. If early relief of airway compression is to be achieved to promote normal development of tracheal cartilage, early bronchoscopy should be considered.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The aortic arch usually passes to the left of the trachea, but in 0.1% of the population it passes to the right of the trachea and this is defined as a right aortic arch (RAA) [1]. The arterial duct can pass to the left or to the right of the trachea and it obliterates after birth and persists as the arterial ligament which cannot be identified on echocardiography, computerized tomography (CT) or magnetic resonance imaging (MRI). Thus, when a RAA and left arterial ligament are present they form a loop around the trachea and oesophagus which has the potential to cause tracheal compression [2]. This is in contrast to a RAA with a right arterial duct where both vessels pass to the right of the trachea but do not encircle it. This formation can easily be overlooked at the time of routine screening, but these vessels would not encircle the trachea and therefore are unlikely to cause trachea compression. The commonest branching patterns of a RAA are either mirror image of a left aortic arch branching pattern or with an aberrant left subclavian artery (ALSA). This ALSA originates from the descending aorta coursing posterior to the trachea and oesophagus and therefore may pulsate on the posterior aspect of the trachea and oesophagus (Fig. 1).

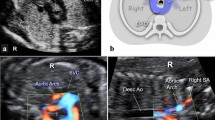
Three vessel and tracheal view on fetal echocardiography. a Left-sided aortic arch as seen during fetal echocardiography. The white arrow indicates the aortic arch which passes to the left of the trachea (T). b Right-sided aortic arch (white arrow) passing to the right of the trachea (T) and the left-sided arterial duct passing to the left of the trachea. These structures form a U-shape around the trachea. c Postnatal MRI reconstruction showing a right-sided aortic arch (RAA) with an aberrant left subclavian artery and the relationship to the trachea. A Kommerell’s diverticulum is also present (white arrow). SVC superior vena cava, PA pulmonary artery, LCA left common carotid artery, RCA right common carotid artery, ALSA aberrant left subclavian artery, RSA right subclavian artery
A RAA is increasingly diagnosed during fetal life due to the incorporation of views of the upper mediastinum into screening of the fetal heart [3,4,5]. The reason for assessment of the upper mediastinum in fetal life is to identify major congenital heart disease such as transposition of the great arteries or tetralogy of Fallot which would be missed if the four chamber view of the heart alone was assessed, but a RAA is also visualized on such views [5,6,7,8,9]. Historically, RAA has been regarded as a largely benign lesion, however, a recent meta-analysis has shown that up to 25% of prenatally diagnosed RAA may require surgical relief of a vascular ring and a meta-analysis has recommended investigation of symptomatic cases [4]. Recently, we have identified significant airways compression in some infants without symptoms who were undergoing general anaesthesia for non-cardiac surgery and in whom bronchoscopy was performed during the same anaesthetic at the request of the anaesthetist to ensure there was no airway compromise which may impact anaesthesia given the diagnosis of a RAA. This lead to an increased awareness and an increase in the number of bronchoscopies performed which prompted us to review our practice and experience of assessment of infants with RAA and left arterial duct (LAD) diagnosed during fetal life. Our objectives were to describe the bronchoscopy findings and to establish if symptoms were a good guide to the presence of airway compression.
Methods
A retrospective review of children (< 16 years) who were prenatally diagnosed with RAA and LAD between January 2005 and December 2015 at King’s College Hospital and Evelina London Children’s Hospital whom underwent bronchoscopy. Patients were identified from departmental databases for this descriptive study. Patients were excluded if the diagnosis was not made prenatally or if there were associated major intracardiac defects (such as tetralogy of Fallot, common arterial trunk and haemodynamically significant ventricular septal defects). Patient records including bronchoscopy reports and MRI or CT imaging were reviewed.
Postnatal Management
In the earlier years (until 2010), many patients with a RAA were not routinely referred for postnatal cardiac evaluation due to the perceived benign nature of the lesion. Thus, patients underwent a neonatal check and no further elective reviews or surveillance were planned. From 2010, more patients underwent a postnatal cardiac review and a respiratory assessment performed in patients symptomatic for a vascular ring. The reason for postnatal cardiac review is to ensure that a double aortic arch or minor forms of congenital heart disease (for example ventricular septal defects) were not overlooked on prenatal echocardiography. With modern ultrasound imaging systems, the prenatal diagnosis of a RAA is easily identified compared to with postnatal echocardiography, whereas a double aortic arch with hypoplasia of one arch can be overlooked on both prenatal and postnatal echocardiography. Some cases underwent a postnatal MRI or CT scan if the laterality/number of aortic arches was not clear after birth. If symptoms of airway compression were present, then further investigation was performed (bronchoscopy and CT/MRI). Patients who were asymptomatic did not undergo any airway investigation in the earlier years apart from when bronchoscopy was requested by anaesthetists who were concerned about the potential impact of a vascular ring in two patients who were requiring a general anaesthetic for elective non-cardiac surgery. Bronchoscopy was performed at the start of the general anaesthetic for these patients following advanced discussion and consent from the parents. Significant tracheal compression was identified in both patients. Given this finding, the approach to management changed such that all prenatally diagnosed cases with a RAA/LAD underwent a postnatal cardiac review and the majority of patients were referred for a respiratory assessment even in the absence of airway symptoms. This occurred in the most recent years from approximately 2013. During an airway consultation further investigations including bronchoscopy were discussed with the parents. Furthermore, experience of the impact of airways compression in cardiac patients was developing and infants with double aortic arch and mild symptoms were identified to have critical tracheal narrowing [10]. These potential implications were discussed with the parents in prenatal counselling and in subsequent postnatal visits such that they were aware of our findings in this evolving area.
Airway Assessment
Dynamic airway studies were performed by flexible bronchoscopy using a 2.8-mm Olympus (BF-XP260F) scope. Bronchoscopy was carried out via endotracheal tube or laryngeal mask airway during spontaneous ventilation using inhalational or intravenous anaesthesia to assess the degree and site of airway compression and exclude other abnormalities such as tracheal stenosis. In some cases, negative suction pressure of − 10 to 20 ccH20 was applied to look for critical airway closing pressure points. Almost all cases had Propofol induction where the airway can also be assessed in a brief period of total apnoea. A detailed description of bronchoscopy methodology from our institute has been previously described [11]. All studies were recorded for video review. The site of airway compression (tracheal, carinal, right or left main bronchus) was noted and the degree of airway compression classified according to the Myer classification [12]: insignificant (< 50%), mild (51–70%), significant: > 70%. The sidedness of the aortic arch, origin of the left subclavian artery and position of the arterial duct were identified on fetal echocardiogram. MRI or CT scan were primarily used to identify vascular anatomy and position of vessels relative to the airway, rather than degree of airway compression.
Statistics
Data is presented as median, range and interquartile range (IQR). Continuous, non-parametric data are analysed using Mann–Whitney test and categorical variables using Chi-squared on SPSS version 23 for MAC and statistical significance was taken as p < 0.05.
Ethical Approval
Given the evolving practice, we sought review of our practice and this was approved by the Guy’s & St Thomas’ NHS Trust Hospitals clinical governance department.
Results
During the study period, 143 cases of isolated RAA with LAD were diagnosed prenatally. All cases were reviewed after birth and 100 cases were subsequently discharged as they were asymptomatic and seven were excluded from this study due to additional congenital heart disease [haemodynamically significant ventricular septal defects (n = 6) and disconnected left pulmonary artery (n = 1)]; thus 36 cases were referred for airways investigation and 34 have completed airway investigations. On detailed questioning, 11/34 patients (32%) demonstrated symptoms suggestive of airways compression and 23/34 were asymptomatic. Bronchoscopy was performed at a median age of 9 months (range: 0.4–123) at a median weight of 7 kg (range: 2–46). Chromosomal analysis was performed in 15/34 cases with four cases of microdeletion of chromosome 22q11, one with 45XO/22q11, one with Noonan syndrome and the remaining ten were normal.
Symptoms
Symptoms of airway compression were present in 11/34 cases from a median of 5 months (range: birth—24 months). Two patients were symptomatic from birth requiring ventilation. Reported symptoms/presentations included: noisy breathing (n = 5), respiratory distress (n = 1), wheeze (n = 1), recurrent croup (n = 1), recurrent lower respiratory tract infections (n = 3) and presentation requiring ventilation (n = 3). Respiratory symptoms were present in the neonatal period in three patients. No gastro-oesophageal symptoms were present in this cohort. The median age at investigation was similar between those with and without symptoms: 8 months (IQR: 4.0–2.9) vs. 11 months (IQR: 6.6–29), respectively (p = 0.3). The patient who was investigated at 123 months age was seen in infancy, but was then lost to follow-up.
Pulsatile compression of the trachea was identified in 32/34 cases on bronchoscopy of which seven were mild (51–70% compression) and 25/34 (74%) demonstrated significant tracheal compression > 70% (Table 1; Fig. 2). Normal airway patency was identified in two cases (Fig. 3). Carinal compression was seen in 22/34 cases of which eight showed mild compression and 14 demonstrated significant compression. There was one case of significant carinal compression which showed only mild tracheal compression in an asymptomatic patient, all others had concomitant significant tracheal compression (Fig. 4; Table 1).

Bronchoscopy findings in a 8-month old with tracheal and carinal compression in the absence of symptoms. a Upper tracheal view with right-sided compression partially occluding distal trachea. b Mid-tracheal view with obvious compression from the right on the tracheal wall which correlated with the position of the aortic arch as it passed to the right of the trachea (moderate compression). c Carinal view with severe posterior compression of both left and right bronchial origins along the course of the Kommerell’s diverticulum behind the airway. d View from the origin of right main bronchus with severe compression at the point of origin of the aberrant left subclavian artery from right aortic arch

Results of investigation and findings in fetuses diagnosed with a right aortic arch

Percentage of cases with significant (> 70%) tracheal, carinal right and left main bronchus compression in asymptomatic (n = 23) and symptomatic (n = 11) patients
The sensitivity and specificity of airway symptoms for significant tracheal compression were 34% and 75%, respectively. The proportion of patients with significant tracheal compression (> 70%) did not differ between those with and without symptoms (82 vs. 69%, p = 0.4). The incidence of significant carinal compression was also similar between symptomatic and asymptomatic patients (36 vs. 43%, p = 0.7). Logistic regression analysis showed no identifiable predictors of symptoms (weight, ALSA or the presence of significant airway compression).
Branching Pattern of the Aortic Arch
An ALSA was present in 29/34 (85%). Significant tracheal compression was identified in 15/21 asymptomatic patients with an ALSA. Five cases of mirror image branching pattern were investigated, two were asymptomatic and one showed mild tracheal compression and the other significant tracheal compression. Significant carinal compression was identified in 13/29 (45%) patients with an ALSA, mild carinal compression in 6/29(21%). Carinal compression was identified in one case without an ALSA.
Management of Cases
All cases were discussed in a multidisciplinary meeting to decide treatment. Surgery was performed in 27 cases (79%) and three cases are awaiting surgery. Median age at surgery was 15 months (range 0.6–128). 10/11 symptomatic cases were operated to relieve the vascular ring and one case did not have significant tracheal or carinal compression despite the presence of recurrent lower respiratory tract infections. Surgery was performed in 17/23 patients with asymptomatic tracheal compression to date and a further two are awaiting surgery. At surgery, an additional finding of an atretic left aortic arch with no luminal continuity to the descending aorta was identified in 11 patients (eight cases with ALSA, three cases with mirror image branching pattern) and this had not been visualized with CT or MRI. Surgical procedures included: ligation of the ALSA (n = 15), re-implantation of ALSA (n = 1), division of ligamentum arteriosum (n = 18), division of patent arterial duct (n = 2), division of atretic left arch (n = 11) and resection of Kommerell’s diverticulum (n = 9). These did not require cardiopulmonary bypass and no surgical mortality occurred. The following early postoperative complications occurred: chylothorax (n = 1), thoracotomy wound infection (n = 1), recurrent laryngeal palsy (n = 1) and Horner’s syndrome (n = 1), small pneumothorax (n = 1). Surgery was not required in five patients as the mild degree of tracheobronchial compression was not deemed sufficient to warrant surgery.
Follow-Up After Surgery
Median follow-up in the asymptomatic group was 1.9 years (range 0.8–10.9) and in the symptomatic group was 2.7 years (0.2–3.4). Five children have ongoing respiratory symptoms following surgery: two with incomplete double arch (partially atretic left aortic arch), two with RAA with ALSA and one with mirror image branching pattern. Three patients had persistence of severe symptoms and required surgical intervention: one required tracheostomy and long-term ventilation, and two required re-operation for aortopexy (10 and 2 years following original surgery). None of the asymptomatic patients have reported new respiratory symptoms.
Discussion
Recommendations for prenatal screening of the fetal heart now include visualization of the aortic and ductal arches and this has led to increased recognition of RAA as an isolated cardiac finding. Together, the LAD and the RAA partially encircle the trachea and oesophagus and the right pulmonary artery lies immediately anterior to the trachea. The management of such cases poses a dilemma; for infants with respiratory symptoms there is a clear indication to proceed to further investigation. However, current consensus documents do not specify how asymptomatic infants should be managed [4].
This is the first reported series of cases with significant tracheal compression in the absence of respiratory symptoms in infants with a RAA. The initial bronchoscopies were performed for other reasons and following the detection of asymptomatic airways compression in a few infants, the threshold for investigation was lowered such that investigation was considered. Tracheal compression was evident in 91% of asymptomatic patients and significant tracheal compression was seen in 69%. Therefore, symptoms cannot be used as a reliable indicator of tracheal compression.
The management of this group of patients poses a clinical predicament. There is understandable reluctance to investigate and potentially operate on infants without symptoms of airways compression, but our data show no mortality and a low rate of morbidity following surgery, in keeping with other data [13]. The view of our surgeons has been that if relief of the vascular ring is to be undertaken, the tissue of infants is more elastic and pliable, making the surgery technically easier in infants rather than older children. Previous reports have suggested that chronic symptoms after treatment of symptomatic vascular rings are likely due to inadequate catch-up growth of the tracheal rings after relief of the chronic compression [14, 15] and this abnormal tracheal development can lead to airways collapse with air trapping and bronchiectasis [16]. The trachea almost triples its length and diameter from birth to adolescence [17] and persisting vascular compression is likely to prevent this normal process of cartilage growth. Thus, a justification for early surgery is to reduce future problems with airway cartilage and poor tracheal growth. Long-term follow-up studies of symptomatic patients with double aortic arch or RAA with ALSA who underwent surgery to relieve tracheal compression from the vascular ring have shown that up to a third of patients have chronic respiratory symptoms [14, 15, 18,19,20,21,22] and persistent postoperative asymptomatic tracheal obstruction has also been reported [15, 23]. This was seen in our cohort of symptomatic patients whereby three had significant tracheomalacia 3 years after the removal of the compression. There are no published longitudinal data of tracheal compression which shows it improves with time and in our cohort only one patient has declined surgery and has not been re-investigated as yet, therefore we cannot confirm that pursing a conservative approach in this asymptomatic cohort leads to irreversible tracheal damage, but the data in symptomatic patients suggest this. Furthermore, the timing of postnatal airways investigation in asymptomatic children needs to be balanced to avoid unnecessary investigations. Symptoms of airway compression in early infancy or in the 1st year of life may be difficult to recognize or not present due to low tidal volumes in small infants, there may be insufficient muscle power to generate clearly obstructive airway symptoms and activity levels are less strenuous. Symptoms include upper airways noise/stridor, recurrent chest infections, asthma and feeding difficulty which are common in childhood and they may not reach a threshold for investigation [24]. If conservative management is pursued, it is not known if these patients would develop symptoms later in childhood/adulthood as the majority of patients are discharged by 2 years of age and therefore longitudinal assessment is required [4].
Our study confirms that immediate respiratory deterioration after delivery is unlikely, occurring in only one patient in our series, our advice for perinatal care is that staff and facilities for endotracheal intubation should be available. Patients remain under cardiology review throughout infancy and the rationale for combined bronchoscopy and CT or MRI imaging is discussed with parents. Our recommended approach is that investigation is performed in the 1st year; however, we are still evolving a protocol regarding the exact timing as there is some preference that this is performed at 6 months of age such that earlier repair can be carried out. The gold standard diagnostic test for extrinsic pulsatile tracheal compression is a free-breathing dynamic bronchoscopy without positive airways pressure to prevent a false-negative assessment. If tracheal compression is confirmed, a CT or MRI scan is undertaken to identify the vascular structures responsible. The advantage of a specialized CT protocol with contrast throughout the respiratory phase compared to MRI is detection of dynamic airway issues with parenchymal lung views. The process is relatively rapid (minutes) compared to MRI, although involves a small dose of radiation. The mortality risks of these two combined procedures in our institution are extremely low (risk < 1:1000); however, bronchoscopy requires skilled operators [11]. A multidisciplinary approach to the analysis of findings is recommended with involvement of an airway service with experience of bronchoscopy in infants and the cardiac team to assist in anatomical assessment of the vascular structures.
Risk Stratification of RAA for Investigation
Currently, prenatal risk stratification for airways compression has been based on the presence of an ALSA and this is based upon classical teaching; however, in our series, infants with mirror image branching pattern were also found to have significant tracheal compression so this cannot be used in isolation. Fetal MRI provides good definition of vessels and could improve risk stratification as it can assist in identifying cases where there is a patent diminutive left aortic arch during prenatal life which becomes atretic and persist as a fibrous cord by infancy [25]. An atretic left arch has been a common finding within our cohort and it cannot be confidently diagnosed on fetal echocardiography or postnatal imaging as the lumen is obliterated. Therefore, cases of mirror image branching pattern, the prevalence of which is lower, should also be investigated and although a RAA may be evident on detailed imaging, this does not exclude a partial double aortic arch. Further assessment of the site at which the descending aorta crosses the midline is also highly relevant, if it crosses the midline in the upper mediastinum this creates additional pulsatility on the posterior aspect of the trachea [26].
Further risk stratification of children who are at risk of maldevelopment of tracheal cartilage may be possible with newer modalities such as optical coherence tomography [27] which is performed during bronchoscopy and can circumferentially identify regions of cartilage within the airway. This may prove a useful tool to understand changes in tracheal cartilage morphology and may further assist in identifying airways which may be most at risk and warrant ongoing surveillance. Risk stratification will become more important as improvements in prenatal screening programmes will result in an increased frequency of diagnosis of isolated RAA [5, 9].
Limitations
This was a retrospective observational study describing our evolving experience of prenatally diagnosed RAA with LAD, thus the true denominator of airway involvement is not known in this condition. In the earlier cases, there was no precedence for detailed investigation of asymptomatic patients. More recently, asymptomatic cases were investigated at the discretion of the attending consultant and therefore without patient-related bias. The identification of symptoms is difficult in pre-ambulant infants due to the reasons discussed.
A further patient group that was not studied is those RAA associated with other forms of congenital heart disease, for example, haemodynamically significant ventricular septal defects and tetralogy of Fallot [28]. The degree of tracheal compression in these cases is not known and screening bronchoscopy to identify asymptomatic tracheal compression should be considered in these cases given the findings of this study.
Conclusion
Significant tracheal compression may be present in infants with a RAA in the absence of symptoms. The absence of an ALSA does not exclude development of significant tracheal compression and in this group the presence of the left arterial ligament alone or the co-existence of an atretic left aortic arch ligament is responsible for completing the anatomical vascular ring. If the potential for normal development of tracheal cartilage is to be optimized, early airway investigation should be considered even in asymptomatic patients. A prospective study is warranted.
Abbreviations
- RAA:
-
Right aortic arch
- ALSA:
-
Aberrant left subclavian artery
- LAD:
-
Left arterial duct
References
Achiron R, Rotstein Z, Heggesh J, Bronshtein M, Zimand S, Lipitz S, Yagel S (2002) Anomalies of the fetal aortic arch: a novel sonographic approach to in-utero diagnosis. Ultrasound Obstet Gynecol 20(6):553–557. https://doi.org/10.1046/j.1469-0705.2002.00850.x
McLaren CA, Elliott MJ, Roebuck DJ (2008) Vascular compression of the airway in children. Paediatr Respir Rev 9(2):85–94. https://doi.org/10.1016/j.prrv.2007.12.008
Vigneswaran TV, Greco E, Simpson JM, Nicolaides KH, Zidere V (2016) P45 is it important to identify an isolated right aortic arch in fetal life? Heart 102(Suppl 1):A23. https://doi.org/10.1136/heartjnl-2016-309377.45
D’Antonio F, Khalil A, Zidere V, Carvalho JS (2015) Fetuses with right aortic arch: a multicentre cohort study and meta-analysis. Ultrasound Obstet Gynecol. https://doi.org/10.1002/uog.15805
Zidere V, Tsapakis EG, Huggon IC, Allan LD (2006) Right aortic arch in the fetus. Ultrasound Obstet Gynecol 28(7):876–881. https://doi.org/10.1002/uog.3841
International Society of Ultrasound in Obstetrics & Gynecology, Carvalho JS, Allan LD, Chaoui R, Copel JA, DeVore GR, Hecher K, Lee W, Munoz H, Paladini D, Tutschek B, Yagel S (2013) ISUOG practice guidelines (updated): sonographic screening examination of the fetal heart. Ultrasound Obstet Gynecol 41(3):348–359. https://doi.org/10.1002/uog.12403
Hunter LE, Simpson JM (2014) Prenatal screening for structural congenital heart disease. Nat Rev Cardiol 11(6):323–334. https://doi.org/10.1038/nrcardio.2014.34
Donofrio MT, Moon-Grady AJ, Hornberger LK, Copel JA, Sklansky MS, Abuhamad A, Cuneo BF, Huhta JC, Jonas RA, Krishnan A, Lacey S, Lee W, Michelfelder EC Sr, Rempel GR, Silverman NH, Spray TL, Strasburger JF, Tworetzky W, Rychik J (2014) Diagnosis and treatment of fetal cardiac disease: a scientific statement from the American Heart Association. Circulation 129(21):2183–2242. https://doi.org/10.1161/01.cir.0000437597.44550.5d
Deaprtment of Health (2015) Fetal anomaly screening programme. https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/421650/FASP_Standards_April_2015_final_2_.pdf
Griffiths B, Lee G, Durward A (2017) Critical airway obstruction in apparently asymptomatic neonates. Pediatr Pulmonol 52(4):E15–E17. https://doi.org/10.1002/ppul.23564
Manna SS, Durward A, Moganasundram S, Tibby SM, Murdoch IA (2006) Retrospective evaluation of a paediatric intensivist-led flexible bronchoscopy service. Intensive Care Med 32(12):2026–2033. https://doi.org/10.1007/s00134-006-0351-y
Myer CM 3rd, O’Connor DM, Cotton RT (1994) Proposed grading system for subglottic stenosis based on endotracheal tube sizes. Ann Otol Rhinol Laryngol 103(4 Pt 1):319–323. https://doi.org/10.1177/000348949410300410
Herrin MA, Zurakowski D, Fynn-Thompson F, Baird CW, Del Nido PJ, Emani SM (2017) Outcomes following thoracotomy or thoracoscopic vascular ring division in children and young adults. J Thorac Cardiovasc Surg. https://doi.org/10.1016/j.jtcvs.2017.01.058
Gross RE (1955) Arterial malformations which cause compression of the trachea or esophagus. Circulation 11(1):124–134. https://doi.org/10.1161/01.cir.11.1.124
McLaughlin RB Jr, Wetmore RF, Tavill MA, Gaynor JW, Spray TL (1999) Vascular anomalies causing symptomatic tracheobronchial compression. Laryngoscope 109(2 Pt 1):312–319. https://doi.org/10.1097/00005537-199902000-00025
Williams HE, Landau LI, Phelan PD (1972) Generalized bronchiectasis due to extensive deficiency of bronchial cartilage. Arch Dis Child 47(253):423–428. https://doi.org/10.1136/adc.47.253.423
Wailoo MP, Emery JL (1982) Normal growth and development of the trachea. Thorax 37(8):584–587
Anand R, Dooley KJ, Williams WH, Vincent RN (1994) Follow-up of surgical correction of vascular anomalies causing tracheobronchial compression. Pediatr Cardiol 15(2):58–61. https://doi.org/10.1007/BF00817607
Backer CL, Ilbawi MN, Idriss FS, DeLeon SY (1989) Vascular anomalies causing tracheoesophageal compression. Review of experience in children. J Thorac Cardiovasc Surg 97(5):725–731
Ruzmetov M, Vijay P, Rodefeld MD, Turrentine MW, Brown JW (2009) Follow-up of surgical correction of aortic arch anomalies causing tracheoesophageal compression: a 38-year single institution experience. J Pediatr Surg 44(7):1328–1332. https://doi.org/10.1016/j.jpedsurg.2008.11.062
Woods RK, Sharp RJ, Holcomb GW 3rd, Snyder CL, Lofland GK, Ashcraft KW, Holder TM (2001) Vascular anomalies and tracheoesophageal compression: a single institution’s 25-year experience. Ann Thorac Surg 72(2):434–438. https://doi.org/10.1016/S0003-4975(01)02806-5 (discussion 438–439)
Yilmaz M, Ozkan M, Dogan R, Demircin M, Ersoy U, Boke E, Pasaoglu I (2003) Vascular anomalies causing tracheoesophageal compression: a 20-year experience in diagnosis and management. Heart Surg Forum 6(3):149–152
Marmon LM, Bye MR, Haas JM, Balsara RK, Dunn JM (1984) Vascular rings and slings: long-term follow-up of pulmonary function. J Pediatr Surg 19(6):683–692
Licari A, Manca E, Rispoli GA, Mannarino S, Pelizzo G, Marseglia GL (2015) Congenital vascular rings: a clinical challenge for the pediatrician. Pediatr Pulmonol 50(5):511–524. https://doi.org/10.1002/ppul.23152
Lloyd DF, van Amerom JF, Pushparajah K, Simpson JM, Zidere V, Miller O, Sharland G, Allsop J, Fox M, Lohezic M, Murgasova M, Malamateniou C, Hajnal JV, Rutherford M, Razavi R (2016) An exploration of the potential utility of fetal cardiovascular MRI as an adjunct to fetal echocardiography. Prenat Diagn 36(10):916–925. https://doi.org/10.1002/pd.4912
Donnelly LF, Fleck RJ, Pacharn P, Ziegler MA, Fricke BL, Cotton RT (2002) Aberrant subclavian arteries: cross-sectional imaging findings in infants and children referred for evaluation of extrinsic airway compression. Am J Roentgenol 178(5):1269–1274. https://doi.org/10.2214/ajr.178.5.1781269
Williamson JP, Armstrong JJ, McLaughlin RA, Noble PB, West AR, Becker S, Curatolo A, Noffsinger WJ, Mitchell HW, Phillips MJ, Sampson DD, Hillman DR, Eastwood PR (2010) Measuring airway dimensions during bronchoscopy using anatomical optical coherence tomography. Eur Respir J 35(1):34–41. https://doi.org/10.1183/09031936.00041809
Miranda JO, Callaghan N, Miller O, Simpson J, Sharland G (2014) Right aortic arch diagnosed antenatally: associations and outcome in 98 fetuses. Heart 100(1):54–59. https://doi.org/10.1136/heartjnl-2013-304860
Acknowledgements
We wish to thank Professor Lindsey Allan and the Late Dr Ian Huggon who instigated training of the fetal medicine team at Kings College Hospital to screen for anomalies of the great arteries and for developing understanding of the laterality of the aortic arch in the prenatal period. The foetal and paediatric cardiology team at Evelina London Children’s Hospital who have managed the patients, the cardiac MRI team who performed the cross-sectional imaging, the multidisciplinary airway team who investigated these patients and the cardiothoracic surgeons who performed the surgery.
Author information
Authors and Affiliations
Contributions
VZ and JS proposed the study. TV, VZ and JS designed the study. TV, EK, AD and AN collated the data. TV and AD analysed the data and wrote the draft manuscript. AB, JS, KP, AD, AN and VZ critically reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Vigneswaran, T.V., Kapravelou, E., Bell, A.J. et al. Correlation of Symptoms with Bronchoscopic Findings in Children with a Prenatal Diagnosis of a Right Aortic Arch and Left Arterial Duct. Pediatr Cardiol 39, 665–673 (2018). https://doi.org/10.1007/s00246-017-1804-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-017-1804-5