
Abstract
The release of silver (Ag) nanoparticles (NPs) from the use of consumer products to the environment has raised concern about the risk to ecosystems because of its unpredictable toxicological impact to microorganisms in terrestrial environment. In this study, the impact of Ag chemical speciation (Ag+ and Ag-NPs [50-nm uncoated and 15-nm polyvinylpyrrolidone (PVP)-coated Ag-NPs]) to soil nitrification kinetics was investigated using a batch soil-slurry nitrification method along with sorption isotherm and dissolution experiments. The results of nitrification potential (i.e., kinetic rate) suggest that Ag+/Ag-NPs, which strongly sorb in soils, suppressed the nitrification processes. Among each chemical species, the degree of suppression increased with increasing [Ag]total. Although ionic Ag (Ag+) species is known to exhibit greater antimicrobial effects than the solid state Ag species, such as Ag-NPs, in most studies, PVP-coated 15-nm Ag-NPs, however, more effectively suppressed the soil nitrification process than did Ag+ under the same dose. Although several physicochemical-based toxicity mechanisms of dispersed Ag-NPs were discussed in pure culture and aquatic systems, it is not clearly understood how PVP-coated Ag-NPs could exhibit greater toxicity to nitrifying bacteria than Ag+ in soils. In assessing the impact of Ag-NPs to microbial mediated processes (e.g., N cycles) in the terrestrial environment, it might be critical to understand the interactions and reactivity of Ag-NPs at the soil–water interface.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Nanotechnology has grown since the mid-1980s (Drexler 2004) and is expected to become a trillion-dollar market by 2015 (Nel et al. 2006). Today, engineered nanoparticles (ENPs) are widely used in household products, biomedical supplies, cosmetics, electronics, and other commercial applications (e.g., Nel et al. 2006; Nowack and Bucheli 2007). Growing attention has been raised about their risk to ecosystems (e.g., Wiesner et al. 2006; Nowack and Bucheli 2007; Klaine et al. 2008). Several studies have been performed to quantify the release of NPs to the environment. A modeling scenario by Gottschalk et al. (2009) identified the primary sinks for other metallic NPs (silver and TiO2) as landfills, soils, and sediments. However, the overall impact of NPs on natural environments remains largely unknown (Guzman et al. 2006; Suresh et al. 2013). Any impact of ENPs as contaminants in terrestrial environments could potentially affect microbially mediated nutrient and trace-element cycles. Although the toxicity of ENPs (e.g., ZnO, CuO, Cu, Fe2O3, CeO2, Ag) to bacteria, including nitrifying bacteria (Nitrosomonas and Nitrobacter), Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, and Bacillus subtilis, has been extensively studied under pure culture media (e.g., Qi et al. 2004; Choi and Hu 2008; Sotiriou et al. 2010; Pelletier et al. 2010; Li et al. 2011; Azam et al. 2012), these toxicological data are often difficult to extrapolate to the ecosystem scale. Natural environments, such as soil and sediment systems, may provide additional sinks to decrease or enhance the bioavailability of ENPs that corresponds to the actual response in heterogeneous environments. Several studies on the toxicity of fullerene (C60) NPs on soil bacteria and microbial community in soil components have already shown a lack of toxicity (Tong et al. 2007; Johansen et al. 2008) whereas its aquatic toxicity is more pronounced (Lyon et al. 2005). This clearly suggests the validity of toxicological assessment in the adsorbent system, such as soils and sediments.
In this study, the impact of Ag-NPs on soil-nitrification process mediated by nitrifying bacteria was further investigated in an agricultural soil. These bacteria are essential players in the soil nitrogen cycle, in which they convert ammonium to nitrite to nitrate. The main objective of the study was to assess the effects of Ag+ and nano-Ag on nitrification kinetics in aerobic soils. Batch sorption and dissolution experiments of Ag-NPs were also performed to understand NP distribution in soils and soil solutions during the nitrification experiments. The results of this study provide valuable evidence to regulating bodies in their difficult task of assessing and regulating new nanotechnologies and the potential release of NPs to ecosystems.
Materials
Surface soils (top 10–30 cm) of Toccoa sandy loam (coarse-loamy, thermic typic Udifluvents) were collected from the Clemson University organic farm (Clemson, South Carolina, USA). The moisture content of soils was kept at field capacity at room temperature before nitrification experiments.
Characterization of soils was reported in the work by VandeVoort and Arai (2012). Cation exchange capacity, % organic matter, and pH are 7.4 cmolc/kg 1.5 %, and 5.2 ± 0.2, respectively. Clay mineralogy is quartz and kaolinite, hydroxyl interlayer vermiculite, gibbsite, and hematite, goethite. The following Ag nanoparticles (NPs) were obtained from commercial companies [Ultrasound Research Nanomaterials and Nanostructured and Amorphous Materials Inc. (both Huston, Texas, USA)]. Physicochemical characteristics are listed in Table 1.
All reagents were prepared in distilled deionized MilliQ water (18.2 MΩ) using the following ACS-grade salts: silver sulfate (Ag2(SO4)), sodium azide (NaN3), ammonium phosphate monobasic (NH4H2PO4), and ammonium sulfate ((NH4)2SO4)). All 0.45-μm membrane microfiltration filters (Millipore) and 1-kDa centrifugal ultrafiltration filters (Pall-Gellman Microsep) were preconditioned with 0.1 M copper nitrate (Cu(NO3)2) before use in dissolution, partitioning, and retention experiments to minimize Ag+ adsorption by membrane surfaces (Cornelis et al. 2010).
Methods
Batch Nitrification Kinetic Experiments
A shaken soil-slurry method by Hart et al. (1994) was used to assess nitrification kinetics in soils. The slurry method was chosen over other soil-nitrification methods (Boast and Robertson 1982; Killham 1987; Robertson and Vitousek 1981) due to its adaptability for our NP-soil system and reproducibility. The method assessed the maximum rate (V max : nitrification potential) of nitrification in a soil, which served as an indicator of the size of ammonium-oxidizer communities. In this method, tested soil samples are incubated under ideal conditions (water content, NH4 +, aeration, and phosphorus availability) (Hart et al. 1994). The procedures described here are specific for assessing soil-nitrification potential using the shaken soil-slurry method, which was adapted from Hart et al. (1994). For the control batch, the following solutions were combined and brought up to the volume in a 1-L volumetric flask: 1 mM of ammonium phosphate monobasic (NH4H2PO4) stock solution and 0.25 mM of ammonium sulfate (NH4)2SO4 stock solution. Two types of Ag-NPs were used in this experiment. First, uncoated superfine Ag powder (50 nm) (hereafter referred to as “uAg50”) was purchased from Inframat Advanced Materials, and polymer-coated Ag (15 nm) (hereafter referred to as “pAg15”) was purchased from Nanostructured and Amorphous Materials. The Ag+ was prepared as silver sulfate (Ag2SO4). In addition, sodium azide (NaN3) (65–100 mg/L) was used as an antimicrobial agent (Skipper and Westerman 1973) to observe the NO3 levels under an abiotically controlled environment. The difference between the control and NaN3 systems also shows the native microbiological activity for nitrification in the soil.
Appropriate amounts of Ag/AgNP stock solutions were added to assure total Ag concentrations of 0–300 mg/L. Due to the high toxicity of Ag+ and pAg15, only two concentrations (1 and 10 mg/L) were used to observed the kinetics, whereas four concentrations (1, 10, 100, and 300 mg/L) were used for the less toxic uAg50. For all solutions, pH was adjusted at 7.2 ± 0.2 using 0.1–1 M NaOH. Then 9 g of field-moist soil (18.44 % by wt%) was placed into 125-mL Erlenmeyer flasks. Each system consists of 12 replicates. The reason for using 12 samples was to increase the accuracy of later statistical analysis due to the heterogeneity of soils. Sixty mL of the above-mentioned combined solution was added to the soil sample and capped with a vented Parafilm (to allow gas exchange). Flasks were then placed on an orbital shaker at 180 rpm for 24 h. Ten mL of soil slurry from each flask was sampled four times during the 24-h period (after 2, 4, 22, and 24 h). These times were found to be the most efficient scheme for estimating nitrification rate in soil by concentrating the sampling at the beginning and end of the incubation. The 24-h period was chosen because it has been found to provide sufficient sensitivity for most soils systems (Hart et al. 1994). At each sampling time, soil suspensions were centrifuged at 8,000×g for 8 min. Five mL of the supernatant from each tube was then placed in a disposable polypropylene culture tube and frozen for later analysis of NO3 −.
Data Analysis and Calculations
Nitrate Analysis
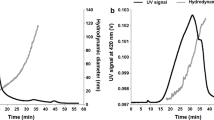
For NO3 − analysis, solutions were thawed and immediately analyzed using salicylic acid colorimetric technique at a wavelength of 420 nm (detection limit 0.4 mg/L NO3 −–N) (Cataldo et al. 1975). The chemical compositions of the two reagents are 5 % salicylic acid (5 g of sodium salicylate dissolved in 100 mL of H2SO4) and 1.7 M NaOH solutions. Eighty μL of the solution was transferred to 8-mL cuvettes (1/100 ratio of sample to reagents). Five percent salicylic acid solution (0.32 mL) was then added. After sufficient time for the solution to cool, 7.6 mL of 1.7 M NaOH was added to the cuvettes. After 30 min of cooling, absorbance values were read in the colorimetric analysis. During these sample times, pH was recorded in the well-mixed soil suspensions using electrodes. The concentrations of NO3 − (mg N/L) were then calculated using a nitrate standard curve. These concentrations were used to calculate nitrate production (mg N/kg soil) using Eq. (1):
The maximum rate (V max) of nitrification in each flask was calculated by measuring the slope of nitrate production over time by way of linear regression analysis, which give us the rate of production: mg N/kg soil h−1.
Statistical Analysis
t Test
Values of (V max) of 11–12 flasks in different Ag systems were compared with the control using a t test function in the Microsoft excel program. The V max in each concentration of the Ag+, coated Ag-NPS, and uncoated Ag-NPs were assessed based on both p < 0.01 and <0.05.
Ag+ and Nano-Ag Sorption Experiments
Sorption of Ag (both and nano-Ag) was evaluated in the same soil used in the nitrification experiments. Two suspension densities (2 g oven-dry soil in 30 mL for Ag+ and 0.1 g oven-dry soil in 30 mL for Ag-NPs), which approximate the suspension density of the nitrification samples, were prepared in the same nutrient solutions used in nitrification experiments. Ionic Ag stock solutions were added to assure the concentration of [Ag]total ranging from 5 to 30 mg/L in 30-mL Nalgene polycarbonate centrifuge tubes. Similarly, Ag-NP stock solutions were added to assure [Ag] total ranging from 10 to 500 mg/L. For the Ag+ experiment, centrifuge tubes were rotated on an end-over-end shaker at 30 rpm for 48 h. Samples were then passed through 0.2-μm PVDF filters and tested for total trace metal (e.g., Ag) by way of inductively coupled plasma-atomic emission spectroscopy (ICP-AES). For the Ag-NP experiments, nanoparticles were added to soils suspended in the same nutrient solution and shaken end-over-end at 30 rpm for 24 h. The samples were then centrifuged at 7,000×g for 8 min, and the supernatants were filtered by centrifugation using microfilter centrifuge tubes (washed with copper nitrate to prevent reaction of Ag-NP with the filter) at 3,750 g for 20 minutes. Five milliliters of 5 M nitric acid was then added to the aliquot for Ag-NP digestion for 1 week. After digestion, the aliquot was analyzed for total metals using ICP-AES. To facilitate data comparison, the distribution coefficient value (K d) was estimated using Eq. (2):
where A i = concentration of adsorbate on the solid at equilibrium (mg/g), and C i = total dissolved adsorbate concentration remaining in solution at equilibrium (mg/mL).
Ag-NP Dissolution Experiments
Ag-NP dissolution is important in that “dissolved Ag+” may exhibit different toxicity mechanisms to bacteria than nano-AG particles. For this reason, each type of Ag-NP listed in Table 1 was suspended in aqueous solution at high concentrations (500 mg/L for uAg50 and 250 mg/L for pAg15). The dissolution experiments were performed under oxic conditions. The pH of the solution was maintained at pH 7.2 using 2-(N morpholino)ethanesulfonic acid sodium salt buffer. Samples were then shaken end-over-end at 30 rpm at sample periods ranging from 1 to 5 days. Each day, one sample was filtered by centrifugation using microfilter centrifuge tubes (washed with copper nitrate) at 3,750 g for 20 minutes to separate Ag-NPs from suspension. The resulting aliquot solutions were then analyzed for total Ag concentration using ICP-AES.
Results and Discussion
Ag+ and Nano-Ag Sorption Experiments
Partitioning coefficient (K d) values are listed in Table 2. When the initial concentration (C i ) is compared with the equilibrium concentration (C eq) in Table 2, it is clear that nearly 100 % of Ag-NP and Ag+ are sorbed to soils suggesting the high affinity of Ag+ and Ag-NPs in these soils. The strong interactions of Ag-NPs in soils and sediments are consistent with previous reports (Cornelis et al. 2012; Park et al. 2013; Schlich et al. 2013; VandeVoort and Arai 2012).
The K d values for Ag-NPs range from 93,137 to 418,088 mL/g for uncoated Ag-NPs and from 3,696 to 7,726 mL/g for PVP-coated Ag-NPs. The results showed a lower partitioning of PVP-coated Ag-NPs (e.g., K d = 7,726.95) into soil particles compared with uncoated Ag-NPs (e.g., K d = 418,088.47 mL/g). This was expected because uncharged PVP capping agent is known to increase the stability of nanoparticles (dispersed) (Huynh and Chen 2011). This might induce the steric repulsion transport process in soil media. Park et al. reported that nearly 100 % of citrate-capped Ag-NPs was retained in sediments and/or loamy soil (K d = 100,000 and 76,433 mL/g, respectively) (Park et al. 2013). Furthermore, K d values (approximately 125,000 mL/g) that derive from the 0.1 % PVP-coated Ag-NP soil system in the work by Cornelis et al. (2012) is comparable with our K d of 93,137 mL/g of uncoated Ag-NPs.
Sorption of Ag+ to Taccoa entisoils yields a K d value (mL/g) of approximately 779–1,771 (Table 2), which is much less than the K d of Ag-NPs, suggesting the strong affinity of Ag-NPs to soils. These K d values are similar to those (average K d 1,791 mL/g with initial Ag concentration of 1.10 mg/kg) reported in 16 types of Australian soils (Cornelis et al. 2012). Many argued that soil organic matter and/or total carbon content is the key in assessing the affinity of Ag+ (soft metal) in soils due to the metal-chelating complexes with thiol functional groups (soft base) in humic substances (Akcay et al. 2003; Bell and Kramer 1999; Cornelis et al. 2012; Jacobson et al. 2005). Besides organic matter, other soil properties, such as pH and ionic strength and clay mineral content (Fe and Al oxides), have also been discussed as playing vital roles in determining the fate and bioavailability of Ag+ and Ag-NPs in natural soils (Cornelis et al. 2010, 2012).
Ag-NP Dissolution Experiments
Dissolution data of uncoated (50-nm) and PVP-coated (15-nm) Ag-NPs in the nutrient solution are showed in Fig. 3c, d. PVP-coated Ag-NPs released more Ag+ than did uncoated Ag-NPs during the dissolution experiments. Although PVP-coated Ag-NPs released 7.9–8.7 mg/L, uncoated Ag-NPs released <0.55 mg/L in 5 days. In the first 24 h, PVP-coated NPs had the highest release of [Ag]total at 3.49 % (8.72 mg/L) compared with uncoated Ag-NP (0.1 % with 0.525 mg/L). The difference was expected because coating agents (such as PVP) have been proven to stabilize Ag-NP against aggregation and increase its dispersion (El Badawy et al. 2010b; Lin et al. 2012). The high surface area of dispersed NPs facilitates dissolution. Furthermore, aerobic condition also plays an important role in Ag-NP dissolution. Oxidative dissolution of elemental Ag(0), such as Ag(0)-NPs, has been frequently documented in the literature. In a study performed recently by Liu and Hurt (2010), the kinetic dissolution of citrate-stabilized Ag-NP was found to be dependent on different environmental conditions, such as dissolved oxygen concentration, pH, temperature, and natural organic matter, when tested under aqueous conditions. In their findings, maximum dissolved Ag+ (approximately 0.3 mg/L) was recorded under oxic conditions in the first 24 h, which correlate well with our findings, whereas no detectable level of dissolved Ag+ was observed under anoxic conditions (Liu and Hurt 2010).
Interestingly, the release of Ag+ from these Ag-NPs decreased with increasing time (days). Although one can suggest the resorption of dissolved Ag+ onto Ag-NPs, kinetically limited ligand sorption on NPs is likely to retard the dissolution process. The major components of nutrient solutions are phosphate and sulfate in the dissolution experiments. It is likely that sorption of these anions is suppressing Ag-NP dissolution. Choi et al. (2009) previously reported the effects of anions on the oxidative dissolution of Ag-NPs. Anions (SO −24 , Cl−, ethylene diamine tetraacetic acid, PO4 3−, S2−) effectively decreased the dissolution of Ag-NPs in oxic aquatic systems (Choi et al. 2009).
Batch Nitrification Kinetic Experiments
The results from the nitrification kinetic experiments are listed in Table 3 and shown Figs. 1, 2, and 3. Graphs present the different treatment conditions with their different concentrations, whereas the table lists and compare all treatments with that of the control. For each treatment, the kinetic rate (V max value) of NO3 − production was calculated from linear fits. The V max values were then compared statistically with control A (buffered) using Student t test. Based on these comparisons, 90 % of the treatments were significantly different from the control (p < 0.01).

Nitrification potential (rate of nitrification) in Taccoa entisol amended with a nutrient solution (1 mM ammonium phosphate monobasic and 0.25 mM ammonium sulfate). a Buffered control system. b Unbuffered control system. c Sixty-five mg/L of NaN3. d Five hundred mg/L of NaN3. e Ag+ = 1 mg/L. f Ag+ = 10 mg/L

Effect of uncoated 50-nm Ag-NPs on the soil-nitrification potential (rate of nitrification) in Taccoa entisol soil amended with a buffered nutrient solution (1 mM ammonium phosphate monobasic and 0.25 mM ammonium sulfate). a [Ag]total = 1 mg/L. b [Ag]total = 10 mg/L. c [Ag]total = 100 mg/L. d [Ag]total = 300 mg/L

Effect of PVP-coated 15-nm Ag-NPs on the nitrification potential (rate of nitrification) in Taccoa entisol soil amended with a buffered nutrient solution (1 mM ammonium phosphate monobasic and 0.25 mM ammonium sulfate). a [Ag]total = 1 mg/L. b [Ag]total = 10 mg/L. Dissolution of Ag NPs in a buffered nutrient solution (some error bars are hidden within symbols). c Uncoated 50-nm Ag-NPs. d PVP-coated 15-nm Ag-NPs
The goodness of linear fit in the present study was variable in the different treatments (NaN3, Ag+, and Ag-NP). This variability in data points might be attributed to (1) the heterogeneous distribution of background nitrate in soils and (2) the variable changes in the microbial community. It is well known that nitrate is mobile in soils and can be readily leached in terrestrial environments. However, some researchers have reported a spatial variability and nonuniformity in nitrate content within the same soils (Allaire-Leung et al. 2001; Linkohr et al. 2000; Robertson et al. 1988). It should also be noted that the kinetics of the microbial community changes could potentially contribute to the variability in the 12 replicates. Carrero-Colón et al. (2006) reported that temporal changes in nutrient availability, growth rate, and substrate affinity can alter the microbial community structure, whereas others showed a shift in microbial community activity and structure in soils that were incubated with N2 gas (Øvreås et al. 1998).
Control Systems
The kinetic rate of the buffered (pH 7.2 ± 0.2) control treatment (Fig. 1a) showed a high rate of NO3 − production over time (1.593 mg NO3 − kg−1 h−1), whereas a low production rate (0.97 mg NO3 − kg−1 h−1) was observed with unbuffered (pH 4.76) control treatment (Fig. 1b). This was expected because the nitrification rate has been recorded to be decreased in acidic soils (Allison and Prosser 1993; De Boer and Kowalchuk 2001). Interestingly, some research evidence recorded a greater nitrate production at pH 4 than that at pH 6 in acidic soils (Martikainen and De Boer 1993). In contrast, it has been stated that nitrifying bacteria require much greater pH values with laboratory-grown pure cultures (Jiang and Bakken 1999) with optimal pH being 8.1 for Nitrosomonas and of 7.9 for Nitrobacter (Grunditz and Dalhammar 2001). For example, no growth was recorded under pH 7 for N. europaea when tested in liquid batch culture (Allison and Prosser 1993). Moreover, growth of nitrifying bacteria has been stated to be much faster under shaken soil suspension (generation time 21.7 h) compared with other techniques such as static incubation of moist soil (generation time 129–140 h) (Belser 1979), which explains the fast growth of nitrifying bacteria in the present study.
One should question whether the rate of nitrification in (un)buffered systems represents the soil-nitrification processes. It could be contributed by the native nitrate before the experiments. To evaluate the nitrification process in (un)buffered systems, sodium azide (antimicrobial agent) was used to monitor the release of background nitrate in soils. Addition of NaN3 (65 and 500 mg/L) (Fig. 2a, b, respectively) significantly decreased nitrate production, especially with the 500 mg/L dose (0.014 mg NO3 − kg−1 h−1). This treatment with 500 mg/L NaN3 exhibited the lowest rate of nitrate production among all other treatments. Because the release of nitrate in (un)buffered systems is substantially greater (0.974 mg NO3 − kg−1 h−1) than that in sodium azide systems, it is reasonable to say that the rate of nitrification observed in (un)buffered systems should represent biotically controlled nitrate release (i.e., nitrification).
Effects of Ag+
In Ag+ treatments, kinetic rates of 1 and 10 mg/L Ag+ (as Ag2SO4) (Fig. 1e, f, respectively) were both statistically different from the rate of control (p < 0.01). For 1 mg/L Ag+, a greater nitrification rate (2.05 mg NO3 − kg−1 h−1) than that of the control was observed. With the addition of 10 mg/L Ag+, the nitrate production rate was decreased by approximately 60 % compared with that observed at a dose of 1 mg/L.
Ag+ is known to exhibit toxicity mainly by reacting with amino acids in proteins, more specifically with the thiol groups (CySH and glutathione) (Russell and Hugo 1994; Liau et al. 1997). However, other mechanisms—such as inhibition of several oxidative enzymes, surface binding and damage to membrane, suppression of DNA replication abilities, and generation of reactive oxygen species (ROS)—have also been documented (Clement and Jarrett 1994; Feng et al. 2000; Park et al. 2009). Studies conducted on nitrifying bacteria also showed similar mechanisms of toxicity. In a study performed by Radniecki et al. (2011) in a broth medium, 0.08 mg/L Ag+ (as AgNO3) decreased nitrification activity (N. europaea) by 50 %. In their study, modes of action were mainly caused by inhibition of both ammonia mono-oxygenase-specific oxygen uptake rates (AMO-SOURs) and hydroxylamine oxidoreductase-specific oxygen uptake rates (HAO-SOURs) and destabilization of the outer membrane of the bacterial cells. As they reported, damage to the outer membrane was due to the decrease in intracellular K+ levels (Radniecki et al. 2011). In a recent study, the toxicity to AMO in N. europaea was found to be more related to specific genes. Yang et al. (2013) found that 2.5 μg/L of Ag+ (as AgNO3) upregulated AMO genes amoA1 and amoC2 by 2.1 by 3.3-fold (Yang et al. 2013). Based on the literature review, it is likely that an Ag+-driven antimicrobial effect is suppressing the nitrification process in soils (Fig. 3a, b).
Effects of Ag-NPs
With the addition of uncoated 50-nm Ag-NPs, k values of 1, 10, 100, and 300 mg/L (Fig. 2a–d, respectively) displayed an inverse relationship with the [Ag]total added concentrations. The treatment at the lower concentration of uncoated Ag-NPs (1 mg/L) was the only one that did not show any difference from the control systems (p > 0.01 with 1.35 mg NO3 − kg−1 h−1). In other treatments with uncoated Ag-NPs (10, 100, and 300 mg/L), the rates of nitrate production decreased (1.13, 0.73, and 0.16 mg NO3 − kg−1 h−1, respectively) with increasing Ag-NP concentrations. In contrast, treatments with PVP-coated 15-nm Ag-NPs (Fig. 3a, b) at both 1 and 10 mg/L exhibited a greater toxicity to nitrifying bacteria (0.89 and 0.057 mg NO3 − kg−1 h−1, respectively) comparing with uncoated Ag-NP treatments. To explain the inhibition effect, several toxicity mechanisms are discussed.
Several studies have showed that toxicity of Ag-NPs is mostly caused by the release of Ag+ by way of dissolution, whereas other mechanisms, such as destabilization of the outer membrane and decrease of intracellular adenosine triphosphate (ATP) levels, were also associated (Lok et al. 2006, 2007; Jin et al. 2010). Lok et al. (2007) showed that the antibacterial activities of Ag-NPs are more dependent on oxidized surfaces, which are more present in well-dispersed suspensions. El Badawy et al. (2010a) showed that Ag-NP toxicity to Bacillus species was surface charge-dependent with the primary mechanism of damaging cellular membrane. In their study, four types of differently charged Ag-NPs were used: uncoated, PVP-coated, citrate-coated, and branched polyethyleneimine Ag-NP (El Badawy et al. 2010a).
However, nitrifying bacteria were not among the tested bacterial strains in the above-mentioned studies, which seem to be slightly different in the perspective of mechanisms of toxicity. Choi and Hu (2008, 2009) reported that nitrification inhibition by Ag-NP, under conditions of a continuously stirred tank reactor media, is dependent on the generation of ROS and particle size. Moreover, they found that 1 mg/L of Ag-NPs coated with polyvinyl alcohol (PVA) significantly inhibited the nitrification process, whereas no disruption of cell membrane integrity was observed under the same concentration (Choi and Hu 2008, 2009). In other studies, toxicity to pure culture of N. europaea found to be related to both the release of dissolved Ag and the impact on important functional proteins, such as ATP synthase, AMO, and HAO (Yuan et al. 2012; Yang et al. 2013). Although the reaction conditions in these studies are different from those of our adsorbent system, it is reasonable to assume that a similar mode of toxicity mechanism can be present to soil-nitrifying bacteria.
Conclusion
This study showed the perturbed soil-nitrification kinetics by Ag+ and Ag-NPs oxic conditions. It is clear that the Ag-based compounds used this study had some inhibitory effect to the soil-nitrification process and that the observed toxicity to the nitrification process was dependent on the concentration and the chemical species. At 1 mg/L of [Ag]total, NPs were far more toxic than Ag+. This difference in toxicity is caused by Ag+-complexation processes with inorganic and organic soil components. In particular, soft basic ligands in soils (e.g., thiol functional groups of organic matter) likely chelated with 1 mg/L of Ag+, thus effectively decreasing the toxicity of Ag+ in soils, whereas Ag(0)NPs do not as readily complex with soft ligands in soils. Within NPs, PVP-coated 15-nm was far more toxic than uncoated 50-nm Ag-NPs. PVP-capped Ag-NPs were highly dispersed and released more Ag+ than uncapped NPs as was evident in the dissolution experiments. The reactivity of PVP-capped Ag-NPs induced greater toxicity to nitrifying bacteria. Interestingly, at 10 mg/L of [Ag]total, PVP-coated NPs were most effective in suppressing the nitrification process than Ag+ in the aerobic system. The oxidative dissolution is the likely cause of Ag-NP toxicity. However there is no straightforward explanation for the decreased toxicity in Ag+ under the same concentration. Although the exact mode of PVP-coated Ag-NP toxicity mechanism is not known, the interactions of Ag-NPs with soils might hold the key to understand the pronounced NP toxicity to bacteria in the adsorbent system.
When the dose–response relationship of Ag+/Ag-NPs to bacteria was compared with the literature values, the toxicity of Ag+ and nano-Ag in this study seems to be much lower than that observed in laboratory-pure culture media (Choi and Hu 2008, 2009; Radniecki et al. 2011). According to these studies, 0.08 mg/L of Ag+ and 1 mg/L of PVA-coated Ag-NP decreased the nitrification process by 50 and 86 %, respectively. Compared with our soil-nitrification study, the toxicity of 1 mg/L of Ag+ was not statistically different from that of the control. This difference in the dose–response relationship is likely caused by the partitioning processes of Ag+ and Ag-NPs in soils. The strong interactions of NPs with soil surfaces, as well as the complex secondary reactions of Ag-NPs (e.g., dissolution and ligand complexation) with inorganic and organic soil components, could potentially suppress toxicity to soil microorganisms.
In re-evaluating the annual loading level of sludge amendment to agricultural soils, the potential pertubation to microbially mediated N cycles, as well as to the microbial community structure, might be important to consider if a similar total amount of Ag is present in sludge. Delayed nitrification process could have an impact in nitrogen-fertilizer management in sludge-amended agricultural soils. The results of this study shed light on the toxicity of Ag-NPs to nitrifying bacteria in heterogeneous adsorbent systems, i.e., soils. To advance our knowledge in the risk assessment of engineered Ag-NPs to the ecosystem, greater research effort is needed to understand the role of environmental media (e.g., soils, sediments, suspended solids) that alter the antimicrobial effect of Ag as Ag+ and Ag-NPs to beneficial microorganisms (e.g., nitrifying bacteria) in the terrestrial environment.
References
Akcay H, Oguz A, Karapire C (2003) Study of heavy metal pollution and speciation in Buyak Menderes and Gediz river sediments. Water Res 374:813–822
Allaire-Leung SE, Wu L, Mitchell JP, Sanden BL (2001) Nitrate leaching and soil nitrate content as affected by irrigation uniformity in a carrot field. Agric Water Manag 48(1):37–50
Allison SM, Prosser JI (1993) Ammonia oxidation at low pH by attached populations of nitrifying bacteria. Soil Biol Biochem 25(7):935–941
Azam A, Arham SA, Mohammad O, Mohammad SK, Sami SH, Adnan M (2012) Antimicrobial activity of metal oxide nanoparticles against gram-positive and gram-negative bacteria: a comparative study. Int J Nanomedicine 7:6003
Bell RA, Kramer JR (1999) Structural chemistry and geochemistry of silver–sulfur compounds: critical review. Environ Toxicol Chem 18(1):9–22
Belser LW (1979) Population ecology of nitrifying bacteria. Annu Rev Microbiol 33(1):309–333
Boast CW, Robertson TM (1982) A micro-lysimeter method for determining evaporation from a bare soil: description and laboratory evaluation. Soil Sci Soc Am J 46:689–696
Carrero-Colón M, Nakatsu CH, Konopka A (2006) Effect of nutrient periodicity on microbial community dynamics. Appl Environ Microbiol 72(5):3175–3183
Cataldo DA, Haroon M, Schrader LE, Youngs VE (1975) Rapid colorimetric determination of nitrate in plant tissue by nitration of salicylic acid 1. Commun Soil Sci Plant Anal 6(1):71–80
Choi O, Hu Z (2008) Size dependent and reactive oxygen species related nanosilver toxicity to nitrifying bacteria. Environ Sci Technol 42(12):4583–4588
Choi O, Hu Z (2009) Nitrification inhibition by silver nanoparticles. Water Sci Technol 59(9):1699–1702
Choi O, Clevenger TE, Deng B, Surampalli RY, Ross L Jr, Hu Z (2009) Role of sulfide and ligand strength in controlling nanosilver toxicity. Water Res 43(7):1879–1886
Clement JL, Jarrett PS (1994) Antibacterial silver. Met Based Drugs 1(5–6):467–482
Cornelis G, Kirby JK, Beak D, Chittleborough D, McLaughlin MJ (2010) A method for determining the partitioning of manufactured silver and cerium oxide nanoparticles in soil environments. Environ Chem 7:98–308
Cornelis G, Doolette C, Thomas M, McLaughlin MJ (2012) Retention and dissolution of engineered silver nanoparticles in natural soils. Soil Sci Soc Am J 76(3):891–902
De Boer W, Kowalchuk GA (2001) Nitrification in acid soils: micro-organisms and mechanisms. Soil Biol Biochem 33(7):853–866
Drexler KE (2004) Nanotechnology: from Feynman to funding. Bull Sci Technol Soc 24(1):1–27
El Badawy AM, Silva RG, Morris B, Scheckel KG, Suidan MT, Tolaymat TM (2010a) Surface charge-dependent toxicity of silver nanoparticles. Environ Sci Technol 45(1):283–287
El Badawy AM, Luxton TP, Silvar BG, Scheckel K, Suidat MT, Tolaymat TM (2010b) Impact of environmental conditions (pH, ionic strength, and electrolyte type) on the surface charge and aggregation of silver nanoparticles suspensions. Environ Sci Technol 44(4):1260–1266
Feng QL, Wu J, Chen GQ, Cui FZ, Kim TN, Kim JO (2000) A mechanistic study of the antibacterial effect of silver ions on Escherichia coli and Staphylococcus aureus. J Biomed Mater Res 52(4):662–668
Gottschalk F, Sonderer T, Ort C, Scholz RW, Nowack B (2009) Modeled environmental concentrations of engineered nanomaterials (TiO2, ZnO, Ag, CNT, fullerenes) for different regions. Environ Sci Technol 43:9216–9222
Grunditz C, Dalhammar G (2001) Development of nitrification inhibition assays using pure cultures of nitrosomonas and nitrobacter. Water Res 35(2):433–440
Guzman KA, Finnegan MP, Banfield JF (2006) Influence of surface potential on aggregation and transport of titania nanoparticles. Environ Sci Technol 40(24):7688–7693
Hart SC, Stark JM, Davidson EA, Firestone MK (1994) Nitrogen mineralization, immobilization, and nitrification. Methods of soil analysis. Methods Soil Anal 2:985–1018
Huynh KA, Chen KL (2011) Aggregation kinetics of citrate and polyvinylpyrrolidone coated silver nanoparticles in monovalent and divalent electrolyte solutions. Environ Sci Technol 45(13):5564–5571
Jacobson AR, McBride MB, Baveye P, Steenhuis T (2005) Environmental factors determining the trace-level sorption of silver and thallium to soils. Sci Total Environ 345(1):191–205
Jiang QQ, Bakken LR (1999) Comparison of nitrosospira strains isolated from terrestrial environments. FEMS Microbiol Ecol 30(2):171–186
Jin X et al (2010) High-throughput screening of silver nanoparticle stability and bacterial inactivation in aquatic media: influence of specific ions. Environ Sci Technol 44(19):7321–7328
Johansen A, Pedersen AL, Jensen KA, Karlson U, Hansen BM, Scott-Fordsmand JJ et al (2008) Effects of C60 fullerene nanoparticles on soil bacteria and protozoans. Environ Toxicol Chem 27(9):1895–1903
Killham K (1987) A new perfusion system for the measurement and characterization of potential rates of soil nitrification. Plant Soil 97(2):267–272
Klaine SJ, Alvarez PJJ, Batley GE, Fernandes TF, Handy RD, Lyon DY et al (2008) Nanomaterials in the environment: behavior, fate, bioavailability, and effects. Environ Toxicol Chem 27(9):1825–1851
Li M, Zhu L, Lin D (2011) Toxicity of ZnO nanoparticles to Escherichia coli: mechanism and the influence of medium components. Environ Sci Technol 45(5):1977–1983
Liau SY, Read DC, Pugh WJ, Furr JR, Russell AD (1997) Interaction of silver nitrate with readily identifiable groups: relationship to the antibacterial action of silver ions. Lett Appl Microbiol 25(4):279–283
Lin S, Cheng Y, Liu J, Wiesner MR (2012) Polymeric coatings on silver nanoparticles hinder autoaggregation but enhance attachment to uncoated surfaces. Langmuir 28(9):4178–4186
Linkohr BI, Williamson LC, Fitter AH, Leser HMO (2000) Nitrate and phosphate availability and distribution have different effects on roots system architecture of Arabidopsis. Plant J 29(6):751–760
Liu J, Hurt RH (2010) Ion release kinetics and particle persistence in aqueous nano-silver colloids. Environ Sci Technol 44(6):2169–2175
Lok C-N, Ho CM, Chen R, He Q-Y, Yu W-Y, Sun H et al (2006) Proteomic analysis of the mode of antibacterial action of silver nanoparticles. J Proteome Res 5(4):916–924
Lok C-N, Ho CM, Chen R, He QY, Yu WY, Sun H et al (2007) Silver nanoparticles: partial oxidation and antibacterial activities. J Biol Inorg Chem 12(4):527–534
Lyon DY, Fortner JD, Sayes CM, Colvin VL, Hughes JB (2005) Bacterial cell association and antimicrobial activity of a C60 water suspension. Environ Toxicol Chem 24(11):1162–2757
Martikainen PJ, de Boer W (1993) Nitrous oxide production and nitrification in acidic soil from a Dutch coniferous forest. Soil Biol Biochem 25(3):343–347
Nel A, Xia T, Madler L, Li N (2006) Toxic potential of materials at the nanolevel. Science 311(5761):622–627
Nowack B, Bucheli TD (2007) Occurrence, behavior and effects of nanoparticles in the environment. Environ Pollut 150(1):5–22
Øvreås L, Jensen S, Daae FL, Torsvik V (1998) Microbial community changes in a perturbed agricultural soil investigated by molecular and physiological approaches. Appl Environ Microbiol 64(7):2739–2742
Park H-J, Kim JY, Kim J, Lee J-H, Hahn J-S, Gu MB et al (2009) Silver–ion-mediated reactive oxygen species generation affecting bactericidal activity. Water Res 43(4):1027–1032
Park S, Kim EH, Eo M, Song HD, Lee S, Roh J-K et al (2013) Effect of dispersion stability on the deposition of citrate-capped silver nanoparticles in natural soils. J Nanosci Nanotechnol 13(3):2224–2229
Pelletier DA, Suresh AK, Holton GA, McKeown CK, Wang W, Gu B et al (2010) Effects of engineered cerium oxide nanoparticles on bacterial growth and viability. Appl Environ Microbiol 76(24):7981–7989
Qi L, Xu Z, Jiang X, Hu C, Zou X (2004) Preparation and antibacterial activity of chitosan nanoparticles. Carbohydr Res 339(16):2693–2700
Radniecki TS, Stankus DP, Neigh A, Nason JA, Semprini L (2011) Influence of liberated silver from silver nanoparticles on nitrification inhibition of Nitrosomonas europaea. Chemosphere 85:43–49
Robertson GP, Vitousek PM (1981) Nitrification in the course of ecological succession. Bioscience 31:141–144
Robertson GP, Huston MA, Evans FC, Tiedje JM (1988) Spatial variability in a successional plant community: patterns of nitrogen availability. Ecology 69:1517–1524
Russell AD, Hugo WB (1994) Antimicrobial activity and action of silver. Prog Med Chem 31:351–370
Schlich K, Klawonn T, Terytze Y, Hund-Rinke K (2013) Hazard assessment of a silver nanoparticle in soil applied via sewage sludge. Environ Sci Eur 25(1):17
Skipper HD, Westerman DT (1973) Comparative effects of propylene oxide, sodium azide and autoclaveing on selected soil properties. Soil Biol Biochem 5:409–414
Sotiriou GA, Pratsinis SE (2010) Antibacterial activity of nanosilver ions and particles. Environ Sci Technol 44(14):5649–5654
Suresh AK, Pelletier DA, Doktycz MJ (2013) Relating nanomaterial properties and microbial toxicity. Nanoscale 5(2):463–474
Tong Z, Bishoff M, Niles L, Applegate B, Turco RF (2007) Impact of fullerene (C60) on a soil microbial community. Environ Sci Technol 41(8):2985–2991
VandeVoort AR, Arai Y (2012) Effect of silver nanoparticles on soil denitrification kinetics. Ind Biotechnol 8(6):358–364
Wiesner MR, Lowry GV, Alvarez PJJ (2006) Assessing the risks of manufactured nanomaterials. Environ Sci Technol 40(14):4336–4345
Yang Y, Wang J, Zongming X, Alvarez PJJ (2013) Impacts of silver nanoparticles on cellular and transcriptional activity of nitrogen-cycling bacteria. Environ Toxicol Chem 32(7):1488–1494
Yuan Z, Li J, Cui L, Xu B, Zhang H, Yu C-P (2012) Interaction of silver nanoparticles with pure nitrifying bacteria. Chemosphere 90(4):1404–1411
Acknowledgments
Portions of this research were supported by Janzan University in Saudi Arabia, Saudi Arabian Cultural Mission, Washington, DC; and the 2011 Agriculture and Food Research Initiative Competitive Grants Program, Nanotechnology for Agriculture and Food Systems (Grant No. 2011-03580).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Masrahi, A., VandeVoort, A.R. & Arai, Y. Effects of Silver Nanoparticle on Soil-Nitrification Processes. Arch Environ Contam Toxicol 66, 504–513 (2014). https://doi.org/10.1007/s00244-013-9994-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00244-013-9994-1