
Abstract
Two different analytical methods for the determination and confirmation of ochratoxin A (OTA) in blood serum, kidney, and liver of pigs have been compared. Sample cleanup was based on liquid–liquid phase extraction. The detection of OTA was accomplished with high-performance liquid chromatography (HPLC) combined either with fluorescence detection (FLD) or electrospray ionization (ESI+) tandem mass spectrometry (MS/MS). The comparative method of evaluation was based on the investigation of 90 samples of blood serum, kidney, and liver per animal originating from different regions of Serbia. The analytical results are discussed in view of the respective method validation data and the corresponding experimental protocols. In general, analytical data obtained with liquid chromatography (LC)-MS/MS detection offered comparable good results in the sub-ppb concentration level indicating that the electrospray tandem mass spectrometric (LC–MS/MS) method was more selective and sensitive for the analysis and confirmation of OTA in pig tissues than the HPLC method after the methylation of OTA.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Ochratoxin A (OTA) is a mycotoxin produced by several Aspergillus and Penicillium species on different agricultural commodities (Miller and Trenholm 1994; Pittet 2001; Weidenbörner 2001). It is a potent nephrotoxin and hepatotoxin with teratogenic, mutagenic, and immunosuppressive effects. In humans, the consumption of OTA-contaminated food has been connected to the occurrence of Balkan endemic nephropathy (Creppy 1999), a disease characterized by severe kidney damage. In 1993, the International Agency for Research on Cancer (IARC) classified OTA as possibly carcinogenic for humans (group 2B) (IARC (International Agency for Research on Cancer) 1993). OTA occurs in a variety of food commodities of which cereals and cereal products, coffee, beer and wine are the most important sources of intake. Carryover from contaminated feedstuff has resulted in the detection of OTA in porcine tissue (liver, kidneys) and pig blood (Gareis 1999; Milićević et al. 2008) while the mycotoxin is rapidly metabolized in ruminants (Weidenbörner 2001). Dietary exposure results in detectable levels in human serum and reaches significantly high levels in patients showing symptoms of ochratoxicosis (Creppy 1999). As the contamination of food and feed with mycotoxins might present a serious hazard to humans and animals who consume them, many countries have established limits for controlling these biocontaminants (Van Egmond 1995). Therefore, the assessment of this contamination in every food where OTA can presumably be present is very important. A good study of human exposure to OTA, and mycotoxins in general, requires the application of accurate analytical methods.
Over the years, a great deal of research has been done attempting to develop more sensitive, selective, and simple methods for mycotoxin detection. Detection and determination of OTA is performed by immunochemical methods as enzyme-linked immunosorbent assay (ELISA) (Barna-Vetro et al. 1996) and chromatographic procedures such as thin-layer chromatography with densitometry detection (Santos and Vargas 2002) and especially by high-performance liquid chromatography with fluorescence (HPLC-FLD) (Eskola et al. 2001, 2002; Visconti et al. 1999), mass spectrometry (LC–MS) (Richard et al. 1999) or tandem mass spectrometry (LC–MS/MS) detection (Jorgensen 1998). Residue analysis must be simple and rapid to prevent distribution of harmful products and economic enough to allow developing programs for monitoring OTA over a wide number of food samples. Additionally, analytical methods for the determination of mycotoxins must be fully validated according to European specification (EC 2002) if they are to be used for implementation of legislation and for monitoring and risk assessment studies. However, only a few of the methods found in the literature comply with these premises.
The purpose of this work was to develop a confirmative method that is sensitive, reliable, cost-effective, rapid, and adaptable to a routine work in the surveillance of OTA in pig tissues.
Materials and Methods
Chemical and Reagents
Ochratoxin A crystalline material was purchased from Sigma (St. Louis, MO, USA). Stock concentrated solution was prepared in toluene–acetic acid (99:1 v/v) at a final concentration of 1 mg/mL and kept in security conditions at −20°C and wrapped in aluminum foil, because OTA gradually breaks down under ultraviolet (UV) light. The OTA working solution was prepared by diluting the stock solution with toluene–acetic acid (99:1 v/v) to ~10 μg/ml. The actual concentration of OTA was calculated using a UV spectrophotometer set at 333 nm (ε 5550). After suitable dilutions in water–methanol–acetic acid (50:49:1 v/v/v), the working solution was used to prepare the external calibration curve. A working standard OTA for HPLC was prepared daily just before starting the injection of a series of samples. Other reagents were HPLC grade. All other chemicals were reagent grade or chemically pure.
Sample Collection
During a 6-month period, samples of blood, kidney, and liver from each animal were collected from healthy slaughtered pigs (n = 90) originating from three different regions of Serbia with a significant swine industry. Slaughtered pigs were randomly sampled in the slaughterhouse during meat inspection. Serum samples were collected from each studied farm and from the liver and kidneys of corresponding animals. About 50 ml blood/pig was sampled when slaughtered pigs were bled by jugular puncture. Blood samples remained at room temperature for 24 h to allow clotting to occur and were then centrifuged at 3000g for 20 min. Serum was decanted and stored at −20°C prior to analysis. About 100 g of liver and whole kidney were sampled from each pig. The whole sample was homogenized and stored at −20°C before analysis. No preservatives were added.
Spiking
Pig tissues (blood, kidney, and liver) were spiked by consecutively adding the appropriate amounts of the five combined working solutions. The samples were subsequently stored for 24 h at room temperature (~20°C) to allow evaporation of the solvent and to establish equilibration between the analytes and the matrix.
Extraction and Cleanup for Ochratoxin Analyses from Serum
Serum (0.8 ml) was extracted according to the method of Curtui and Gareis (2001) with 15% trichloroacetic acid (0.2 ml) and dichloromethane (1 ml) by vigorous vortexing for 30 s in a 2-ml safe-lock polypropylene conical-bottom centrifuge tube. The mixture was allowed to stand for 24 h at room temperature and then centrifuged at 14,000g for 10 min. The lower dichloromethane phase was carefully withdrawn by a Pasteur pipette and transferred to a 1.5-ml safe-lock polypropylene conical-bottom centrifuge tube. The acidic phase and the compact precipitate layer formed between the two phases were reextracted with dichloromethane (0.5 ml) for 30 s on a vortex mixer and then centrifuged for 5 min at 14,000g. The pooled dichloromethane extract was evaporated to dryness at 40°C under a gentle nitrogen flow. The remaining residue was dissolved in methanol (80 μl) and transferred to a 300-μl HPLC vial.
Extraction and Cleanup for Ochratoxin Analyses from the Kidney and Liver
Kidney and liver analyses were performed by the method of Matrella et al. (2006), which briefly includes a double extraction with acidic ethyl acetate. The organic phase was removed and extracted with 0.5 M NaHCO3, pH 8.4. The aqueous extract was acidified to pH 2.5 with 7 M H3PO4. OTA was finally back extracted into ethyl acetate (3 ml). The organic phase was evaporated to dryness under N2 steam and reconstituted in 150-μl mobile phases and a 20-μl aliquot was injected.
Chromatographic Conditions (HPLC)
An aliquot of 20 μl for serum samples and 50 μl for kidney and liver samples were injected onto a Waters Symmetry Shield RP (reverse phase) 18, HPLC column (length and inner diameter, 150 × 4.6 mm; particle size, 5 μm) on a Waters Alliance HPLC system. The column was eluted with 4% acetic acid and acetonitrile (32:68 v/v) at 25°C and a flow rate of 1 ml/min. Measurements were performed by fluorescence detection at wavelengths of 334 nm (excitation) and 460 nm (emission) gains 10. A volume of 10 μl was injected for the standards and 20 μl was injected for the samples. For more accuracy, 40 μl were reinjected in the case of the samples with an amount of OTA near the detection limit.
Confirmation Procedures
Extraction and cleanup for ochratoxin analyses from serum, liver, and kidney samples were performed by the methods described earlier. Sample extracts were evaporated to dryness under a gentle stream of nitrogen and stored at −18°C prior to analysis.
HPLC-FLD After Methylation of OTA
The methyl esters of OTA were prepared by adding 50 mL methanol containing 14% boron trifluoride (Sigma) to the evaporated sample extracts and by keeping them at 80°C for 15 min (Gareis 1999). Sample extracts were then evaporated to dryness under a gentle stream of nitrogen and stored at −18°C prior to analysis. Under the chromatographic conditions described earlier, the retention times of the methyl esters of OTA were in the range of 3.455 min.
Quantitation for HPLC-FLD
Quantification for the HPLC–FLD method was performed using an external calibration curve from OTA and quantified using the standard addition method with calibration points set at 0.05, 0.1, 0.5, 1, and 3 ng/g.
Confirmation of OTA by LC–MS/MS
The HPLC system was coupled to a MicroMass Quattro Micro triple-quadrupole mass spectrometer (Micromass Ltd, UK) equipped with an electrospray ionization (ESI) probe operating in the positive mode. The multiple reactions monitoring (MRM) mode was used for the identification and quantification of the compounds. Protonated molecular ions (M + H)+ of the analytes were fragmented within the collision cell to their product ions, which were monitored for a dwell time of 0.40 s with an interchannel delay of 0.05 s. Data acquisition, data processing, and instrument control were performed through Microsoft Windows-based Mass Lynx software (Micro mass) on a Pentium Pro (Digital Equipment) computer. The following operating parameters were used: The electrospray capillary was set at 3.2 kV, the cone voltage was 30 V, and the collision energy was 18 eV. The ion source temperature was set at 115°C and the desolvation temperature was 300°C. Nitrogen was used as the nebulizer gas, the cone gas (flow = 700 l/h), and the desolvation gas (flow = 55 l/h), and argon was used as the collision gas. The transitions reactions monitored by LC–MS/MS were as follows: molecular ion (m/z) = 404.1 (M + H)+, quantifying ions (m/z) = 358.5, and confirmatory ion 239.4 of the OTA.
Quantitation for the External Standard Method
Calibration curves were constructed from the responses of each ion monitored plus the total ion current for OTA.
Validation
For the final validation of the method, the whole procedure was carried out at different concentration levels (five levels with a relative concentration of 0.05, 0.1, 0.5, 1, and 3 ng/g, each in triplicate). These experiments included the spiking of blank extracts (also performed in triplicate) on one concentration level after extraction to differentiate between extraction efficiency and matrix-induced signal suppression/enhancement.
Statistical Analysis
The arithmetic mean, standard deviation, coefficient of variation (CV%), and the variance were calculated. The t-test for double-sided paired data was used to determine the significance between treatments.
Results and Discussion
The results from these LC–MS/MS quantitation methods were compared with those from the conventional HPLC-Fl method and are summarized in Tables 1 and 2 and Figs. 1 and 2.

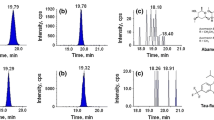
HPLC-FLD (A, B, C) and LC–MS/MS (A1, B1, C1) chromatograms of positive samples of serum, liver, and kidney samples containing 220.8 ng/ml and 14.5 and 52.5 ng/g OTA, respectively

HPLC-FLD chromatograms of positive samples of serum, liver, and kidney containing 220.8 ng/ml and 14.5 and 17 ng/g OTA, respectively, with methylation
Analytical Method Performance
Selectivity
In the case of biomedical samples, the selectivity of the method can be tested by the analysis of six blank samples of different tissues. If no peak is detected at the analyte’s expected retention time, the method would be considered selective (Causon 1997). In this type of sample, where OTA can appear as an endogenous substance, it is important to carry out an additional confirmation of positive samples. For this confirmation, a small amount of OTA was added to the blank sample and it was reanalyzed under same chromatographic conditions. The result was confirmed if the peak appeared at the same retention time as the standard and without shoulders or interfering peaks.
Linearity, Detection Limit, Relative Standard Deviation, and Recovery
Fifteen blank samples were spiked with OTA standards at five different levels (0.05, 0.1, 0.5, 1, and 3 ng OTA/g). From the obtained responses (the area of the mycotoxin divided by the area of the standard), the correlation coefficient (R) and limit of detection (LOD) were calculated. Three replicate calibration standards were chromatographed for each of the five data points included in the curve (equivalent to 0.05, 0.1, 0.5, 1, and 3 ng OTA/g). The parameters of the calibration curve y = 1104.76x − 114.51 (r 2 = 0.999) show a good linear relationship between peak area (y) and equivalent OTA concentration (x).
This LOD was verified by the signal-to-noise (S/N) ratio approach, measuring the chromatographic response of the compound and the chromatographic noise, which should be more than 3 for each compound. The coefficient of variation (CV) was calculated from the analysis of five samples spiked at the same concentration level (the highest spiking level), performed under the same conditions. The overall recoveries of toxins were expressed as the mean recovery obtained of five standards level (n = 5) on two different days. The term recovery in this article means the amount of substance obtained in the last quantification step (after extraction) in relation to the amount of substance added to the material before extraction and is expressed as a percentage.
Method Performance by HPLC-FLD
The LOD (S/N, 3:1) and limit of quantification (LOQ) (S/N, 7:1) were estimated at 0.05, 0.1, 0.5, 1, and 3 ng OTA/ml and were 0.1 and 0.2 ng/ml for serum samples; the recoveries were 86.8% (CV = 9.6%). The LOD (S/N, 3:1) and LOQ (S/N, 7:1) for kidney and liver samples were 0.14 and 0.25 ng/g, respectively, with a 71% (CV = 12%) mean recovery from artificially contaminated samples (n = 5).
Method Performance by LC–MS/MS
The LOD and LOQ for serum were 0.20 and 0.40 ng/ml, respectively, whereas for the kidney and liver they were 0.25 and 0.50, respectively. Recoveries obtained for kidney and liver samples spiked at five levels (n = 5), analyzed with the proposed method, and ranged from 77% to 89% (CV = 13.9%).
Application to the Real Sample
The proposed method of extraction followed by HPLC–FLD and LC–MS/MS has been successfully employed in the study of OTA levels in samples of serum, liver, and kidney of slaughtered swine (n = 90). For tissue samples, the obtained results are shown in Tables 1 and 2. OTA contamination assessment showed that 28 (31%) of the analyzed serum samples were contaminated in a very wide range from 0.22 to 220.8 ng/ml (mean levels = 3.70 ± 23.59 ng/ml). In contrast, our results showed that the frequency of contamination of OTA was higher in the kidney than in the serum and liver (33.3%). The majority of samples (16.6%) contained OTA at low levels (0.01–1 ng/g). The concentrations in 10 samples (11.1%) ranged between 1 and 5 ng/g, whereas OTA in five (5.5%) samples was greater than 5 ng/g (Table 2). In 2.2% of the samples of kidneys, OTA levels were considerably higher and greatly exceeded the permissible levels of this toxin established in Serbia and included those proposed (10 ng/g) by the JECFA (Joint FAO/WHO Expert Committee on Food Additives) (2001). In the present study, OTA was detected in 24 (26.6%) out of 90 liver samples with a much lower mean value (0.63 ng/g) than in kidney samples (1.26 ng/g) (Table 2).
The presence of OTA by HPLC-FLD was detected in 82 (30%) out of 270 tissue samples. The identity of OTA in samples was further confirmed by two analytical methods: HPLC-FLD with methylation and LC–MS/MS. Methylation of OTA in positive samples yielded 11% of the OTA found in tissues samples, whereas confirmation of OTA by LC–MS/MS yielded 95.12% (78) of the OTA found in positive samples. The concentrations in five samples were below of the LOD of LC-MS/MS (0.25 ng/g). The presence of OTA by HPLC-FLD after methylation of OTA was not detected in a positive sample that contained low levels of OTA (<10 ppb) (Fig. 2b, c). The peak width for OTA (<10 ppb) was reduced from expected retention time, resulting in an increased signal to-noise ratio. Thus quantification of OTA in this concentration range was more difficult. The presence of problems due to the coextractive substances in the matrix has been also observed by Wood et al. (1995); thus, a confirmation of the positive analysis is necessary. This problem was resolved with the procedures of confirmation by LC–MS/MS.
The main differences in the performance of the detection techniques can be broken down into three aspects: (1) sensitivity, (2) unambiguous identification of the analytes, and (3) necessary selectivity of sample cleanup to reveal nonbiased results. The excellent detection limits are certainly an advantage of HPLC-FLD. While the LOD for MS/MS detection is 0.2 ng/ml, and 0.25 ng/g which is comparable with an earlier LC–MS/MS investigation of OTA in serum samples, liver and kidney. Triple-quadrupole analyzers display high sensitivity when working in the MRM mode and are thus best suited to obtain the strict maximum residue levels (MRLs) regulated for various toxic compounds in different food matrices. However, the MS method could be made more sensitive by using more samples for the liquid–liquid or solid phase extraction (SPE) procedure. The higher concentration of matrix constituents should not pose any problem because no interfering peaks were observed in the LC–MS/MS chromatograms (Figs. 1 and 2). Furthermore, MS sensitivity achieved in this and the previous study (Lau et al. 2000; Losito et al. 2004; Monaci et al. 2004) can be considered sufficient to measure OTA concentrations in contaminated samples that could pose a threat to human health, taking WHO guidelines into account (WHO/FAO 1995).
Considering the unambiguous analyte identification, the fluorescence properties of OTA might not provide a sufficient degree of certainty. To counteract this disadvantage, confirmation procedures are mainly performed by derivatization of OTA into its methyl ester using BF3/MeOH and the subsequent determination of this compound. This method, however, presents two problems: First, the purity of the reagent employed is frequently uncertain, so it could give rise to interferences itself (Zimmerli and Dick 1995). Second, this method is not efficient for the confirmation of low levels of OTA (Bauer et al. 1984; Miraglia et al. 1995; Valenta 1998). In the HPLC-FLD chromatograms (Fig. 2b), such signals are completely absent. Because of the very narrow chromatographic peak widths obtained, MS scan times had to be reduced to avoid problems with quantification at lower analyte concentrations (sub-ppb range). The confirmation through the hydrolysis of OTA into OTα by the enzyme carboxypeptidase’s A is also employed in some studies and we have already used it for the confirmation of OTA in blood plasma samples (Milićević et al. 2007). This procedure is no longer used in our laboratory because the identification of OTα is problematic, appearing at a low retention time along with other substances in the sample. With MS/MS as a detection system, no further confirmation step is necessary, avoiding any error-prone derivatization steps and any further chromatographic analyses. The high selectivity of MS/MS offers the further advantage that it can be used successfully and very effectively with less selective but less expensive cleanup sorbent materials such as RP-18. On the other hand, analyte selective and time-saving immunoaffinity sorbent materials are needed for the less selective FL, otherwise matrix compounds would interfere with the analyte signal. In conclusion, the proposed method is an inexpensive, rapid, straightforward cleanup procedure for determining OTA in tissues for around 10 samples processed per day.
Conclusions
The relatively high number of publications on the analysis of toxic substances in food samples by LC coupled to MS/MS shows that this technique has become a powerful tool in the quality control of food products and the safeguarding of human health. This article has demonstrated the excellent sensitivity and specificity of the LC–MS/MS method in the quantitative determination and confirmation of OTA in samples of pig tissues at low- and sub-ppb levels. Despite a lower sensitivity of the LC–MS/MS technique, this methodology has several advantages in terms of sample preparation, easy automatization, and unambiguous analyte identification without any further time-consuming and error-prone confirmation steps. From this point of view, it seems that sample preparation and chromatographic separation might be simplified or even eliminated to achieve the highest possible sample throughput as, for example, in drug screening, pharmacokinetics, and other areas of the life sciences. Evaluation of the method with naturally contaminated samples demonstrated that the accuracy of the results is comparable to the most reliable HPLC-FLD procedures and that MS sensitivity is sufficiently high, taking into account proposed OTA tolerance levels within the European Union. Because the detection of OTA in tissues of Serbian slaughtered swine that we analyzed serve as an end-point indicator of the widespread occurrence of OTA, it is important to determine which foods would be contaminated with this potent natural toxin. With the present analytical procedure, it should be possible to carry out surveillance on grains, animal feeds, meat, meat products, and other foodstuffs to determine contamination levels of this toxin. As these techniques improve and become standardized, less controversial and more scientific evaluation of the human risks from exposure to mycotoxins will be possible.
References
Barna-Vetro I, Solti L, Teren J, Gyongyosi A, Szabo E, Wolfling A (1996) Sensitive ELISA test for determination of ochratoxin A. J Agric Food Chem 44(12):4071–4074
Bauer J, Gareis M, Gedek B (1984) Detection and occurrence of ochratoxin A in pigs for slaughter. Berl Muench Tieraerztl Wsch 97:279–283 (in German)
Causon R (1997) Validation of chromatographic methods in biomedical analysis. Viewpoint and discussion. J Chromatogr B 689:175–180
Creppy EE (1999) Human ochratoxicosis. J Toxicol Toxin Rev 18(3–4):277–293
Curtui VG, Gareis M (2001) A simple HPLC method for the determination of the mycotoxins ochratoxin A and B in blood serum of swine. Food Addit Contam 18(7):635–643
EC (2002) Commission Directive (EC) No. 2002/26/EC of 13 March 2002. Laying down the sampling methods and the methods of analysis for the official control of the levels of ochratoxin A in foodstuffs L 75, 38
Eskola M, Parikka P, Rizzo A (2001) Trichothecenes, ochratoxin A and zearalenone contamination and Fusarium infection in Finnish cereal samples in 1998. Food Addit Contam 18(8):707–718
Eskola M, Kokkonen M, Rizzo A (2002) Application of manual and automated Systems for purification of ochratoxin A and zearalenone in cereals with immunoaffinity columns. J Agric Food Chem 50(1):41–47
Gareis M (1999) Vorkommen der Mykotoxine Ochratoxin A und B in deutscher Braugerste und daraus hergestelltem Malz. Arch Lebensmittelhyg 50:83–87
IARC (International Agency for Research on Cancer) (1993) Monographs on the evaluation of carcinogenic risks to humans, vol 56. IARC, Geneva
JECFA (Joint FAO/WHO Expert Committee on Food Additives) (2001) Toxicological evaluation of certain food additives. In Proceedings of the fifty-sixth meeting of JECFA. WHO Food Additives Series. World Health Organization, Geneva, Switzerland
Jorgensen K (1998) Survey of pork, poultry, coffee, beer and pulses for ochratoxin A. Food Addit Contam 15:550–554
Lau BPY, Scott PM, Lewis DA, Kanhere SR (2000) Quantitative determination of ochratoxin A by liquid chromatography/electrospray tandem mass spectrometry. J Mass Spectrom 35:23–32
Losito I, Monaci L, Palmisano F, Tantillo G (2004) Determination of ochratoxin A in meat products by high-performance liquid chromatography coupled to electrospray ionisation sequential mass spectrometry. Rapid Commun Mass Spectrom 18:1965–1971
Matrella R, Monaci L, Milillo MA, Palmisano F, Tantillo MG (2006) Ochratoxin A determination in paired kidneys and muscle samples from swines slaughtered in southern Italy. Food Contam 117:14–117
Milićević D, Jurić V, Mandić M, Đorđević M (2007) The presence of ochratoxin A residue in blood plasma of slaughtered swine. Matica Srpska Proc Nat Sci 113:55–62
Milićević D, Verica Jurić, Stefanović S, Jovanović M, Janković S (2008) Survey of slaughtered pigs for occurrence of ochratoxin A and porcine nephropathy in Serbia. Int J Mol Sci 9:2169–2183
Miller JD, Trenholm HL (eds) (1994) Mycotoxins in grain: compounds other than aflatoxin. Eagan Press, St. Paul, MN
Miraglia M, De-Dominicis A, Brera C, Corneli S, Cava E, Menghetti E, Miraglia E (1995) Ochratoxin A levels in human milk and related food samples: an exposure assessment. Nat Toxins 3:436–444
Monaci L, Tantillo G, Palmisano F (2004) Determination of ochratoxin A in pig tissues by liquid–liquid extraction and clean-up and high-performance liquid chromatography. Anal Bioanal Chem 378:1777–1782
Pittet A (2001) Natural occurrence of mycotoxins in foods and feeds: a decade review. In: de Koe WJ, Samson RA, van Egmond HP, Gilbert J, Sabino M (eds) Mycotoxins and phycotoxins in perspective at the turn of the millennium. Proceedings of the Xth international IUPAC symposium on mycotoxins and phycotoxins. Wageningen Academic Publishers, Wageningen, The Netherlands, pp 153–172
Richard JL, Plattner RD, May J, Liska SL (1999) The occurrence of ochratoxin A in dust collected from a problem household. Mycopathologia 146:99–103
Santos EA, Vargas EA (2002) Immunoaffinity column clean-up and thin layer chromatography for determination of ochratoxin A in green coffee. Food Addit Contam 19(5):447–458
Valenta H (1998) Chromatographic methods for the determination of ochratoxin A in animal and human tissues and fluids. J Chromatogr A 815:75–92
Van Egmond HP (1995) Mycotoxins: regulations, quality assurance and reference materials. Food Addit Contam 12:321–330
Visconti A, Pascale M, Centonze G (1999) Determination of ochratoxin A in wine by means of immunoaffinity column clean-up and high-performance liquid chromatography. J Chromatogr A 864:89–101
Weidenbörner M (2001) Encyclopedia of food mycotoxins. Springer, Berlin
WHO/FAO (1995) Evaluation of certain food additives and contaminants In: The 37th Report of the Joint FAO/WHO Expert Committee on Food Additives. WHO Technical Report Series No. 859. WHO, Geneva
Wood GM, Entwisle AC, Patel S, Hald B, Boenke A (1995) Ochratoxin A in cereals and the BCR-M&T-Projects. Nat Toxins 3:275–279
Zimmerli B, Dick R (1995) Determination of ochratoxin A at the ppt level in human blood, serum, milk and some foodstuffs by high-performance liquid chromatography with enhanced fluorescence detection and immunoaffinity column cleanup methodology and Swiss data. J Chromatogr B 666:85–99
Acknowledgment
This study was funded by the Ministry of Science and Technological development, Belgrade, Serbia (project code TP-20207A). We are grateful to Ministry for their understanding and support to veterinary development.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Milićević, D., Jurić, V., Stefanović, S. et al. Evaluation and Validation of Two Chromatographic Methods (HPLC-Fluorescence and LC–MS/MS) for the Determination and Confirmation of Ochratoxin A in Pig Tissues. Arch Environ Contam Toxicol 58, 1074–1081 (2010). https://doi.org/10.1007/s00244-009-9436-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00244-009-9436-2