
Abstract
The aim of this paper is to give an overview of our current state of knowledge with respect to genotyping for the primary hyperoxalurias and the role of molecular genetics alongside the more traditional biochemical and enzymatic tests for the diagnosis and prognosis of these disorders. The published literature was reviewed to establish the frequency of different mutations and thus the value of testing for a limited number of these mutations in patients with clinical suspicion of primary hyperoxaluria (PH). This approach was compared with whole gene sequencing of the AGXT and GRHPR genes. A limited genetic screen can provide a first line test for PH1 and PH2 in symptomatic patients and can provide a full diagnosis in approximately a third of cases. Molecular genetic analysis is essential for carrier testing and prenatal diagnosis. The value of molecular genetics in prognosis requires a wider evidence base.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The primary hyperoxalurias are inherited disorders of endogenous oxalate overproduction from glyoxylate. Two enzymes, alanine:glyoxylate aminotransferase (AGT) and glyoxylate reductase (GRHPR), which are both present in the hepatocyte are able to metabolise glyoxylate to the less toxic intermediates glycine and glycolate, respectively. However, deficiency of these enzymes leads to the inherited disorders primary hyperoxaluria type 1 (PH1) and type 2 (PH2), respectively (reviewed by [1]).
Diagnosis of these diseases requires a combination of complex biochemical tests including measurement of urine oxalate and related metabolites glycolate and L-glycerate, and measurement of the AGT and GRHPR catalytic activity in a liver biopsy [2]. Urine metabolites do not have a sufficiently high specificity to be regarded as diagnostic but direct the clinician toward the confirmatory test of specific enzyme analyses. With the identification of the genes encoding the enzymes, molecular genetics has been suggested as a less invasive alternative strategy.
The AGXT and GRHPR genes encoding the AGT and GRHPR enzymes have been cloned and mapped to the long arm of chromosomes 2 and the centromeric region of chromosome 9, respectively, and a number of mutations have been described in both genes (see [3] for references). Some of these mutations are relatively common with three, c.33_34insC, c.508A and c.731C, accounting for approximately 45% mutant alleles in PH1 [4] and c.103delG accounting for a similar frequency of mutant alleles in PH2 [5].
This paper reviews the strategies available for the diagnosis of PH and the interaction of biochemical and genetic analyses.
Methods
PubMed was searched with the following terms: primary hyperoxaluria type 1, primary hyperoxaluria type 2, AGXT, GRHPR, alanine:glyoxylate aminotransferase, glyoxylate reductase, hyperoxaluria and mutations.
Results and discussion
The strategy used by our laboratory before 2004 was to measure AGT and GRHPR catalytic and immunoreactivity in liver biopsies from patients with documented hyperoxaluria or end stage renal failure with suspicion of primary hyperoxaluria. This technique has a sensitivity of greater than 95%, but some problems have been encountered in the interpretation of results. First, some patients with PH2 were found to have reduced AGT activity. This is not a problem if GRHPR activity is measured concomitantly but may lead to misdiagnosis as PH1 if AGT alone is measured. Heterozygotes for PH1 may also have reduced AGT activity. Ordinarily this should not cause a diagnostic dilemma as they should be asymptomatic but may be a problem if siblings of patients with PH1, particularly those with equivocal urine oxalate measurements, are tested. We have recently also noticed that AGT activity in the normal range has been found in three cases of PH1 who are homozygous for the missense mutation, Gly170Arg (c.508G>A) which causes mistargeting of AGT to the mitochondrion. This observation may arise from differences in mitochondrial content of a particular biopsy which can not be controlled. It has, however, introduced a requirement to test for this mutation in any symptomatic patient apparently normal for AGT and GRHPR enzyme activity.
Turning our original strategy on its head, molecular genetics was used as the primary test for the diagnosis of primary hyperoxaluria after confirmation of hyperoxaluria. Two approaches were assessed, namely selective mutation testing and sequencing of the entire gene. The results of the first strategy have been recently published [4] and show that by testing for the c.33_34insC, c.508A and c.731C mutations in PH1 and the c.103delG mutation in PH2, a diagnosis (i.e. two mutations) could be made in 34% of cases. In a further third, one mutation only was detected and in the final third, no mutation was identified. The last two groups would therefore require a liver biopsy to confirm the diagnosis but overall, the limited mutation detection approach has a test sensitivity of 62% for PH1 and 40% for PH2. Although the above mutations in the AGXT gene have been seen in all ethnic groups, the c.731C mutation is particularly common in those of Spanish and North African descent [6]. The c.103delG mutation in GRHPR has to date only been identified in Caucasians [5, 7].
Sequencing of the whole gene did not identify all mutations as shown by three studies [8, 9, 10]. It is possible that this is due to mis-calling of heterozygous mutations by automated sequencing software. Restricting sequence analysis to exons may also miss significant changes in promoter or intronic sequences or major deletions. Even when a potential sequence variant is identified, its significance in disease pathology cannot be automatically assumed. For example, there are several known sequence variants (polymorphisms) in AGXT, some of which have no functional effect e.g.Val326Ile [11]. Ideally, the functionality of mutations should always be demonstrated by expression studies, but this is not always possible and the pathological effect may be assumed by family studies only. A database has recently been assembled documenting all published mutations and how their functionality has been demonstrated, whether by expression or family studies (available from the author).
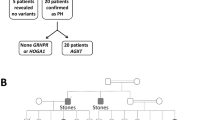
Molecular genetics is undoubtedly the method of choice for prenatal diagnosis and carrier testing. Prenatal diagnosis can use mutation analysis or linkage analysis with intragenic and extragenic polymorphisms [12]. The great advantage of DNA analysis is the ability to perform the diagnosis in the first trimester of pregnancy. We have now performed over 50 prenatals using these techniques. Similar techniques can be used to make the diagnosis of other affected siblings within a family. This technique has the advantage of avoiding a liver biopsy in a patient at high risk of disease or in a sibling with equivocal oxalate results (Fig. 1). However, it makes the assumption that the parents are both carriers of the disease and are not themselves affected. This assumption may be false and there may be three disease alleles tracking within a family such as we described some years ago [13]. It therefore pays to take a careful history from all family members including parents.

Genetic studies on a family with a history of primary hyperoxaluria type 2. N denotes normal allele, M mutant allele (c.403_405+2delAAGT). II3 was initially identified by urine oxalate analysis as affected, but mutation analysis clearly shows that she is heterozygous for the mutation. Subsequent urine oxalate measurements were found to be normal
The role of genotyping in prognosis is a more contentious area, mainly because there is insufficient evidence as yet available. We recently demonstrated that there was little difference in age of onset of disease with any of the common mutations in PH1 [4]. The peak age of onset occurred before the age of 10 years, but some patients, with mutations causing complete ablation of AGT activity, e.g. c.33_34insC, presented as late as 40 years of age, suggesting that other genes or environmental influences were also playing a part in the presentation of disease. Other aspects of prognosis such as genotype relationship to pyridoxine sensitivity are under investigation but studies must be designed carefully to avoid bias.
In conclusion, a limited genetic screen can provide a first line test for PH1 and PH2 and provides a diagnosis in approximately a third of cases. This strategy can limit the requirement for liver biopsy but is dependent on a full work-up being performed on the patient prior to genetic analysis. Molecular genetics continues to be the method of choice for prenatal diagnosis and carrier testing for both PH1 and PH2. The value of genotyping in prognosis needs a wider evidence base and will best be achieved by collaboration of interested clinicians and laboratories.
References
Danpure CJ (2001) Primary hyperoxaluria. In: Scriver C, Beaudet AL, Sly WS, Valle D (eds) The metabolic bases of inherited disease, 8th edn. McGraw-Hill, New York, p 3323
Giafi CF, Rumsby G (1998) Kinetic analysis and tissue distribution of human D-glycerate dehydrogenase/glyoxylate reductase and its relevance to the diagnosis of primary hyperoxaluria type 2. Ann Clin Biochem 35: 104
Danpure CJ, Rumsby G (2004) Molecular aetiology of primary hyperoxaluria and its implications for clinical management. Expert Rev Mol Med 9: 1
Rumsby G, Williams E, Coulter-Mackie MB (2004) Evaluation of mutation screening as a first line test for the diagnosis of the primary hyperoxalurias. Kidney Int 66: 959
Cregeen DP, Williams EL, Hulton SA, Rumsby G (2003) Molecular analysis of the glyoxylate reductase (GRHPR) gene and description of mutations underlying primary hyperoxaluria type 2. Hum Mutat: Mutations in Brief, no. 671 online
Santana A, Salido E, Torres A, Shapiro LJ (2003) Primary hyperoxaluria type 1 in the Canary Islands: a conformational disease due to I244T mutation in the P11L-containing alanine:glyoxylate aminotransferase. Proc Natl Acad Sci U S A 100: 7277
Webster K, Ferree PM, Holmes RP, Cramer SD (2000) Identification of missense, nonsense and deletion mutations in the GRHPR gene in patients with primary hyperoxaluria type II (PH2). Hum Genet 107: 176
Milosevic D, Rinat C, Batinik D, Frishberg Y (2002) Genetic analysis—a diagnostic tool for primary hyperoxaluria type 1. Pediatr Nephrol 17: 896
Van Woerden CS, Groothoff JW, Wanders RJ, Davin J, Wijburg FA (2003) Primary hyperoxaluria type 1 in The Netherlands: prevalence and outcome. Nephrol Dial Transplant 18: 273
Amoroso A, Pirulli D, Florian F, Puzzer D, Boniotto M, Crovella S, Zezlina S, Spano A, Mazzola G, Savoldi S, Ferrettini C, Berutti S, Petrarulo M, Marangella M (2001) AGXT gene mutations and their influence on clinical heterogeneity of type 1 primary hyperoxaluria. J Am Soc Nephrol 12: 2072
Coulter-Mackie MB, Tung A, Henderson HE, Toone JR, Applegarth DA (2003) The AGT gene in Africa: a distinctive minor allele haplotype, a polymorphism (V326I), and a novel PH1 mutation (A112D) in Black Africans. Mol Genet Metab 78: 44
Rumsby G (1998) Experience in prenatal diagnosis of primary hyperoxaluria type 1. J Nephrol 11 [Suppl 1]: 13
Hoppe B, Danpure CJ, Rumsby G, Fryer P, Jennings PR, Blau N, Schubiger G, Neuhaus T, Leumann E (1997) A vertical (pseudodominant) pattern of inheritance in the autosomal recessive disease primary hyperoxaluria type 1: lack of relationship between genotype, enzymic phenotype, and disease severity. Am J Kidney Dis 29: 36
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rumsby, G. An overview of the role of genotyping in the diagnosis of the primary hyperoxalurias. Urol Res 33, 318–320 (2005). https://doi.org/10.1007/s00240-005-0494-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00240-005-0494-2