
Abstract
It has been claimed that synonymous sites in mammals are under selective constraint. Furthermore, in many studies the selective constraint at such sites in primates was claimed to be more stringent than that in rodents. Given the larger effective population sizes in rodents than in primates, the theoretical expectation is that selection in rodents would be more effective than that in primates. To resolve this contradiction between expectations and observations, we used processed pseudogenes as a model for strict neutral evolution, and estimated selective constraint on synonymous sites using the rate of substitution at pseudosynonymous and pseudononsynonymous sites in pseudogenes as the neutral expectation. After controlling for the effects of GC content, our results were similar to those from previous studies, i.e., synonymous sites in primates exhibited evidence for higher selective constraint that those in rodents. Specifically, our results indicated that in primates up to 24 % of synonymous sites could be under purifying selection, while in rodents synonymous sites evolved neutrally. To further control for shifts in GC content, we estimated selective constraint at fourfold degenerate sites using a maximum parsimony approach. This allowed us to estimate selective constraint using mutational patterns that cause a shift in GC content (GT ↔ TG, CT ↔ TC, GA ↔ AG, and CA ↔ AC) and ones that do not (AT ↔ TA and CG ↔ GC). Using this approach, we found that synonymous sites evolve neutrally in both primates and rodents. Apparent deviations from neutrality were caused by a higher rate of C → A and C → T mutations in pseudogenes. Such differences are most likely caused by the shift in GC content experienced by pseudogenes. We conclude that previous estimates according to which 20–40 % of synonymous sites in primates were under selective constraint were most likely artifacts of the biased pattern of mutation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The efficiency of selection against deleterious mutations depends on the selection coefficient and effective population size. As effective population size increases, selection becomes more efficient in purging deleterious mutations (Ohta 1973). Because mammals have small effective population sizes, synonymous sites were thought for many years to evolve neutrally (Graul and Sadee 1997). The question of whether synonymous sites are under selection is important because the ratio of nonsynonymous to synonymous substitution (\(d_{\text{N}} /d_{\text{S}}\)) is commonly used to infer selection in protein-coding genes, and this test assumes that synonymous sites evolve neutrally. If synonymous sites are under selection, then using the \(d_{\text{N}} /d_{\text{S}}\) ratio may overestimate positive selection and underestimate purifying selection. Furthermore, under the assumption of neutrality, synonymous sites are used to compare rates of mutation within genomes and differences in mutation rates among species (Kumar 2005; Kumar and Subramanian 2002; Subramanian and Kumar 2003).
The assumption of neutral evolution at synonymous sites has been challenged by many studies that identified signals of selection at these sites (Bustamante et al. 2002; Doherty and McInerney 2013; Eory et al. 2010; Hellmann et al. 2003b; Ophir et al. 1999; Pagani and Baralle 2004; Smith and Hurst 1998; The Chimpanzee Sequencing and Analysis Consortium 2005). The suggested functional constraints on synonymous sites are (1) signals for the splicing machinery (Caceres and Hurst 2013; Fairbrother et al. 2004; Parmley et al. 2006; Romiguier et al. 2013; Yuan et al. 2012) (2) maintaining mRNA stability (Capon et al. 2004; Duan et al. 2003; Green 2007) (3) binding sites for miRNAs and transcription factors (Gu et al. 2012; Hurst 2006; Stergachis et al. 2013), and (4) efficiency and accuracy of translation (Akashi and Eyre-Walker 1998; Capon et al. 2004; Drummond and Wilke 2008; Duret 2002; Ikemura 1985; Stoletzki and Eyre-Walker 2007; Wright et al. 2004).
Codon usage bias has been primarily observed in species with large effective population sizes such as Drosophila melanogaster, Saccharomyces cerevisiae, Arabidopsis thaliana, and Caenorhabditis elegans (Akashi and Eyre-Walker 1998; Lawrie et al. 2013; Wright et al. 2004; Zhou et al. 2010). In mammals, the correlation between codon bias and tRNA abundance ranges from very weak (Chamary et al. 2006; Doherty and McInerney 2013; Lander et al. 2001) to nonexistent (dos Reis et al. 2004; Duret 2002; Kanaya et al. 2001). Studies using evolutionary comparisons between the divergence at synonymous sites and that at regions that are assumed to be nonfunctional and hence evolve neutrally (e.g., introns, ancestral repeats, and processed pseudogenes) (Berglund et al. 2009; Bustamante et al. 2002; Eory et al. 2010; Hellmann et al. 2003b; Ophir et al. 1999; Smith and Hurst 1998) identify a range of 11–39 % synonymous sites as being under selection. Approximately 1–9 % of synonymous sites at exon–intron boundaries are more conserved than synonymous sites in other regions of the coding sequence and have been proposed to function as exon splice enhancers (Caceres and Hurst 2013; Parmley et al. 2006). Parts of exons have also been identified as exon splice silencers (Wang et al. 2004) and have been shown to be conserved between human and mouse (Wang et al. 2006).
Recent studies that have compared selective constraint between primates and rodents have found that selective constraint is ~2-fold higher in primates than in rodents (Eory et al. 2010; Keightley et al. 2011). Specifically, the percent of synonymous sites under selective constraint in primates and rodents was ~20 and 10 %, respectively. This is contrary to the theoretical expectation, in which the efficiency of selection is expected to be higher in species with large long-term effective population sizes than in species with low effective population sizes. Rodents are estimated to have effective population sizes that are between 2- and 10-fold higher than primates (Charlesworth 2009; Phifer-Rixey et al. 2012). In accordance to theoretical expectations, rodents exhibit a lower \(d_{\text{N}} /d_{\text{S}}\) than either primates or any other mammalian taxon whose genome has been sequenced (Nikolaev et al. 2007; Popadin et al. 2013). To explain the higher selective constraint on synonymous sites in primates than rodents, some authors suggested that the number of exon splice enhancers or exon splice silencers might be higher in primates than in rodents, or that such sequences are under higher selection in primates to maintain functionality over longer life spans (Eory et al. 2010).
To effectively estimate selection on synonymous sites one must control for the effects of neutral processes on the rate of substitution. Some factors affecting the neutral rate of substitution are (1) regional GC content (Hellmann et al. 2005; Tyekucheva et al. 2008; Wolfe et al. 1989), CpG sites (Hwang and Green 2004; Keightley et al. 2011; Siepel and Haussler 2004) (2) DNA replication time (Chen et al. 2010; Pink and Hurst 2010; Stamatoyannopoulos et al. 2009), and (3) recombination rate (Duret and Arndt 2008; Hellmann et al. 2003a; Tyekucheva et al. 2008). Methylated CpG sites in mammals are highly mutagenic because methylated cytosine is unstable and undergoes deamination to thymine (Coulondre et al. 1978). The higher rate of substitution in regions of high GC content can been attributed not only to G and C nucleotides being more mutable than A and T nucleotides (Gojobori et al. 1982) but also to the higher fixation of G and C nucleotides due to biased gene conversion (Duret and Arndt 2008; Meunier and Duret 2004; Ratnakumar et al. 2010). GC-biased gene conversion is a recombination-associated process that results in the biased fixation of G and C nucleotides; as a result hotspots of meiotic recombination have higher rates of substitution than the genomic average. Finally, it has been shown that DNA that replicates late during the S phase undergoes a higher rate of replication-dependent mutation than early replicating DNA (Stamatoyannopoulos et al. 2009). To an extent, these factors have been found to correlate with each other; recombination rate has been found to positively correlate with GC content and to explain a large amount (~47 %) of the variation in GC content (Duret and Arndt 2008). DNA replication time has also been found to vary with GC content, with regions of high GC content replicating early and regions of lower GC content replicating late (Koren et al. 2012; Rhind and Gilbert 2013; Schmegner et al. 2007; Woodfine et al. 2004).
In this study, we estimate selection on synonymous sites using processed pseudogenes as models of strictly neutral evolution. Processed pseudogenes originate through the reverse transcription of mRNAs and their random insertion in to the genome. These sequences are considered “dead on arrival” and are thought to evolve under strict neutrality (Ophir et al. 1999). The use of processed pseudogenes in estimating selection on synonymous sites offers some advantages over the use of other sequences such as introns, intergenic regions, or ancestral repeats. Differences in rates due to base composition are largely constrained since genes and processed pseudogenes share similar sequences. Furthermore, the number of synonymous sites in coding sequences and pseudosynonymous sites in processed pseudogenes is very similar therefore estimates of substitution rate share a similar amount of sampling error.
After assembling a set of orthologous genes and processed pseudogenes in primates and rodents we compared rates of evolution at synonymous sites \((d_{{{\text{S}}_{f} }} )\), at pseudosynonymous sites in pseudogenes \((d_{{{\text{S}}_{\psi } }} )\), as well as the ratio \(d_{{{\text{S}}_{f} }} /d_{{{\text{S}}_{\psi } }}\). If synonymous sites are under selective constraint \(d_{{{\text{S}}_{f} }} /d_{{{\text{S}}_{\psi } }}\) is expected to be significantly lower than one. Because shifts in GC content at synonymous sites in genes or pseudogenes can lead to an increase or decrease in the ratio \(d_{{{\text{S}}_{f} }} /d_{{{\text{S}}_{\psi } }}\), we used a maximum parsimony approach and estimated selective constraint using the six bidirectional mutation patterns (AT ↔ TA, CG ↔ GC, GT ↔ TG, CT ↔ TC, GA ↔ AG, and CA ↔ AC) at each codon site and at fourfold degenerate sites. This allowed us to estimate selective constraint using patterns of mutation that do not affect GC content (AT ↔ TA and CG ↔ GC) versus patterns that can change GC content (GT ↔ TG, CT ↔ TC, GA ↔ AG, and CA ↔ AC).
Materials and Methods
Identification of Processed Pseudogenes and Their Parent Genes
The method used to identify processed pseudogenes is outlined in Section I of the Supplementary Material. Processed pseudogenes from human were used to identify orthologs in chimpanzee (Pan troglodytes), orangutan (Pongo abelii), Rhesus macaque (Macaca mulatta), and common marmoset (Callithrix jacchus). Processed pseudogenes from mouse were used to identify orthologs in rat (Rattus norvegicus). The procedure is explained in Section II of the Supplementary Material.
The parent genes of human processed pseudogenes were used to identify orthologs in chimpanzee, orangutan, macaque, and marmoset, while, mouse parent genes were used to identify orthologs in rat. Orthologs were downloaded from ENSEMBL 72 (Flicek et al. 2013). Parent coding sequences (CDSs) were downloaded for the majority of orthologous processed pseudogenes (4118 human-chimpanzee, 3723 human-orangutan, 2606 human-macaque, 1075 human-marmoset, 1606 mouse-rat). In a few cases there were more than one possible candidate parents (107 human-chimp, 15 human-orangutan, 351 mouse-rat). To identify the parent CDS in such cases, the processed pseudogenes were aligned with their possible parent CDSs using MAFFT (Katoh et al. 2002). Poorly aligned nucleotides were masked using the evaluation mode in T-COFFEE (Notredame et al. 2000) and a CORE score of 5 (Notredame and Abergel 2003). (For more details of the method, see section “Alignment of Processed Pseudogenes and Parent Genes by Codon Positions.”) After alignment refinement the percent similarity was estimated between the processed pseudogene and each possible parent. The CDS with the highest similarity was chosen to be the parent. Because orthologous relations are continuously updated, we further filtered the data according to the most recent version of ENSEMBL (ENSEMBL 77). After alignment refinement, we used the reading frame of the functional coding sequences to determine the positions of homologous pseudocodons in the processed pseudogenes. Aligned codons were discarded if they included gaps, poorly aligned nucleotides, or included stop codons.
Model of Gene and Pseudogene Evolution
We estimated the rate of evolution at synonymous and nonsynonymous sites in genes (\(d_{{{\text{N}}_{f} }} , d_{{{\text{S}}_{f} }} )\) and processed pseudogenes (\(d_{{{\text{N}}_{\psi } }} , d_{{{\text{S}}_{\psi } }} )\) using a maximum likelihood codon model (CODEML) developed by Nielsen and Yang (1998) and implemented in the PAML phylogenetic analysis package (Yang 1997). Specifically, we used the free ratios model to estimate divergences along each lineage (Fig. 1). The free ratios model was implemented using a fixed transition/transversion of 4 (κ = 4) and allowing the κ ratio to be estimated. Equilibrium codon frequencies were estimated using the base composition frequencies at the three codon positions (CodonFreq = 2). To reduce the amount of random error in our estimates, we only used orthologous sets where the number of synonymous sites between orthologous genes and pseudogenes was at least one hundred. This resulted in 1246 estimates from human-chimpanzee comparisons, 950 in human-orangutan, 667 in human-macaque, 566 in human-marmoset, and 392 in mouse-rat. Given that a protein-coding gene may give rise to more than one pseudogene and such cases may bias the data set, a single pair of orthologous genes and corresponding orthologous pseudogenes was chosen. This reduced the data set to 664 orthologous sets in the human-chimpanzee comparison, 547 in human-orangutan, 423 in human-macaque, 427 in human-marmoset, and 217 in mouse-rat. Orthologous genes and pseudogenes were removed if the genes or pseudogenes resided on the X-chromosomes as the X-chromosome has been shown to evolve slower than autosomes (Vicoso and Charlesworth 2006).

In the above phylogeny \(f_{1}\) and \(f_{2}\) depict the codon sequences of a pair of orthologous genes, while \({\psi}_{1}\) and \({\psi}_{2}\) represent the codon sequences of the corresponding pseudogenes. Subscripts 1 and 2 indicate two different species
Effects of GC Content on Rate of Synonymous Substitution
To estimate GC content at fourfold degenerate sites (Fig. 1), we only used orthologous genes and pseudogenes that shared at least 40 fourfold degenerate sites. The GC content of fourfold degenerate sites in genes is designated as \({\text{GC}}_{{4{\text{D}}_{f} }}\). For the same set of genes and pseudogenes, GC contents was estimated for each codon site in genes and corresponding “codon” sites in pseudogenes. GC content at these sites is depicted as \({\text{GC}}_{{{\text{codon}}i}}\) were i is the codon position.
To estimate the GC content of sequences flanking the transcriptional start and end sites of genes, we retrieved 5000 nucleotides upstream and downstream of the transcription start site and end site. The notations for GC content at upstream and downstream sequences will be \({\text{GC}}_{{{\text{flank}}_{{f_{\text{up}} }} }}\) \({\text{GC}}_{{{\text{flank}}_{{f_{\text{down}} }} }}\) for genes.
To examine the effects of GC content on \(d_{{{\text{S}}_{f} }},\) we used estimates of GC content from three different regions: (1) fourfold degenerate sites in genes \(({\text{GC}}_{{4{\text{D}}_{f} }} )\) (2) sequences flanking the transcriptional start sites of genes \(({\text{GC}}_{{{\text{flank}}f_{\text{up}} }} )\); and (3) the sequences flanking transcriptional end sites of genes \({\text{GC}}_{{{\text{flank}}_{{f_{\text{down}} }} }}\). Using the lowess function implemented in R we performed regression analyses of \({\text{GC}}_{{4{\text{D}}_{f} }}\), \(GC_{{{\text{flank}}f_{\text{up}} }}\) and \({\text{GC}}_{{{\text{flank}}f_{\text{down}} }}\) against \(d_{{{\text{S}}_{f} }}\). lowess is a locally weighted regression. Because of the limited number of genes in our set, we performed the above analysis using a set of 3059 genes in 13 mammals downloaded from the OMA browser (http://omabrowser.org/; Schneider et al. 2007). The phylogeny (in Newick format) used to estimate \(d_{{{\text{N}}_{f} }}\) and \(d_{{{\text{S}}_{f} }}\) was: (elephant(dog((cow,pig),horse))((rabbit(mouse,rat))(marmoset(macaque(orangutan(chimp,human))))));
Evolution of Pseudononsynonymous and Pseudosynonymous Sites in Pseudogenes
Under the assumption that processed pseudogenes evolve under strict neutrality, the rate of substitution at pseudononsynonymous sites (\(d_{{{\text{N}}_{\psi } }}\)) and pseudosynonymous sites (\(d_{{{\text{S}}_{\psi } }}\)) should be approximately equal. For each species, we compared the log likelihood of the model in which \(d_{{{\text{N}}_{\psi } }} /d_{{{\text{S}}_{\psi } }}\) could be less or equal to 1 to the log-likelihood of the model in which \(d_{{{\text{N}}_{\psi } }} /d_{{{\text{S}}_{\psi } }}\) was equal to 1. Assuming that the former model represents the alternative hypothesis (H1) and the latter model represents the null hypothesis (H0), we can test if \(d_{{{\text{N}}_{\psi } }} /d_{{{\text{S}}_{\psi } }}\) is significantly different from 1 by considering two times the difference between the log-likelihoods of the two models Ln1–Ln0 to be asymptotically distributed as a χ 2 random variable with one degree of freedom.
To further test if pseudogenes are under any selective constraint, we compared mean divergence at pseudononsynonymous (\(\overline{{d_{{{\text{N}}_{\psi } }} }}\)) and pseudosynonymous (\(\overline{{d_{{{\text{S}}_{\psi } }} }}\)) sites using the ratio \(\overline{{d_{{{\text{N}}_{\psi } }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\). 95 % confidence intervals were estimated using a bootstrap approach (For further details, see “Bootstrap Analysis” section). To study the effects of the initial GC content at synonymous and nonsynonymous sites on \(d_{{{\text{N}}_{\psi } }}\) and \(d_{{{\text{S}}_{\psi } }},\) we used the GC content at the second codon position (\({\text{GC}}_{{{\text{codon}}2}}\)) as a proxy of the GC content at nonsynonymous sites and the GC content at fourfold degenerate sites (\({\text{GC}}_{{4{\text{D}}_{f} }}\)) as a proxy of the GC content at synonymous sites.
Estimating Selective Constraint on Synonymous and Nonsynonymous Sites
To estimate selection on synonymous and nonsynonymous sites we used two methods: (1) we estimated selective constraint on synonymous sites using the ratio \(\overline{{d_{{{\text{S}}_{f} }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\) and selective constraint on nonsynonymous sites using the ratios \(\overline{{d_{{{\text{N}}_{f} }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\) and \(\overline{{d_{{{\text{N}}_{f} }} }} /\overline{{d_{{{\text{S}}_{f} }} }}\). By using a bootstrap method, 95 % confidence intervals were estimated. Furthermore, using the lowess function in R we studied the change in \(d_{{{\text{S}}_{f} }}\) and \(d_{{{\text{S}}_{\psi } }}\) with \({\text{GC}}_{{4{\text{D}}_{f} }}\); (2) Using a maximum parsimony approach we estimated the number of substitutions along the gene and pseudogene lineages (Fig. 1). After concatenating the gene-pseudogene codon alignments, we counted the number of substitutions for each codon site and fourfold degenerate sites. Substitutions were separated into the six bidirectional mutation patterns (AT ↔ TA, CG ↔ GC, GT ↔ TG, CT ↔ TC, GA ↔ AG, and CA ↔ AC). To test whether the number of changes along the gene (\(f\)) and pseudogene lineages (\(\psi\)) were significantly different we used a one tail Fisher’s exact test. In instances where the probability was less than 0.05, we used the proportion (\(f/{\psi}\)) as a proxy to selective constraint.
Bootstrap Analysis
To estimate 95 % confidence intervals for the ratios \(\overline{{d_{{{\text{N}}_{\psi } }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\), \(\overline{{d_{{{\text{S}}_{f} }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\),\(\overline{{d_{{{\text{N}}_{f} }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }},\) and \(\overline{{d_{{{\text{N}}_{f} }} }} /\overline{{d_{{{\text{S}}_{f} }} }}\) we used 10,000 bootstrap samples. Each bootstrap replicate is a random sample of the numerator and denominator of the same size as the original data set.
Results
Alignment Refinement and Estimating Selection at Synonymous Sites
To test the effect of alignment refinement on estimating \(\overline{{d_{{{\text{S}}_{f} }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }},\) we compared the results when using all sites and after removing codons sites masked by different CORE scores. Although there were slight decreases, (<5 %) in both \(d_{{{\text{S}}_{f} }}\) and \(d_{{{\text{S}}_{\psi } }}\), there were no significant changes in the ratios (Fig. 2).

\(\overline{{d_{{{\text{S}}_{f} }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\) and 95 % bootstrap CI’s when using different levels of stringency during alignment refinement \(\overline{{d_{{{\text{S}}_{f} }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\) does not change by increasing the CORE score
Do Processed Pseudogenes Evolve Neutrally?
Processed pseudogenes are formed through the reverse transcription of an mRNA and are randomly inserted in the genome. Because genic regions are found within regions of higher GC content than the genome average, processed pseudogenes are expected to experience a shift in GC content after their formation. As shown in Table 1, the first and third codon positions that include synonymous sites have the highest GC content since they are under weaker selection and are more likely to be influenced by regional GC content. After pseudogene formation, these sites experience the highest shift in GC content. On the other hand, the GC content at the second codon position does not experience a major shift. This is expected if we assume that equilibrium GC content is close to the mammalian average of ~40 % (Arndt et al. 2003; Duret and Arndt 2008).
Assuming that the GC content at second codon sites (\({\text{GC}}_{{{\text{codon}}2}}\)) represents the GC content at nonsynonymous sites and the GC content at fourfold degenerate sites (\({\text{GC}}_{{4{\text{D}}f}}\)) the GC content at synonymous sites, Fig. 3 shows how \(d_{{{\text{N}}_{\psi } }}\) and \(d_{{{\text{S}}_{\psi } }}\) change with GC content. As expected, because the initial GC content at fourfold degenerate sites is higher than the GC content at the second codon sites \(d_{{{\text{S}}_{\psi } }} > d_{{{\text{N}}_{\psi } }}\) when GC content is high. This may occur because processed pseudogenes experience a mutational pressure to lower their GC content after formation. The difference in \(d_{{{\text{S}}_{\psi } }}\) and \(d_{{{\text{N}}_{\psi } }}\) leads to a ratio \(\overline{{d_{{{\text{N}}_{\psi } }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\) that is significantly lower than 1 (Table 2 “All”). However when using genes with \({\text{GC}}_{{{\text{codon}}2}}\) and \({\text{GC}}_{{4{\text{D}}f}}\) between 0.3 and 0.5, thus limiting nonequilibrium at pseudosynonymous sites in pseudogenes, \(\overline{{d_{{{\text{N}}_{\psi } }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }} \approx 1\) (Table 2). In conclusion, after controlling for the effect of GC on \(d_{{{\text{S}}_{\psi } }}\), out results indicate that the processed pseudogenes evolve under strict neutrality.

Lowess curves of \({\text{GC}}_{{{\text{codon}}2}}\) against \(d_{{{\text{N}}_{\psi } }}\), and \({\text{GC}}_{{4{\text{D}}_{f} }}\) against \(d_{{{\text{S}}_{\psi } }}\) (shaded areas represent the 95 % confidence intervals). As \({\text{GC}}_{{4{\text{D}}_{f} }}\) content increases, \(d_{{{\text{S}}_{\psi } }}\) increases, and becomes greater than \(d_{{{\text{N}}_{\psi } }}\)
The Relationship Between GC Content and the Rate of Synonymous Substitution
A positive relation between GC content and the rate of substitution at synonymous sites has been previously identified (Bielawski et al. 2000; Eory et al. 2010; Hurst and Williams 2000). To study how the rate of substitution at synonymous sites (\(d_{{{\text{S}}_{f} }}\)) covaries with GC content, we used the GC content at fourfold degenerate sites (\({\text{GC}}_{{4{\text{D}}_{f} }}\)) and the GC content upstream and downstream of the transcriptional start and end site, respectively \({\text{GC}}_{{{\text{flank}}f_{\text{up}} }}\), \({\text{GC}}_{{{\text{flank}}f_{\text{down}} }}\)) (Fig. 4). As shown in Fig. 4, when \({\text{GC}}_{{4{\text{D}}_{f} }}\), \({\text{GC}}_{{{\text{flank}}f_{\text{up}} }}\) and \({\text{GC}}_{{{\text{flank}}f_{\text{down}} }}\) are at ~0.40 \(d_{{{\text{S}}_{f} }}\) is approximately the same (i.e., regression lines intersect). However, as GC content increases, the trajectories of \(d_{{{\text{S}}_{f} }}\) start to differ (Fig. 4). \(d_{{{\text{S}}_{f} }}\) experiences a sharper increase when using the GC content of sequences upstream or downstream of the transcriptional start and end site of genes. This relation is further verified by a much larger set of genes (Figure S3 in the Supplementary Material).

Variation in the rate of substitution at synonymous sites with GC content at fourfold degenerate sites (\({\text{GC}}_{{4{\text{D}}_{f} }}\), and GC content upstream and downstream of the transcriptional start site (\({\text{GC}}_{{{\text{flank}}_{{f{\text{up}}}} }}\), \({\text{GC}}_{{{\text{flank}}_{{f{\text{down}}}} }}\)). When \({\text{GC}}_{{4{\text{D}}_{f} }}\), \({\text{GC}}_{{{\text{flank}}_{{f{\text{up}}}} }}\), \({\text{GC}}_{{{\text{flank}}_{{f{\text{down}}}} }}\) is ~0.40 \(d_{{{\text{S}}_{f} }}\) is approximately the same, however as GC content at these the location increases the trajectory of \(d_{{{\text{S}}_{f} }}\) start to differ. This is confirmed by a much larger set of mammalian genes (Supplementary Figure S3)
Do Synonymous Sites Evolve Under Strict Neutrality?
To estimate selective constraint on synonymous sites we used the ratio \(\overline{{d_{{{\text{S}}_{\text{f}} }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\). Under the assumption of strict neutrality and similar mutation patterns at synonymous and pseudosynonymous sites in pseudogenes (i.e., when both sites are evolving under equilibrium conditions), \(d_{{{\text{S}}_{f} }}\) should evolve at approximately the same rate as \(d_{{{\text{S}}_{\psi } }}\). As shown in Fig. 4, \(d_{{{\text{S}}_{f} }}\) increases when \({\text{GC}}_{{4{\text{D}}_{f} }}\), \({\text{GC}}_{{{\text{flank}}f_{\text{up}} }},\) or \({\text{GC}}_{{{\text{flank}}f_{\text{down}} }}\) are greater than ~0.4. If we assume that the value of \(d_{{{\text{S}}_{f} }}\) is similar to what is expected under equilibrium and if we further assume that nonequilibrium evolution at pseudosynonymous sites in pseudogenes is limited when \({\text{GC}}_{{4{\text{D}}_{f} }}\) and \({\text{GC}}_{{{\text{codon}}2}}\) is ~0.40 (Fig. 3), then any deviation of \(d_{{{\text{S}}_{f} }} < d_{{{\text{S}}_{\psi } }}\) from 1 could be the result of selective constraint.
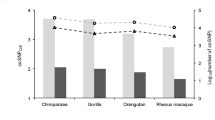
As shown in Fig. 5, when \({\text{GC}}_{{4{\text{D}}_{f} }} \approx 0.4\) in most primates \(d_{{{\text{S}}_{f} }} < d_{{{\text{S}}_{\psi } }}\); while in rodents \(d_{{{\text{S}}_{f} }} \approx d_{{{\text{S}}_{\psi } }}\). This is further demonstrated by the ratios \(\overline{{d_{{{\text{S}}_{f} }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\) in Table 3. Contrary to the higher selective constraint on nonsynonymous sites exhibited by rodents (\(\overline{{d_{{{\text{N}}_{f} }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\) or \(\overline{{d_{{{\text{N}}_{f} }} }} /\overline{{d_{{{\text{S}}_{f} }} }}\)), primates exhibit a higher selective constraint on synonymous sites. This is contrary to the theoretical expectation in which the efficiency of selection against deleterious mutations is higher in species with larger long-term effective population sizes than species smaller long-term effective population sizes (Ohta 1973).

Variation of \(d_{{{\text{S}}_{f} }}\) and \(d_{{{\text{S}}_{\psi } }}\) with \({\text{GC}}_{{4{\text{D}}_{f} }}\). When \({\text{GC}}_{{4{\text{D}}_{f} }}\) content is ~0.40, \(d_{{{\text{S}}_{\psi } }}\) > \(d_{{{\text{S}}_{f} }}\) in all primates except chimpanzee; while in rodents \(d_{{{\text{S}}_{f} }} \approx d_{{{\text{S}}_{\psi } }}\).When \({\text{GC}}_{{4{\text{D}}_{f} }}\) content increases, in all species except rat \(d_{{{\text{S}}_{f} }}\) can become larger than \(d_{{{\text{S}}_{\psi } }}\). This change in rate between \(d_{{{\text{S}}_{f} }}\) and \(d_{{{\text{S}}_{\psi } }}\) is most prominent along the mouse lineage
A possible contribution to the larger deviation from neutrality in primates could be nonequilibrium evolution at pseudosynonymous sites in pseudogenes. To further control for such effects, we used a maximum parsimony approach and estimated selection on nonsynonymous and synonymous sites in genes using six bidirectional mutational patterns. This would allow us to study how selective constraint is affected by mutational patterns that lead to a shift in GC content (GT:TG, CT:TC, GA:AG, CA:AC) and ones that do not (AT:TA, CG:GC). Selective constraint was estimated for each codon position and fourfold degenerate sites. As shown in Table 4, rodents show higher selective constraint on the first and second position all across the different mutational patterns, while at the third codon position selective constraint becomes similar. As expected, on average, selective constraint on the third codon position is the lowest when using GA or CT changes because most of them code for the same amino acid. When using fourfold degenerate sites almost all mutational patterns do not show a significant difference between genes and pseudogenes. However, in the case of macaque and marmoset, we see some significant deviations in the case of CA and CT mutations (Table 4). These can be caused by pseudogenes experiencing a mutational pressure to lower their GC content. Because of the large sample of changes we looked at unidirectional changes (C → A, A → C, C → T, T → C) in macaque and marmoset genes (\(f\)) and pseudogenes (\(\psi\)). As shown in Table 5, only C → A and C → T mutations show a significant difference between genes and pseudogenes. This supports our hypothesis that the significant deviations in the rates of synonymous substitutions between genes and pseudogenes observed in macaque and marmoset (Fig. 5; Table 4) are most likely caused by pseudogenes experiencing nonequilibrium to lower their GC content. The significant difference in total changes observed in orangutan can be caused by the cumulative effect of mutational patterns that are close to being significant. As in the case of macaque and marmoset, the largest differences are observed by CA and CT changes.
Discussion
A frequently used method of estimating selection at synonymous sites in genes is comparing the rate of substitution at regions assumed to be nonfunctional and under no selection, to the rate at synonymous sites (Bustamante et al. 2002; Eory et al. 2010; Hellmann et al. 2003b; Ophir et al. 1999; Subramanian and Kumar 2003). Because the frequency of insertions and deletions in nonfunctional regions is much higher than in protein-coding genes, alignment errors can have a significant impact on the estimate of selective constraint on synonymous sites. Using different levels of alignment quality, we show that the quality level does not have a significant impact on \(\overline{{d_{{{\text{S}}_{f} }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\) (Fig. 2).
When testing for selective constraint on synonymous sites we assume that pseudosynonymous sites in pseudogenes are under no selective constraint. To test the above assumption, we compared the rate of substitution at pseudononsynonymous and pseudosynonymous sites in pseudogenes \(\overline{{d_{{{\text{N}}_{\psi } }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\). Assuming that processed pseudogenes are evolving under no selective constraint \(\overline{{d_{{{\text{N}}_{\psi } }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }} \approx 1\). Although our initial estimates refuted the null hypothesis of processed pseudogenes evolving under strict neutrality (\(\overline{{d_{{{\text{N}}_{\psi } }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }} < 1\)) (Table 2 “All”). However, when controlling for the difference in GC content at synonymous and nonsynonymous sites in pseudogenes \(\overline{{d_{{{\text{N}}_{\psi } }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\) becomes approximately equal to 1 (Table 2 “0.3 < GC < 0.5”).
Under the assumption that processed pseudogenes evolve under strict neutrality selective constraint on synonymous sites can be estimated using the ratio \(\overline{{d_{{{\text{S}}_{f} }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\). Because selection at synonymous sites is weak, differences in mutation patterns or rates of biased gene conversion between genes and pseudogenes can cause complications in estimating the extent of selective constraint on synonymous sites (Comeron 2006; Lawrie et al. 2011; McVean and Charlesworth 1999). For example, if synonymous sites are located within hot spots of recombination, \(d_{{{\text{S}}_{f} }}\) can be equal or even larger than \(d_{{{\text{S}}_{\psi } }}\), therefore masking any conservation due to purifying selection. To the opposite effect, because processed pseudogenes usually move from regions of high GC content to areas of low GC content they experience a mutational pressure to lower their GC content and therefore this can cause \(d_{{{\text{S}}_{\psi } }}\) to be larger than \(d_{{{\text{S}}_{f} }}\), therefore creating the false impression of selective constraint on synonymous sites.
As shown in Fig. 4 and Figure S3 (Supplementary Material), any deviations from a GC content of ~0.4, \(d_{{{\text{S}}_{f} }}\) experiences a significant increase. Assuming an equilibrium GC content similar to the average GC content of primate and rodent genomes (~0.40) (Arndt et al. 2003; Duret and Arndt 2008), an increase in \(d_{{{\text{S}}_{f} }}\) with GC content could be caused by CpG deamination, biased gene conversion, or an increase in the mutation rate from AT to GC nucleotides (Piganeau et al. 2002). If we assume that the value of \(d_{{{\text{S}}_{f} }}\) when \({\text{GC}}_{{4{\text{D}}_{f} }} \approx 0.4\) is the least affected by the above factors, when comparing it to \(d_{{{\text{S}}_{\psi } }}\) it seems that synonymous sites are evolving at a significantly lower rate in primates, while in rodents, they evolve at the same rate as the neutral expectation (Table 3; Fig. 5). On the contrary, rodents exhibit a higher selective constraint on nonsynonymous sites (Table 3) which is in accordance to the theoretical expectation in which species with larger effective population sizes are more efficient in purging deleterious mutations (Ohta 1973).
To further ensure that the significant difference between \(d_{{{\text{S}}_{f} }}\) and \(d_{{{\text{S}}_{\psi } }}\) in primates is not due to pseudogenes experiencing a higher rate of GC to AT mutations after formation, we estimated selective constraint on each codon position and at fourfold degenerate sites using a maximum parsimony approach. Specifically, we separated mutations along the gene and pseudogene lineages into six patterns (GT:TG, CT:TC, GA:AG, CA:AC, AT:TA, CG:GC). We did not look at unidirectional changes because of the very small number of changes in certain patterns. As shown in Table 4 selective constraint on the first and second position is higher in rodents; but when selective constraint becomes weaker such as the third codon position the difference in selective constraint disappears. When it comes to fourfold degenerate sites in which all mutational changes do not alter the amino acid, almost all mutation patterns indicate no selective constraint on synonymous sites. In the case of macaque and marmoset, however, we observe a very large difference in CA:AC and CT:TC mutations between genes and pseudogenes. If these differences are caused by pseudogenes experiencing a mutational pressure to lower their GC content, we expect to see a significant difference in patterns C → T and C → A. As expected, only patterns C → T and C → A show a significant difference (Table 5). This difference between primates and rodents can be caused by the higher mutability of CpG sites in primates (Keightley et al. 2011).
Our results help resolve previous puzzling findings (Eory et al. 2010; Keightley et al. 2011) in which synonymous sites in rodents are under lower selective constraint than primates, despite having larger effective population sizes (Charlesworth 2009). According to the results of the present study, synonymous sites are under no selective constraint. Although we can conclude with high confidence that the percent of synonymous sites under selective constraint is not significantly greater than 10 %, our method may not be sensitive enough to detect if 10 % or less of synonymous sites are under selection as suggested by human population data (Keightley and Halligan 2011) or as exon spice site studies (Caceres and Hurst 2013; Hurst 2006). Some of the shortcomings of our study is that our sample of genes is limited to ones giving rise to pseudogenes, and furthermore, our study did not control for any effects associated with transcription coupled repair. If transcription-associated processes are mutagenic (Green et al. 2003; Majewski 2003) then \(\overline{{d_{{{\text{S}}_{f} }} }} /\overline{{d_{{{\text{S}}_{\psi } }} }}\) can be overestimated if processed pseudogenes are at large transcriptionally inactive.
References
Akashi H, Eyre-Walker A (1998) Translational selection and molecular evolution. Curr Opin Genet Dev 8:688
Arndt PF, Petrov DA, Hwa T (2003) Distinct changes of genomic biases in nucleotide substitution at the time of Mammalian radiation. Mol Biol Evol 20:1887
Berglund J, Pollard KS, Webster MT (2009) Hotspots of biased nucleotide substitutions in human genes. PLoS Biol 7:e26
Bielawski JP, Dunn KA, Yang Z (2000) Rates of nucleotide substitution and mammalian nuclear gene evolution. Approximate and maximum-likelihood methods lead to different conclusions. Genetics 156:1299
Bustamante CD, Nielsen R, Hartl DL (2002) A maximum likelihood method for analyzing pseudogene evolution: implications for silent site evolution in humans and rodents. Mol Biol Evol 19:110
Caceres EF, Hurst LD (2013) The evolution, impact and properties of exonic splice enhancers. Genome Biol 14:R143
Capon F, Allen MH, Ameen M, Burden AD, Tillman D, Barker JN, Trembath RC (2004) A synonymous SNP of the corneodesmosin gene leads to increased mRNA stability and demonstrates association with psoriasis across diverse ethnic groups. Hum Mol Genet 13:2361
Chamary JV, Parmley JL, Hurst LD (2006) Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet 7:98
Charlesworth B (2009) Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat Rev Genet 10:195
Chen CL, Rappailles A, Duquenne L, Huvet M, Guilbaud G, Farinelli L, Audit B, d’Aubenton-Carafa Y, Arneodo A, Hyrien O, Thermes C (2010) Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res 20:447
Comeron JM (2006) Weak selection and recent mutational changes influence polymorphic synonymous mutations in humans. Proc Natl Acad Sci USA 103:6940
Coulondre C, Miller JH, Farabaugh PJ, Gilbert W (1978) Molecular basis of base substitution hotspots in Escherichia coli. Nature 274:775
Doherty A, McInerney JO (2013) Translational selection frequently overcomes genetic drift in shaping synonymous codon usage patterns in vertebrates. Mol Biol Evol 30:2263
dos Reis M, Savva R, Wernisch L (2004) Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res 32:5036
Drummond DA, Wilke CO (2008) Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134:341
Duan J, Wainwright MS, Comeron JM, Saitou N, Sanders AR, Gelernter J, Gejman PV (2003) Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet 12:205
Duret L (2002) Evolution of synonymous codon usage in metazoans. Curr Opin Genet Dev 12:640
Duret L, Arndt PF (2008) The impact of recombination on nucleotide substitutions in the human genome. PLoS Genet 4:e1000071
Eory L, Halligan DL, Keightley PD (2010) Distributions of selectively constrained sites and deleterious mutation rates in the hominid and murid genomes. Mol Biol Evol 27:177
Fairbrother WG, Holste D, Burge CB, Sharp PA (2004) Single nucleotide polymorphism-based validation of exonic splicing enhancers. PLoS Biol 2:E268
Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, Gil L, Garcia-Giron C, Gordon L, Hourlier T, Hunt S, Juettemann T, Kahari AK, Keenan S, Komorowska M, Kulesha E, Longden I, Maurel T, McLaren WM, Muffato M, Nag R, Overduin B, Pignatelli M, Pritchard B, Pritchard E, Riat HS, Ritchie GR, Ruffier M, Schuster M, Sheppard D, Sobral D, Taylor K, Thormann A, Trevanion S, White S, Wilder SP, Aken BL, Birney E, Cunningham F, Dunham I, Harrow J, Herrero J, Hubbard TJ, Johnson N, Kinsella R, Parker A, Spudich G, Yates A, Zadissa A, Searle SM (2013) Ensembl 2013. Nucleic Acids Res 41:D48
Gojobori T, Li WH, Graur D (1982) Patterns of nucleotide substitution in pseudogenes and functional genes. J Mol Evol 18:360
Graul RC, Sadee W (1997) Evolutionary relationships among proteins probed by an iterative neighborhood cluster analysis (INCA). Alignment of bacteriorhodopsins with the yeast sequence YRO2. Pharm Res 14:1533
Green P (2007) 2x genomes–does depth matter? Genome Res 17:1547
Green P, Ewing B, Miller W, Thomas PJ, Green ED (2003) Transcription-associated mutational asymmetry in mammalian evolution. Nat Genet 33:514
Gu W, Wang X, Zhai C, Xie X, Zhou T (2012) Selection on synonymous sites for increased accessibility around miRNA binding sites in plants. Mol Biol Evol 29:3037
Hellmann I, Ebersberger I, Ptak SE, Paabo S, Przeworski M (2003a) A neutral explanation for the correlation of diversity with recombination rates in humans. Am J Hum Genet 72:1527
Hellmann I, Zollner S, Enard W, Ebersberger I, Nickel B, Paabo S (2003b) Selection on human genes as revealed by comparisons to chimpanzee cDNA. Genome Res 13:831
Hellmann I, Prufer K, Ji H, Zody MC, Paabo S, Ptak SE (2005) Why do human diversity levels vary at a megabase scale? Genome Res 15:1222
Hurst LD (2006) Preliminary assessment of the impact of microRNA-mediated regulation on coding sequence evolution in mammals. J Mol Evol 63:174
Hurst LD, Williams EJ (2000) Covariation of GC content and the silent site substitution rate in rodents: implications for methodology and for the evolution of isochores. Gene 261:107
Hwang DG, Green P (2004) Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc Natl Acad Sci USA 101:13994
Ikemura T (1985) Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol 2:13
Kanaya S, Yamada Y, Kinouchi M, Kudo Y, Ikemura T (2001) Codon usage and tRNA genes in eukaryotes: correlation of codon usage diversity with translation efficiency and with CG-dinucleotide usage as assessed by multivariate analysis. J Mol Biol 53:290
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059
Keightley PD, Halligan DL (2011) Inference of site frequency spectra from high-throughput sequence data: quantification of selection on nonsynonymous and synonymous sites in humans. Genetics 188:931
Keightley PD, Eory L, Halligan DL, Kirkpatrick M (2011) Inference of mutation parameters and selective constraint in mammalian coding sequences by approximate Bayesian computation. Genetics 187:1153
Koren A, Polak P, Nemesh J, Michaelson JJ, Sebat J, Sunyaev SR, McCarroll SA (2012) Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet 91:1033
Kumar S (2005) Molecular clocks: four decades of evolution. Nat Rev Genet 6:654
Kumar S, Subramanian S (2002) Mutation rates in mammalian genomes. Proc Natl Acad Sci USA 99:803
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ (2001) Initial sequencing and analysis of the human genome. Nature 409:860
Lawrie DS, Petrov DA, Messer PW (2011) Faster than neutral evolution of constrained sequences: the complex interplay of mutational biases and weak selection. Genome Biol Evol 3:383
Lawrie DS, Messer PW, Hershberg R, Petrov DA (2013) Strong purifying selection at synonymous sites in D. melanogaster. PLoS Genet 9:e1003527
Majewski J (2003) Dependence of mutational asymmetry on gene-expression levels in the human genome. Am J Hum Genet 73:688
McVean ATG, Charlesworth B (1999) A population genetic model for the evolution of synonymous codon usage: patterns and predictions. Genet Res 74:145
Meunier J, Duret L (2004) Recombination drives the evolution of GC-content in the human genome. Mol Biol Evol 21:984
Nikolaev SI, Montoya-Burgos JI, Popadin K, Parand L, Margulies EH, Antonarakis SE (2007) Life-history traits drive the evolutionary rates of mammalian coding and noncoding genomic elements. Proc Natl Acad Sci USA 104:20443
Notredame C, Abergel C (2003) Using multiple alignment methods to assess the quality of genomic data analysis. In: Andrade M (ed) Bioinformatics and genomes: current perspectives. Horizon Scientific Press, Wymondham (United Kingdom), pp 30–50
Notredame C, Higgins DG, Heringa J (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205
Ohta T (1973) Slightly deleterious mutant substitutions in evolution. Nature 246:96
Ophir R, Itoh T, Graur D, Gojobori T (1999) A simple method for estimating the intensity of purifying selection in protein-coding genes. Mol Biol Evol 16:49
Pagani F, Baralle FE (2004) Genomic variants in exons and introns: identifying the splicing spoilers. Nat Rev Genet 5:389
Parmley JL, Chamary JV, Hurst LD (2006) Evidence for purifying selection against synonymous mutations in mammalian exonic splicing enhancers. Mol Biol Evol 23:301
Phifer-Rixey M, Bonhomme F, Boursot P, Churchill GA, Pialek J, Tucker PK, Nachman MW (2012) Adaptive evolution and effective population size in wild house mice. Mol Biol Evol 29:2949
Piganeau G, Mouchiroud D, Duret L, Gautier C (2002) Expected relationship between the silent substitution rate and the GC content: implications for the evolution of isochores. J Mol Evol 54:129
Pink CJ, Hurst LD (2010) Timing of replication is a determinant of neutral substitution rates but does not explain slow Y chromosome evolution in rodents. Mol Biol Evol 27:1077
Popadin KY, Nikolaev SI, Junier T, Baranova M, Antonarakis SE (2013) Purifying selection in mammalian mitochondrial protein-coding genes is highly effective and congruent with evolution of nuclear genes. Mol Biol Evol 30:347
Ratnakumar A, Mousset S, Glemin S, Berglund J, Galtier N, Duret L, Webster MT (2010) Detecting positive selection within genomes: the problem of biased gene conversion. Philos Trans R Soc Lond B 365:2571
Rhind N, Gilbert DM (2013) DNA replication timing. Cold Spring Harb Perspect Biol 5:a010132
Romiguier J, Ranwez V, Douzery EJ, Galtier N (2013) Genomic evidence for large, long-lived ancestors to placental mammals. Mol Biol Evol 30:5
Schmegner C, Hameister H, Vogel W, Assum G (2007) Isochores and replication time zones: a perfect match. Cytogenet Genome Res 116:167
Schneider A, Dessimoz C, Gonnet GH (2007) OMA Browser–exploring orthologous relations across 352 complete genomes. Bioinformatics 23:2180
Siepel A, Haussler D (2004) Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol Biol Evol 21:468
Smith NG, Hurst LD (1998) Sensitivity of patterns of molecular evolution to alterations in methodology: a critique of Hughes and Yeager. J Mol Evol 47:493
Stamatoyannopoulos JA, Adzhubei I, Thurman RE, Kryukov GV, Mirkin SM, Sunyaev SR (2009) Human mutation rate associated with DNA replication timing. Nat Genet 41:393
Stergachis AB, Haugen E, Shafer A, Fu W, Vernot B, Reynolds A, Raubitschek A, Ziegler S, LeProust EM, Akey JM, Stamatoyannopoulos JA (2013) Exonic transcription factor binding directs codon choice and affects protein evolution. Science 342:1367
Stoletzki N, Eyre-Walker A (2007) Synonymous codon usage in Escherichia coli: selection for translational accuracy. Mol Biol Evol 24:374
Subramanian S, Kumar S (2003) Neutral substitutions occur at a faster rate in exons than in noncoding DNA in primate genomes. Genome Res 13:838
The Chimpanzee Sequencing and Analysis Consortium (2005) Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437:69
Tyekucheva S, Makova KD, Karro JE, Hardison RC, Miller W, Chiaromonte F (2008) Human-macaque comparisons illuminate variation in neutral substitution rates. Genome Biol 9:R76
Vicoso B, Charlesworth B (2006) Evolution on the X chromosome: unusual patterns and processes. Nat Rev Genet 7:645
Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB (2004) Systematic identification and analysis of exonic splicing silencers. Cell 119:831
Wang Z, Xiao X, Van Nostrand E, Burge CB (2006) General and specific functions of exonic splicing silencers in splicing control. Mol Cell 23:61
Wolfe KH, Sharp PM, Li WH (1989) Mutation rates differ among regions of the mammalian genome. Nature 337:283
Woodfine K, Fiegler H, Beare DM, Collins JE, McCann OT, Young BD, Debernardi S, Mott R, Dunham I, Carter NP (2004) Replication timing of the human genome. Hum Mol Genet 13:191
Wright SI, Yau CB, Looseley M, Meyers BC (2004) Effects of gene expression on molecular evolution in Arabidopsis thaliana and Arabidopsis lyrata. Mol Biol Evol 21:1719
Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555
Yuan Q, Zhou Z, Lindell SG, Higley JD, Ferguson B, Thompson RC, Lopez JF, Suomi SJ, Baghal B, Baker M, Mash DC, Barr CS, Goldman D (2012) The rhesus macaque is three times as diverse but more closely equivalent in damaging coding variation as compared to the human. BMC Genet 13:52
Zhou T, Gu W, Wilke CO (2010) Detecting positive and purifying selection at synonymous sites in yeast and worm. Mol Biol Evol 27:1912
Acknowledgments
We thank Kiyoshi Ezawa and the reviewers for their useful comments and suggestions. We also thank Jeff Sarlo for his technical support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Price, N., Graur, D. Are Synonymous Sites in Primates and Rodents Functionally Constrained?. J Mol Evol 82, 51–64 (2016). https://doi.org/10.1007/s00239-015-9719-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-015-9719-3