
Abstract
Gene duplication is a major force for generating evolutionary novelties that lead to adaptations to environments. We previously identified two paralogs encoding phytochrome A (phyA), GmphyA1 and GmphyA2, in soybean, a paleopolyploid species. GmphyA2 is encoded by the E4 locus responsible for photoperiod sensitivity. In photoperiod insensitive lines, GmphyA2 is inactivated by the insertion of a retrotransposon in exon 1. Here, we describe the detailed characterization of the element and its evolutionary significance inferred from the distribution of the allele that harbors the element. Structural characteristics indicated that the element, designated SORE-1, is a novel Ty1/copia-like retrotransposon in soybean, which was phylogenetically related to the Sto-4, BARE-1, and RIRE1 elements. The element was transcriptionally active, and the transcription was partially repressed by an epigenetic mechanism. Sequences homologous with SORE-1 were detected in a genome sequence database of soybean, most of which appeared silent. GmphyA2 that harbors the SORE-1 insertion was detected only in cultivated soybean lines grown in northern regions of Japan, consistent with the notion that photoperiod insensitivity caused by the dysfunction of GmphyA2 is one of genetic changes that allowed soybean cultivation at high latitudes. Taking into account that genetic redundancy is conferred by the two phyA genes, we propose a novel model for the consequences of gene duplication and transposition of retrotransposons: when the gene is duplicated, retrotransposon insertion that causes the loss of a gene function can lead to adaptive evolution while the organism is sustained by the buffering effect brought about by gene duplication.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gene duplication is a major source of evolutionary novelties and can occur through duplication of individual genes, chromosomal segments, or entire genomes (polyploidization). Under the classic model of duplicate gene evolution, one of the duplicated genes is free to accumulate mutations, which results in either the inactivation of transcription and/or a function (pseudogenization or nonfunctionalization) or the gain of a new function (neofunctionalization) as long as another copy retains the requisite physiological functions (Lynch and Conery 2000, and references therein). However, empirical data suggest that a much greater proportion of gene duplicates is preserved than predicted by the classic model (Force et al. 1999).
Recent advances in genome study have led to the formulation of several evolutionary models: a model proposed by Hughes (1994) suggests that gene sharing, whereby a single gene encodes a protein with two distinct functions, precedes the evolution of two functionally distinct proteins; the duplication–degeneration–complementation model suggests that duplicate genes acquire debilitating yet complementary mutations that alter one or more subfunctions of the single gene progenitor, an evolutionary consequence for duplicated loci referred to as subfunctionalization (Force et al. 1999; Lynch and Force 2000; reviewed by Moore and Purugganan 2005). In addition to this notion, models involving epigenetic silencing of duplicate genes (Rodin and Riggs 2003) or purifying selection for gene balance (Freeling and Thomas 2006; Birchler and Veitia 2007) have also been proposed.
Because the vast majority of mutations affecting fitness are more or less deleterious and because gene duplicates are generally assumed to be functionally redundant at the time of origin, virtually all models predict that the usual fate of a pair of duplicate genes is the nonfunctionalization of one copy (Lynch and Conery 2000). Gene nonfunctionalization can be caused by point mutations, insertions, deletions, or epigenetic modifications.
Transposable elements (TEs) are a primary DNA source that causes insertion-mediated dysfunction of a gene. Of these TEs, retrotransposons in particular can generate stable mutations when they are inserted within or near genes because they transpose via replication and the sequence at the insertion site is retained (Kumar and Bennetzen 1999). In addition to the irreversible nature of the insertion, a characteristic of the transposition of a retrotransposon is its inducible nature depending on environmental conditions: in plants, activation of a retrotransposon is under the influence of environmental factors such as cold, pathogen infection, microbial elicitors, tissues culture, protoplast production, and wounding (Hirochika 1993; Mhiri et al. 1997; Pouteau et al. 1994; Takeda et al. 1999; Ivashuta et al. 2002). These observations have led to the hypothesis that retrotransposons may contribute to the environmental adaptation of an organism by creating novel phenotypes through insertional mutagenesis.
Polyploidization is a well-known mechanism of gene duplication in plants. Approximately 70–80% of angiosperm species have undergone polyploidization at some point in their evolutionary history (Moore and Purugganan 2005). Soybean, Glycine max (L.) Merr., is considered to be a typical paleopolyploid species with a complex genome (Lackey 1980; Hymowitz 2004; Shoemaker et al. 2006). The soybean genome actually possesses a high level of duplicate sequences, and furthermore, possesses homoeologous duplicated regions, which are scattered across different linkage groups (Lohnes et al. 1997; Zhu et al. 1994; Shoemaker et al. 1996; Lee et al. 1999). Based on the genetic distances estimated by synonymous substitution measurements for the pairs of duplicated transcripts from EST collections of soybean and Medicago truncatula, Schlueter et al. (2004) estimated that soybean probably underwent two major genome duplication events: one that took place 15 million years ago (MYA) and another 44 MYA. Differential patterns of expression have often been detected between homoeologous genes in soybean (Schlueter et al. 2006, 2007), which indicate that subfunctionalization has occurred in these genes.
We have identified multiple homologs of the gene that encodes phytochrome A (phyA), one of the red light- and far-red light-absorbing photoreceptors, in the soybean genome (Liu et al. 2008). These include two phyA paralogs designated GmphyA1 and GmphyA2. GmphyA2 was mapped on locus E4, which confers photoperiod insensitivity (Liu et al. 2008). The E4 locus was originally identified by extending day length to 20 h with incandescent lamps (Buzzell and Voldeng 1980). The e4 allele does not influence the photoperiod-sensitivity by itself, but in combination with the e3 allele at one of the other loci that control flowering, it conditions photoperiod insensitivity, a trait adaptive to high-latitude environments (Saindon et al. 1989; Cober et al. 1996). Analysis of the GmphyA2 gene from photoperiod-insensitive lines with the recessive allele e4 revealed the insertion of a retrotransposon in exon 1 of the gene, which resulted in dysfunction of the gene (Liu et al. 2008). In contrast to plants homozygous for the E4 allele, which responded to red light and far-red light similarly, near-isogenic lines (NILs) carrying the e4 allele (SORE-1-inserted GmphyA2) produced longer hypocotyls when grown in far-red light than they did when grown in red light, but their hypocotyls were shorter than when grown in complete darkness (Liu et al. 2008), indicating that the mutation alone did not cause a complete loss of phyA function. This genetic redundancy suggests that the presence of duplicate copies of the phyA genes accounts for the generation of photoperiod insensitivity while protecting against the deleterious effects of mutation (Liu et al. 2008).
In the present study, we characterized in detail the retrotransposon inserted in the GmphyA2 gene. We found that the element is a novel Ty1/copia-like retrotransposon and is transcriptionally active. We also showed that the distribution of the element at the locus is confined to cultivated soybean accessions having an early maturing trait, which confers adaptation to high-latitude environments. These results indicate that dysfunction of one copy of duplicate genes via insertion of the retrotransposon has led to the acquisition of adaptive traits for organisms, a novel consequence of nonfunctionalization of duplicate genes by a retrotransposon.
Materials and Methods
Plant Materials
Cultivated soybean line #130I (Abe et al. 2003; Liu et al. 2008) was used to characterize the SORE-1 element (for designation of the element, see Results). Cultivated soybean line Kariyutaka was used for transcriptional analysis of SORE-1. These soybean lines are both homozygous for the e4 allele. Three hundred thirty-two cultivated soybean (G. max) accessions and 85 wild soybean (ssp. soja) accessions were used to analyze the distribution of SORE-1 inserted in the GmphyA2 gene.
Analysis of Phylogenetic Relationships Between SORE-1 and Other Retrotransposons
The amino acid sequences of the reverse transcriptase (RT) of retroelements from various organisms were retrieved from the DDBJ/EMBL/GenBank database through a search for conserved motifs. The exceptions were Mag, Tgmr, and Del, which did not have amino acid sequences in the database. The RT sequences of Mag and Tgmr were obtained by translating the nucleotide sequences from the database. The RT sequence of Del was obtained from the original report (Smyth et al. 1989). Alignment of the protein sequence was done using the CLUSTAL W Multiple Sequence Alignment Program version 1.8 (Thompson et al. 1994). A phylogenetic tree was constructed using the neighbor-joining (NJ) method (Saitou and Nei 1987) based on protein sequences deduced from the nucleotide sequences of retrotransposons. Estimates of evolutionary distance were obtained using Kimura’s method (Kimura 1980) and bootstrap values were calculated with 1,000 replicates.
DNA Gel-Blot Analysis
Total DNA was isolated from mature leaves according to the method of Doyle and Doyle (1987). DNA gel-blot analysis was done as described previously (Liu et al. 2008). A 1.7-kb region in the 5′-portion of the open reading frame (ORF) of SORE-1 was amplified by PCR with primers 5′-CTCCGCACCATGTCCAATAA-3′ and 5′-GACATAGATTATGCTATAAGG-3′ and was used as a probe.
Treatment of Plants with 5-Azacytidine and RT-PCR Analysis
Seeds of cv. Kariyutaka were germinated in a plate on filter paper soaked with 500-μM 5-azacytidine solution. RNA was isolated from root tissues of 3-day-old plants. Isolation of RNA, cDNA synthesis, and RT-PCR were done as described previously (Nagamatsu et al. 2007). A reaction mixture without reverse transcriptase was used as a control to confirm that no amplification occurred from genomic DNA contaminants in the RNA sample. Primers 5′-GGACATAGATTATGCTATAAGG-3′ and 5′-TGGTGAGCCGAAGAGAAGAA-3′ were used to amplify SORE-1 transcripts. Transcripts of the β-tubulin gene were amplified by PCR with primers β-tub-For (5′-GACCCGATAACTTCGTGTTC-3′) and β-tub-Rev (5′-GAGCTTGAGTGTTCGGAAAC-3′) as a control for the RT-PCR.
Analysis of SORE-1 Homologs in the William 82 Genome
To detect SORE-1 homologs, we ran a homology-based search of the genome sequence database of soybean cv. William 82 (http://www.phytozome.net/index.php) using the entire ORF sequence of SORE-1 as a query sequence. Parameters for homology search were set as follows: output format: gapped alignments; comparison matrix: BLOSUM62; word length: 11; expected threshold: 0.1; number of alignments to show: 200; and filter options: off. Sequence comparisons in detail between SORE-1 and SORE-1 homologs were done using BioEdit (Hall 1999). Phylogenetic relationships between SORE-1 and SORE-1 homologs were analyzed using the nucleotide sequence of the region corresponding to the ORF of SORE-1 by the NJ method as described.
Analysis of the Distribution of SORE-1 in GmphyA2
The presence or absence of SORE-1 in exon 1 of GmphyA2 was analyzed by PCR as described previously for analyzing segregation of the E4/e4 alleles in genetic experiments (Liu et al. 2008) using a common forward primer in exon 1 (PhyA2-For; 5′-AGACGTAGTGCTAGGGCTAT-3′) and allele-specific reverse primers in the retrotransposon (PhyA2-Rev/e4; 5′-GCTCATCCCTTCGAATTCAG-3′) or in exon 1 of GmphyA2 (PhyA2-Rev/E4; 5′-GCATCTCGCATCACCAGATCA-3′). PCRs performed in the presence of the three primers resulted in the amplification of a 837-bp fragment when SORE-1 is inserted in exon 1 of GmphyA2 and of a 1,229-bp fragment when SORE-1 is not inserted in the gene. The amplified products were separated by electrophoresis on a 0.8% agarose gel and visualized under UV light.
Results
Identification of a Ty1/copia-Like Retrotransposon in Exon 1 of GmphyA2
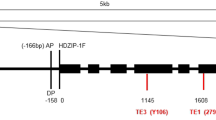
We have found that a sequence of 6,238 bp is inserted at nucleotide 692 from the start codon in exon 1 of the GmphyA2 gene in #130I, a photoperiod-insensitive line of soybean (Fig. 1a; Liu et al. 2008; nucleotide sequence data have been deposited in the DDBJ/EMBL/GenBank database as accession AB370254). The element comprised two 383-bp long terminal repeats (LTRs) and an internal domain of 5,472 bp (Fig. 1b) and was flanked by a 5-bp target-site duplication sequence (5′-AAAAC-3′; according to the orientation of the element described later). The nucleotide sequences of the two LTRs were 100% identical each other and contained 2-bp inverted repeats (5′-TG…CA-3′) in their ends (Liu et al. 2008). These features are canonical for retrotransposons (reviewed by Kumar and Bennetzen 1999). We designated the inserted sequence as SORE-1 (SOybean RetroElement 1) in line with the naming of BARE-1 and RIRE1, which were closely associated with this element in the subsequent phylogenetic analysis (see Fig. 2). Here, we describe the detailed characterization of the ORF of SORE-1.

Characterization of SORE-1. a Position of SORE-1 insertion in the GmphyA2 gene. The open arrow indicates the position and direction of the ORF in SORE-1. The hatched boxes and filled boxes indicate LTRs of SORE-1 and exons of GmphyA2, respectively. Orientation of transcription of the GmphyA2 gene is indicated by an arrow. b Organization of SORE-1. Gray boxes and hatched boxes indicate the ORF and LTRs of the element, respectively. Orientation of the ORF is indicated by an arrow. LTR long terminal repeat, Gag Gag protein, Prot protease, Int integrase, RT reverse transcriptase, RH RNaseH, TSD target site duplication, PBS primer-binding site, PPT polypurine tract. c Nucleotide sequences of PBS and PPT among SORE-1, BARE-1, Tgmr, and Tnt1-94. Identical nucleotides are shown in bold. d Amino acid sequences across conserved domains of proteins encoded by retrotransposons among SORE-1, BARE-1, SIRE-1, Tgmr, Tnt1-94, copia, and Ty1. Amino acid sequences of domains that encompass the RNA-binding motif of Gag, the D(S/T)G motif of protease, the GKGY domain of integrase, and the domains 3–5 of RT are shown. Note that Ty1 does not contain the RNA-binding motif (Petersoon-Burch and Voytas 2002). Amino acid residues highly conserved among aligned sequences (5 or more of the 7 sequences) are in bold. Highly conserved motifs of Gag, protease, and integrase of Pseudoviridae retroelements (Petersoon-Burch and Voytas 2002), and those of RT in retroelements from widely diverse organisms (Xiong and Eickbush 1990) are indicated by asterisks below the sequences

Phylogenetic relationships between SORE-1 and Ty1/copia-like retrotransposons isolated from various organisms. The phylogenetic tree was constructed using the NJ method based on protein sequences deduced from the nucleotide sequences of the RT domains. Protein sequences of RTs of four Ty3/gypsy-like retrotransposons (gypsy, Mag, Ty3-2, and Del) were used as the outgroup to root the tree. Bootstrap values of 1,000 replicates (>500) are shown. A scale bar represents branch length. The following sequences were included in the analysis: Drosophila melanogaster gypsy (M12927), Bombyx mori Mag (X17219), Saccharomyces cerevisiae Ty3-2 (M23367), Lilium henryi Del (X13886), Zea mays Opie-2 (U68408), Arabidopsis thaliana Endovir1-1 (AY016208), G. max SIRE-1 (AF053008), Z. mays Hopscotch (U12626), Oryza longistaminata Retrofit (U72726), A. thaliana AtRE1 (AB021265), A. thaliana Evelknievel (AF039373), G. max Tgmr (U96748), Solanum tuberosum Tst1 (X52387), S. cerevisiae Ty1 (M08706), G. max SORE-1 (AB370254), Z. mays Sto-4 (AF082133), Hordeum vulgare BARE-1 (Z17327), Oryza australiensis RIRE1 (D85597), A. thaliana Ta1-3 (X13291), Nicotiana tabacum Tnt1-94 (X13777), N. tabacum Tto1 (D83003), D. melanogaster copia (M11240), S. cerevisiae Ty5-6p (U19263), and A. thaliana Art1 (Y08010)
In the internal sequence of the element, the sequences of the primer-binding sites (PBSs) and polypurine tract (PPT) were identified adjacent to the LTRs (Fig. 1c). A single, large ORF comprising 3,966 bp was also identified. The amino acid sequence deduced from the nucleotide sequence of the ORF in SORE-1 contained various features that are common to proteins encoded by various retrotransposons as follows. The RNA-binding motif (Cx2Cx4Hx4C) is characteristic of Gag protein and is widespread among retrotransposons (Petersoon-Burch and Voytas 2002). The RNA-binding motif of the protein encoded by SORE-1 was actually CFFCKKKGHMKKNC (Fig. 1d). Similarly, the D(S/T)G motif is characteristic of the catalytic site of protease and was also detected in the protein encoded by the ORF in SORE-1 (Fig. 1d). The integrase and RNaseH of retrotransposons are known to contain protein domains comprising conserved amino acid residues located at intervals. The conserved N-terminal HHCC domain, the catalytic DD35E domain (Haren et al. 1999; Petersoon-Burch and Voytas 2002), and the GKGY motif (Petersoon-Burch and Voytas 2002; Fig. 1d) of integrase were all found in SORE-1. The conserved D10, E48, D70, and D134 residues of the RNaseH (Malik and Eickbush 2001) were also found at corresponding positions in SORE-1 (data not shown). Previous studies have shown that RT is the most conserved coding region in retrotransposons (Xiong and Eickbush 1988). The deduced amino acid sequence of the RT of SORE-1 also had high similarity to the RTs of other retrotransposons (Fig. 1d) throughout the seven conserved domains of this protein (Xiong and Eickbush 1990). The RT sequence was present between the integrase and RNaseH sequences. Both sequence similarity with other retrotransposons and the allocation of motifs in the ORF revealed the presence of Gag–protease–integrase–RT–RNaseH domains in this order (Fig. 1b), which indicated that SORE-1 belongs to the Ty1/copia-like retrotransposon. These analyses also indicated that the element was inserted in exon 1 of the GmphyA2 gene in an orientation opposite to the transcription of the GmphyA2 gene (Fig. 1a).
Based on the protein sequences of the RTs, a phylogenetic analysis of SORE-1 and other Ty1/copia-like retrotransposons was conducted (Fig. 2). The relationships among the Ty1/copia-like retrotransposons revealed on the phylogenetic tree were largely consistent with previous reports of a similar analysis (Laten et al. 1998; Petersoon-Burch and Voytas 2002; Xiao et al. 2007). SORE-1 was grouped with Sto-4 of maize, BARE-1 of barley, and RIRE1 of rice, and was located on a clade that was distinct from the clades with the Ty1/copia-like retrotransposons previously identified in soybean, namely, SIRE-1 (Laten et al. 1998) or Tgmr (Bhattacharyya et al. 1997). Branch formation of SORE-1, Sto-4, BARE-1, and RIRE1 was supported by high bootstrap values. These results indicate that SORE-1 is a novel class of retrotransposon in soybean.
The Presence of SORE-1-Related Elements in the Soybean Genome
The presence or absence of sequences homologous with SORE-1 in the soybean genome was examined by a gel-blot analysis of total DNA isolated from the line #130I (e4e4), which harbors SORE-1 in the GmphyA2 gene, using the 5′-portion of the ORF of SORE-1 as a probe (Fig. 3a). Ten to twenty hybridization signals, excluding weakly hybridized ones, were detected per lane, indicating the presence of multiple sequences homologous with SORE-1 in the genome.

The presence of SORE-1 homologs in the soybean genome and expression of SORE-1. a DNA gel-blot analysis of DraI- or EcoRI-digested #130I genomic DNA with a probe containing the 5′-region of the SORE-1 ORF. Arrows indicate hybridized fragments containing SORE-1 in exon 1 of GmphyA2. b Phylogenetic relationships between SORE-1, SORE-1 homologs present in Williams 82, and elements closely related with SORE-1 in other plants. Phylogenetic tree was constructed based on nucleotide sequence of the region corresponding to the SORE-1 ORF using the NJ method. Nucleotide sequences of Sto-4, BARE-1, and RIRE1 were used as the outgroup to root the tree. Bootstrap values of 1,000 replicates (>500), the presence (+) or absence (−) of intact ORF, and sequence identity between 5′- and 3′-LTRs are shown. c Detection of SORE-1 transcripts by RT-PCR. Total RNA was isolated from root tissues of 3-day-old plants that were grown in the presence or absence of 5-azacytidine (5-aza). Transcripts of β-tubulin were amplified as a control. A reaction mixture without reverse transcriptase (RT) was used as a negative control
The presence of nucleotide sequences similar to SORE-1 in the soybean genome was also examined using a recently released genome sequence database of soybean cv. Williams 82 (http://www.phytozome.net/index.php). Homology search of the database identified 98 sequences that were very similar to SORE-1 over the entire ORF region (Supplementary Table S1). These sequences were dispersed in the genome (Supplementary Table S1). We randomly chose 20 sequences from the 98 sequences and characterized them in detail (Table 1). Unlike SORE-1 in GmphyA2 of plant lines homozygous for the e4 allele, most of these elements harbored termination codon(s) just after the start codon, which results in short ORFs. In addition, there was variation in the extent of sequence identity between 5′- and 3′-LTRs, and only two of them contained LTRs with 100% identity (Table 1). These results suggest that most of these elements underwent sequence changes after insertion at the respective locus and are silent in the genome of Williams 82. A phylogenetic analysis based on nucleotide sequences corresponding to the ORF region of SORE-1 indicated that SORE-1 is closely related to the elements containing both intact ORF and LTRs with 100% or almost 100% identity (Fig. 3b).
The SORE-1 Is Transcriptionally Active and Is Partially Silenced
Transcription of SORE-1 was analyzed by RT-PCR. Gel-blot analysis of DNA using methylation-sensitive restriction enzymes indicated that all three HpaII sites in the SORE-1 ORF were methylated (data not shown), which suggests that transcription of SORE-1 is epigenetically suppressed. We therefore examined whether the level of mRNA from SORE-1 was affected by treating plants with the demethylating agent 5-azacytidine. Seeds of cv. Kariyutaka were germinated in the presence or absence of 5-azacytidine, and RNA was extracted from young roots 3 days after germination. Transcripts of SORE-1 were detected by RT-PCR in plants that were not treated with 5-azacytidine (Fig. 3c), and the level of transcripts prominently increased after treatment with 5-azacytidine (Fig. 3c). These results indicate that SORE-1 is transcribed at least in root tissues, but transcription is partially suppressed by an epigenetic mechanism(s) involving cytosine methylation.
Soybean Lines Carrying the SORE-1-Inserted GmphyA2 Allele Are Distributed Within a Restricted Region of Northern Japan
Soybean is basically a short-day plant, and soybean cultivars adapted to high latitudes are insensitive to photoperiods, which allows flowering under long days and seed production during a limited growing season. Our analyses indicated that inactivation of GmphyA2, which constitutes the e4 allele that confers insensitivity to long days, is caused by the insertion of SORE-1 in exon 1 of the gene (Liu et al. 2008). Based on these findings, we hypothesized that the insertion of SORE-1 in exon 1 of GmphyA2 is one of the major genetic changes that allowed soybeans to grow well at high latitudes. To test this hypothesis, we analyzed the presence or absence of SORE-1 at this locus in various cultivated and wild soybean accessions.
A region encompassing a portion of SORE-1 and the region flanking it was amplified by PCR using DNA isolated from 332 cultivated soybean accessions from various East Asian countries over a wide range of latitude and including regions where cultivated soybean originated (Supplementary Table S2). We also analyzed 85 wild soybean (ssp. soja) accessions that were collected from natural populations in various regions of Japan (Tozuka et al. 1998). While no plants that harbor SORE-1 at the locus were found in the wild soybean lines examined, the SORE-1 insertion at the locus was detected in 10 accessions of cultivated soybean, all of which are grown in northern Japan (Fig. 4). Nine of the ten accessions are ‘Ohyachi 2’ (a pure-line selection from ‘Ohyachi’), ‘Bekkai Zairai’, ‘Fusakushirazu’, ‘Gokuwase Kamishunbetu’, ‘Gonjiro Daizu’, ‘Karafuto 1’, ‘Miharu Daizu’, ‘Ohsodefuri 50’, and ‘Urayama Wase’, which were collected from Hokkaido Island. The remaining accession (‘Col/Aomori/1981/L145’) is from Aomori Prefecture, in northeastern Honshu (the main island of Japan) and the nearest to Hokkaido Island (Hokkaido Prefectural Tokachi Agricultural Experiment Station 1988). All these accessions were photoperiod-insensitive and early maturing (Abe et al. 2003, unpublished data). In addition, historical record indicates that the local variety Ohyachi, introduced by an immigrant from northeastern Japan (Fig. 4), enabled soybean cultivation to expand in the late nineteenth century into the inland, northern, and eastern areas of Hokkaido Island that have harsher environments for soybean cultivation and where various landraces, including those tested in the present study, had been established (Nakamura and Tsuchiya 1991). Thus, the distribution of SORE-1-inserted GmphyA2 in the northern regions of Japan is consistent with the notion that disruption of GmphyA2 by the insertion of SORE-1 contributed to the expansion of cultivated region of soybean toward regions of higher latitude.

Distribution of SORE-1-inserted GmphyA2 in cultivated soybean accessions. The presence or absence of SORE-1-inserted GmphyA2 in soybean accessions cultivated in various regions of East Asia was analyzed by PCR. Geographic categories: northeastern China and far eastern Russia; central China; southern China; Korean Peninsula; and four areas of Japan including Hokkaido, northeastern, central, and western (from north to south). Frequency of SORE-1-inserted GmphyA2 is indicated as a filled area in each chart, and numerals indicate the number of accessions with SORE-1-inserted GmphyA2 versus total number of accessions analyzed
Discussion
Identification of SORE-1 and Its Potential Use as a Source of Insertional Mutagenesis
Retrotransposons are ubiquitous in plants and, in many cases, comprise over 50% of nuclear DNA content (reviewed by Kumar and Bennetzen 1999). In some plants, they represent up to 80% of the genome (Feschotte et al. 2002). In soybean, Ty1/copia-like retrotransposon families, Tgmr (Bhattacharyya et al. 1997) and SIRE-1 (Laten et al. 1998), and a Ty3/gypsy-like retrotransposon family, Diaspora (Yano et al. 2005), have been identified. Sequence comparisons revealed that SORE-1 does not belong to any of these families and thus is a novel retrotransposon. In addition, SORE-1 is the first dicot retrotransposon that grouped with Sto-4, BARE-1, and RIRE1, all of which have been identified in monocot plants.
In plants, retrotransposons are often transcriptionally silenced via epigenetic modifications involving cytosine methylation, thereby suppressing transposition of the retrotransposon (Feschotte et al. 2002). Our expression analyses of SORE-1 in plants with or without 5-azacytidine treatment revealed that SORE-1 was transcriptionally active, although transcription was partially suppressed by such an epigenetic mechanism. Transcriptionally active retrotransposons have been shown to be capable of inducing random disruption of genes in various plants, which is most typically evidenced by Tos17 of rice (Hirochika 2001; Miyao et al. 2007). The presence of transcriptional activity together with the fact that SORE-1 actually disrupted a gene suggests that SORE-1 may provide a transposon-based tool for functional genomics in soybean, in addition to a system using the Ds transposon (Mathieu et al. 2009).
Genetic Redundancy of the PhyA Gene Revealed by the Mutation Caused by Insertion of SORE-1
We have previously reported that there are four copies of the phyA gene in the soybean genome. Two of these, designated GmphyA1 and GmphyA2, were active, while the other two copies were inactive in a photoperiod-sensitive line #130S (Liu et al. 2008). The two phyA copies were found to be not only paralogs, but also homoeologs that resulted from ancient chromosomal duplications and rearrangements. In photoperiod-insensitive lines, GmphyA2 was also inactivated by the insertion of SORE-1, so that GmphyA1 was the only active copy of phyA. The effect of the disruption of GmphyA2 has been further characterized by analyzing plant response to light quality. In contrast to the soybean plants carrying the E4 allele (intact GmphyA2), which responded to red light and far-red light similarly, NILs carrying the e4 allele (SORE-1-inserted GmphyA2) produced longer hypocotyls when grown in far-red light than they did when grown in red light, but their hypocotyls were shorter than when grown in complete darkness (Liu et al. 2008). These observations indicated that the phyA function was lost partially, not completely, in the e4 homozygotes, which led to the notion that the phyA functions involved in the de-etiolation response are genetically redundant. This phenomenon is in contrast to a complete loss of the de-etiolation response under continuous far-red light that is observed for phyA mutants of Arabidopsis, pea and rice, in which the phyA gene is present as a single copy gene (Weller et al. 1997; Neff and Chory 1998; Takano et al. 2001; Weller et al. 2001; Takano et al. 2005). The observed genetic redundancy in soybean is attributed to the presence of multiple copies of the active phyA gene (Liu et al. 2008). Under the presence of the genetic redundancy of phyA, disruption of GmphyA2 through insertion of SORE-1 resulted in a novel phenotype in terms of plant response to both photoperiod and light quality.
Insensitivity to photoperiod allows soybean plants to flower under long day lengths and produce seeds before frost at high latitudes. In fact, the distribution of SORE-1 in the GmphyA2 gene was confined to soybean accessions that are grown only in northern regions of Japan (Fig. 4). Mutant plants carrying the disrupted GmphyA2 were probably selected by local farmers in these regions because of an increase in fitness in a particular environment, namely, ability of plants to mature under a restricted cropping season. Saindon et al. (1989) reported that under the genetic background of e3, plants homozygous for the e4 allele started flowering several days earlier than those homozygous for the E4 allele at Ottawa, Canada (45°25′N). We also obtained similar results at Sapporo, Japan (43°25′N) (Abe et al., unpublished data). It has been known that mutation of GmphyA2 is not the only genetic change that allowed cultivation of soybean plants at high latitudes (Abe et al. 2003). Although loci other than the E4 locus may account for the photoperiod insensitivity of cultivated soybean plants harboring no SORE-1 insertion at the locus, both phenotypic changes and allelic distribution indicate that disruption of GmphyA2 is advantageous for adaptation of soybean plants to higher latitudes.
Evolutionary Significance of Retrotransposon Insertion into a Duplicate Gene
Retrotransposons can destabilize the genome through insertional mutagenesis, deletions, gene rearrangements, introducing polyadenylation signals, or providing a substrate for illegitimate homologous recombination (reviewed by Muotri et al. 2007). In addition to a deleterious effect caused by insertion into a gene, TE-induced characters that may benefit the host organism, e.g., modification of regulatory functions of gene expression, replacing the function of damaged chromosomal ends, and repair of double-strand chromosome breaks, have been reported in eukaryotes (reviewed by McDonald 1995; Kidwell and Lisch 1997). In plants, correlations between the copy number of the BARE-1 retrotransposon, genome size, and local environmental conditions have been detected in naturally grown wild barley, which suggest that retrotransposon integrational activity, by increasing genome size, may be adaptive (Kalendar et al. 2000). Similarly, stress activation of retrotransposon (Hirochika 1993; Mhiri et al. 1997; Pouteau et al. 1994; Takeda et al. 1999; Ivashuta et al. 2002) may reflect the hosts’ response and adaptive process to an environment. Silencing or activation of genes adjacent to retrotransposons by readout transcription from the LTRs synthesizing antisense or sense RNA of the genes, respectively (Kashkush et al. 2003), may also potentially benefit the host. Nonetheless, a mechanistic relationship between insertion of TE and an increase in fitness to an environment has been substantiated in very few eukaryotes: the examples include increased resistance of Drosophila to a pesticide via gene truncation mediated by a long, interspersed element-like TE (Aminetzach et al. 2005) or generation of an early flowering phenotype associated with an increase in the mRNA level of the TaFT gene, an ortholog of Arabidopsis FLOWERING LOCUS T, by insertion of a retrotransposon in the gene promoter in wheat (Yan et al. 2006).
The direct effect of SORE-1 insertion into GmphyA2 is a simple disruption of gene function by the creation of a premature stop codon and the interference of transcription (Liu et al. 2008). However, the most intriguing aspect of this insertion is a resulting increase in fitness in a particular environment, established because of the genetic redundancy brought about by the gene duplication. Thus, these observations consequently revealed a novel fate of the insertion of a retrotransposon into a gene region. Based on these findings, we propose the following model to explain an evolutionary relationship between gene duplication and transposition of retrotransposons. When a retrotransposon is inserted in or around a single-copy gene, the insertion most likely confers more or less deleterious effects to the host organism. On the other hand, when a retrotransposon is inserted in a duplicated gene, the insertion may have a weaker effect on the phenotype, which may even be, in some cases, beneficial to the host organism. On such an occasion, a set of duplicated genes, one of which is disrupted, can contribute to adaptive evolution via natural or artificial selection. In these processes, the inducible nature of the transposition of retrotransposons, which depends on environmental conditions, facilitates the occurrence of mutation and allows retrotransposons to play a more significant role in adaptive evolution compared with other mutagenic events that are not necessarily dependent on the particular environment.
Genes involved in the evolution of domestication traits of plants have been isolated (reviewed by Doebley 2006). Interestingly, none of the genes that contributed to the domestication of diploid and ancient polyploid species discovered so far are null alleles: the mutations in these genes caused changes in protein function and/or gene expression rather than a loss of function of the protein, which led to the notion that domestication involved “tinkering” rather than “crippling” of precisely tuned wild species (Doebley 2006). Dubcovsky and Dvorak (2007) further proposed that, in contrast to the case of diploid species, null mutations of one of the duplicate or triplicate homologous gene copies may have only subtle effects and thus may appear as “tinkering” mutations with a potential to generate adaptive variation in a young polyploid species like wheat. Our results in soybean, a paleopolyploid plant, are consistent with this idea on the point that disruption of one of duplicated genes is involved in adaptation to a particular environment.
Overall, our results thus illustrate that a retrotransposon insertion that causes loss of function of a gene product can be involved in adaptive evolution when the gene is duplicated. The environmental factor(s) that activate transposition of SORE-1 remains to be examined. It is tempting to speculate that plant cells positively regulate transposition of retrotransposons because of their potential advantages to the host and utilize them as a means of diversification, although plant cells normally suppress their transposition by epigenetic mechanisms. In this context, the utility of the retrotransposon as a mutagen is expanded by gene duplication because of the buffering effect brought about by gene duplication.
References
Abe J, Xu DH, Miyano A, Komatsu K, Kanazawa A, Shimamoto Y (2003) Photoperiod-insensitive Japanese soybean landraces differ at two maturity loci. Crop Sci 43:1300–1304
Aminetzach YT, Macpherson JM, Petrov DA (2005) Pesticide resistance via transposition-mediated adaptive gene truncation in Drosophila. Science 309:764–767
Bhattacharyya MK, Gonzales RA, Kraft M, Buzzell RI (1997) A copia-like retrotransposon Tgmr closely linked to the Rps1-k allele that confers race-specific resistance of soybean to Phytophthora sojae. Plant Mol Biol 34:255–264
Birchler JA, Veitia RA (2007) The gene balance hypothesis: from classical genetics to modern genomics. Plant Cell 19:395–402
Buzzell RI, Voldeng HD (1980) Inheritance of insensitivity to long daylength. Soybean Genet Newslett 7:26–29
Cober ER, Tanner JW, Voldeng HD (1996) Soybean photoperiod-sensitivity loci respond differentially to light quality. Crop Sci 36:606–610
Doebley J (2006) Unfallen grains: how ancient farmers turned weeds into crops. Science 312:318–319
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Dubcovsky J, Dvorak J (2007) Genome plasticity a key factor in the success of polyploidy wheat under domestication. Science 316:1862–1866
Feschotte C, Jiang N, Wessler SR (2002) Plant transposable elements: where genetics meets genomics. Nat Rev Genet 3:329–341
Force A, Lynch M, Pickett FB, Amores A, Yan Y, Postlethwait J (1999) Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151:1531–1545
Freeling M, Thomas BC (2006) Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome Res 16:805–814
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Haren LB, Ton-Hoang B, Chandler M (1999) Integrating DNA: transposases and retroviral integrases. Annu Rev Microbiol 53:245–281
Hirochika H (1993) Activation of tobacco retrotransposons during tissue culture. EMBO J 12:2521–2528
Hirochika H (2001) Contribution of the Tos17 retrotransposon to rice functional genomics. Curr Opin Plant Biol 4:118–122
Hokkaido Prefectural Tokachi Agricultural Experiment Station (1988) The origins and characteristics of soybean accessions. Misc Pub Hokkaido Pref Tokachi Agric Expt Sta 11:1–175
Hughes AL (1994) The evolution of functionally novel proteins after gene duplication. Proc R Soc Lond B 256:119–124
Hymowitz T (2004) Speciation and cytogenetics. In: Boerma HR, Specht JE (eds) Soybeans: improvement, production, and uses, Ed 3, Agronomy Monograph No. 16. American Society of Agronomy, Inc., Crop Science Society of America, Inc., Soil Science Society of America, Inc. Madison, WI, pp 97–136
Ivashuta S, Naumkina M, Gau M, Uchiyama K, Isobe S, Mizukami Y, Shimamoto Y (2002) Genotype-dependent transcriptional activation of novel repetitive elements during cold acclimation of alfalfa (Medicago sativa). Plant J 31:615–627
Kalendar R, Tanskanen J, Immonen S, Nevo E, Schulman AH (2000) Genome evolution of wild barley (Hordeum spontaneum) by BARE-1 retrotransposon dynamics in response to sharp microclimatic divergence. Proc Natl Acad Sci USA 97:6603–6607
Kashkush K, Feldman M, Levy AA (2003) Transcriptional activation of retrotransposons alters the expression of adjunct genes in wheat. Nat Genet 33:102–106
Kidwell MG, Lisch D (1997) Transposable elements as sources of variation in animals and plants. Proc Natl Acad Sci USA 94:7704–7711
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kumar A, Bennetzen JL (1999) Plant retrotransposons. Annu Rev Genet 33:479–532
Lackey JA (1980) Chromosome numbers in the Phaseoleae (Fabaceae: Faboideae) and their relation to taxonomy. Am J Bot 67:595–602
Laten HM, Majumdar A, Gaucher EA (1998) SIRE-1, a copia/TY1-like retroelement from soybean, encodes a retroviral envelope-like protein. Proc Natl Acad Sci USA 95:6897–6902
Lee JM, Bush AL, Specht JE, Shoemaker RC (1999) Mapping of duplicate genes in soybean. Genome 42:829–836
Liu B, Kanazawa A, Matsumura H, Takahashi R, Harada K, Abe J (2008) Genetic redundancy in soybean photoresponses associated with duplication of the phytochrome A gene. Genetics 180:995–1007
Lohnes DG, Specht JE, Cregan PB (1997) Evidence for homoeologous linkage groups in the soybean. Crop Sci 37:254–257
Lynch M, Conery JS (2000) The evolutionary fate and consequences of duplicate genes. Science 290:1151–1155
Lynch M, Force A (2000) The probability of duplicate gene preservation by subfunctionalization. Genetics 154:459–473
Malik HS, Eickbush TH (2001) Phylogenetic analysis of ribonuclease H domains suggests a late, chimeric origin of LTR retrotransposable elements and retroviruses. Genome Res 11:1187–1197
Mathieu M, Winters EK, Kong F, Wan J, Wang S, Eckert H, Luth D, Paz M, Donovan C, Zhang Z, Somers D, Wang K, Nguyen H, Shoemaker RC, Stacey G, Clemente T (2009) Establishment of a soybean (Glycine max Merr. L) transposon-based mutagenesis repository. Planta 229:279–289
McDonald JF (1995) Transposable elements: possible catalysts of organic evolution. Trends Ecol Evol 10:123126
Mhiri C, Morel JB, Vernhettes S, Casacuberta JM, Lucas H, Grandbastien MA (1997) The promoter of the tobacco Tnt1 retrotransposon is induced by wounding and by abiotic stress. Plant Mol Biol 33:257–266
Miyao A, Iwasaki Y, Kitano H, Itoh J, Maekawa M, Murata K, Yatou O, Nagato Y, Hirochika H (2007) A large-scale collection of phenotypic data describing an insertional mutant population to facilitate functional analysis of rice genes. Plant Mol Biol 63:625–635
Moore C, Purugganan MD (2005) The evolutionary dynamics of plant duplicate genes. Curr Opin Plant Biol 8:122–128
Muotri AR, Marchetto MCN, Coufal NG, Gage FH (2007) The necessary junk: new functions for transposable elements. Human Mol Genet 16:R159–R167
Nagamatsu A, Masuta C, Senda M, Matsuura H, Kasai A, Hong JS, Kitamura K, Abe J, Kanazawa A (2007) Functional analysis of soybean genes involved in flavonoid biosynthesis by virus-induced gene silencing. Plant Biotechnol J 5:778–790
Nakamura S, Tsuchiya T (1991) Soybean. In: Nomura N, Sasaki K, Sanbuichi K, Nakamura S, Minami T, Tsuchiya T, Chiba K, Iida S, Okuyama T, Tsukada Y (eds) Legume varieties in Hokkaido. Japan Beans and Peas Foundation, Tokyo, pp 37–158
Neff MM, Chory J (1998) Genetic interactions between phytochrome A, phytochrome B, and cryptochrome 1 during Arabidopsis development. Plant Physiol 118:27–36
Petersoon-Burch BD, Voytas DF (2002) Genes of the Pseudoviridae (Ty1/copia retrotransposons). Mol Biol Evol 19:1832–1845
Pouteau S, Grandbastien M-A, Boccara M (1994) Microbial elicitors of plant defence responses activate transcription of a retrotransposon. Plant J 5:532–542
Rodin SN, Riggs AD (2003) Epigenetic silencing may aid evolution by gene duplication. J Mol Evol 56:718–729
Saindon G, Voldeng HD, Beversdorf D, Buzzell RI (1989) Genetic control of long daylength response in soybean. Crop Sci 29:1436–1439
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Schlueter JA, Dixon P, Granger C, Grant D, Clark L, Doyle JJ, Shoemaker RC (2004) Mining EST databases to resolve evolutionary events in major crop species. Genome 47:868–876
Schlueter JA, Scheffler BE, Schlueter SD, Shoemaker RC (2006) Sequence conservation of homoeologous bacterial artificial chromosomes and transcription of homoeologous genes in soybean (Glycine max L. Merr). Genetics 174:1017–1028
Schlueter JA, Vasylenko-Sanders IF, Deshpande S, Yi J, Siegfried M, Roe BA, Schlueter SD, Scheffler BE, Shoemaker RC (2007) The FAD2 gene family of soybean: insights into the structural and functional divergence of a paleopolyploid genome. Crop Sci 47:S-14–S-26
Shoemaker RC, Polzin K, Labate J, Specht JE, Brummer EC, Olson T, Young N, Concibido V, Wilcox J, Tamulonis JP, Kochert G, Boerma HR (1996) Genome duplication in soybean (Glycine subgenus Soja). Genetics 144:329–338
Shoemaker RC, Schlueter JA, Doyle JF (2006) Paleopolyploidy and gene duplication in soybean and other legumes. Curr Opin Plant Biol 9:104–109
Smyth DR, Kalitsis P, Joseph JL, Sentry JW (1989) Plant retrotransposon from Lilium henryi is related to Ty3 of yeast and the gypsy group of Drosophila. Proc Natl Acad Sci USA 86:5015–5019
Takano M, Kanegae H, Shinomura T, Miyano A, Hirochika H, Furuya M (2001) Isolation and characterization of rice phytochrome A mutants. Plant Cell 13:521–534
Takano M, Inagaki N, Xie X, Yuzurihara N, Hihara F, Ishizuka T, Yano M, Nishimura M, Miyao A, Hirochika H, Shinomura T (2005) Distinct and cooperative functions of phytochromes A, B, and C in the control of deetiolation and flowering in rice. Plant Cell 17:3311–3325
Takeda S, Sugimoto K, Otsuki H, Hirochika H (1999) A 13-bp cis-regulatory element in the LTR promoter of the tobacco retrotransposon Tto1 is involved in response to tissue culture, wounding, methyl jasmonate and fungal elicitors. Plant J 18:383–393
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tozuka A, Fukushi H, Hirata T, Ohara M, Kanazawa A, Mikami T, Abe J, Shimamoto Y (1998) Composite and clinal distribution of Glycine soja in Japan revealed by RFLP analysis of mitochondrial DNA. Theor Appl Genet 96:170–176
Weller JL, Murfet IC, Reid JB (1997) Pea mutants with reduced sensitivity to far-red light define an important role for phytochrome A in day-length detection. Plant Physiol 114:1225–1236
Weller JL, Beauchamp N, Kerckhoffs LHJ, Platten JD, Reid JB (2001) Interaction of phytochromes A and B in the control of de-etiolation and flowering in pea. Plant J 26:283–294
Xiao W, Su Y, Sakamoto W, Sodmergen (2007) Isolation and characterization of TY1/copia-like retrotransposons in mung bean (Vigna radiata). J Plant Res 120:323–328
Xiong Y, Eickbush TH (1988) Similarity of reverse transcriptase-like sequences of viruses, transposable elements, and mitochondrial introns. Mol Biol Evol 5:675–690
Xiong Y, Eickbush TH (1990) Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J 9:3353–3362
Yan L, Fu D, Li C, Blechl A, Tranquilli G, Bonafede M, Sanchez A, Valarik M, Yauda S, Dubcovsky J (2006) The wheat and barley vernalization gene VRN3 is an orthologue of FT. Proc Natl Acad Sci USA 103:19581–19586
Yano ST, Panbehi B, Das A, Laten HM (2005) Diaspora, a large family of Ty3-gypsy retrotransposons in Glycine max, is an envelope-less member of an endogenous plant retrovirus lineage. BMC Evol Biol 5:30
Zhu T, Schupp JM, Oliphant A, Keim P (1994) Hypomethylated sequences: characterization of the duplicated soybean genome. Mol Gen Genet 244:638–645
Acknowledgments
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Kanazawa, A., Liu, B., Kong, F. et al. Adaptive Evolution Involving Gene Duplication and Insertion of a Novel Ty1/copia-Like Retrotransposon in Soybean. J Mol Evol 69, 164–175 (2009). https://doi.org/10.1007/s00239-009-9262-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-009-9262-1