
Abstract
The unexpected presence of archaea-like genes for tetrahydromethanopterin (H4MPT)-dependent enzymes in the completely sequenced genome of the aerobic marine planctomycete Pirellula sp. strain 1 (“Rhodopirellula baltica”) and in the currently sequenced genome of the aerobic freshwater planctomycete Gemmata obscuriglobus strain UQM2246 revives the discussion on the origin of these genes in the bacterial domain. We compared the genomic arrangement of these genes in Planctomycetes and methylotrophic proteobacteria and performed a phylogenetic analysis of the encoded protein sequences to address the question whether the genes have been present in the common ancestor of Bacteria and Archaea or were transferred laterally from the archaeal to the bacterial domain and therein. Although this question could not be solved using the data presented here, some constraints on the evolution of the genes involved in archaeal and bacterial H4MPT-dependent C1-transfer may be proposed: (i) lateral gene transfer (LGT) from Archaea to a common ancestor of Proteobacteria and Planctomycetes seems more likely than the presence of the genes in the common ancestor of Bacteria and Archaea; (ii) a single event of interdomain LGT can be favored over two independent events; and (iii) the archaeal donor of the genes might have been a representative of the Methanosarcinales. In the bacterial domain, the acquired genes evolved according to distinct environmental and metabolic constraints, reflected by specific rearrangements of gene order, gene recruitment, and gene duplication, with subsequent functional specialization. During the course of evolution, genes were lost from some planctomycete genomes or replaced by orthologous genes from proteobacterial lineages.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Planctomycetes (order Planctomycetales) comprise a distinct phylum within the bacterial domain by 16S rRNA analysis (Garrity 2001). Four cultured genera have been described within the order Planctomycetales: Pirellula, Planctomyces, Gemmata, and Isosphaera. Consistent with their independent phylogenetic position, members of the Planctomycetes show several characteristics that are unusual for Bacteria (Fuerst 1995), such as the lack of peptidoglycan as a component of their cell wall (Konig et al. 1984; Liesack et al. 1986), a budding reproduction, and the presence of intracellular compartments (Lindsay et al. 1997), some of which are membrane-bounded nucleoids reminiscent of the eukaryotic nucleus. Furthermore, there is increasing evidence from cultivation-independent studies that Planctomycetes are of considerable ecological importance: they frequently account for a significant part of the microbial communities of diverse marine and terrestrial habitats (Neef et al. 1998; Strous et al. 1999; Wang et al. 2002), which also indicates a broad physiological potential in the planctomycete lineage. The first whole-genome analysis of a member of the Planctomycetes, the aerobic marine organism Pirellula sp. strain 1 (currently in the process of being validly described as “Rhodopirellula baltica” [Schlesner et al. 2004], has been completed recently (Glöckner et al. 2003). This allowed us to explore the physiological repertoire of a planctomycete model organism on a molecular level. Whole-genome sequencing is currently in process also for three further representatives of the Planctomycetes, Gemmata obscuriglobus UQM2246 (Ward et al. 2003), Gemmata sp. strain Wa1-1 (IntegratedGenomics; GOLD: Genomes online database), and “Kuenenia stuttgartiensis” (www.anammox.com).
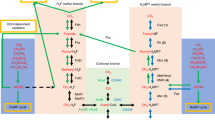
Surprisingly, the annotation of the “R. baltica” genome revealed the presence of genes encoding proteins with significant similarity to tetrahydromethanopterin (H4MPT)-dependent enzymes (Glöckner et al. 2003). Archaea-like genes encoding these enzymes were noticed only some years ago to exist in the bacterial domain as part of the C1 metabolism in aerobic methylotrophic proteobacteria (Chistoserdova et al. 1998; Vorholt et al. 1999). Prior to their discovery in methylotrophic proteobacteria, H4MPT-dependent enzymes had been considered unique for the ecologically specialized energy metabolism of strictly anaerobic methanogenic (Thauer 1998) and sulfate-reducing archaea (Klenk et al. 1997). In methylotrophic proteobacteria, proteins encoded by the archaea-like genes function in a detoxifying pathway which oxidizes formaldehyde to formate and is especially important for the organisms during growth on C1 compounds when high amounts of the cytotoxin formaldehyde are generated (Chistoserdova et al. 1998; Marx et al. 2003). In Fig. 1 the reactions comprising this pathway are schematically depicted in comparison to those involved in archaeal methanogenesis. Reactions in this methylotrophic pathway are catalyzed in part by the archaea-like enzymes (Fae, Mch, Ftr/Fmd) and in part by uniquely bacterial enzymes (MtdB/A). Fae is the formaldehyde-activating enzyme (Vorholt et al. 2000), MtdA is a methylene-H4MPT/methylene-tetrahydrofolate (H4F) dehydrogenase (NADP+ dependent; EC 1.5.1.5 [Vorholt et al. 1998]), MtdB is a methylene-H4MPT dehydrogenase (NAD+/NADP+ dependent; EC 1.5.1.— [Hagemeier et al. 2000]), and Mch is the methenyl-H4MPT cyclohydrolase (EC 3.5.4.27 [Pomper et al. 1999]). Ftr is the archaeal formylmethanofuran:H4MPT-formyltransferase (formyltransferase; EC 2.3.1.101) and Fmd is the archaeal formylmethanofuran dehydrogenase (formyl-MFR-DH; EC 1.2.99.5 [Vorholt and Thauer 2002]). It has been shown in methylotrophic proteobacteria that Ftr together with three subunits of Fmd (FmdA, FmdB, and FmdC) forms the formyltransferase/hydrolase complex (Fhc), which transfers the H4MPT-bound formyl group to a second cofactor, a compound analogous to the archaeal cofactor methanofuran, and concomitantly hydrolyzes it to formate (Pomper et al. 2002).

Schematic representation of the archeal and proteobacterial pathways involving H4MPT-dependent enzymes. CH2=H4MPT: methylene-H4MPT. CH≡H4MPT+: methenyl-H4MPT. CHO-H4MPT: Formyl-H4MPT. MFR: methanofuran. Enzyme abbreviations are as given in Table 1. Mtd (archaeal pathway): F420-dependent-H4MPT dehydrogenase (EC 1.5.99.9). Fhc: formyltransferase/hydrolase complex (Pomper et al. 2002). Letters A, B, and P in squares: enzyme present in (domain) Archaea, (phylum) Proteobacteria, and (phylum) Planctomycetes. Filled squares: protein has been isolated and characterized. Open squares: gene product not yet isolated or characterized.
To account for the fact that H4MPT-dependent enzymes outside the archaeal domain have only been found in Proteobacteria so far, lateral gene transfer (LGT) from an archaeal donor to a proteobacterium has been suggested as the most likely explanation (Chistoserdova et al. 1998; Vorholt et al. 1999). Moreover, the clear separation of proteobacterial sequences from their archaeal counterparts observed in a previous phylogenetic analysis of Mch has been taken as an indication of a single interdomain gene transfer event rather than multiple independent events (Vorholt et al. 1999).
The intriguing finding that “R. baltica” as a representative of the independent phylum of the Planctomycetes possesses the archaea-like genes led us to inspect further planctomycete genome data available so far for the presence of these genes and to reconstruct their phylogeny in order to get a deeper insight into the mechanisms putatively involved in the evolution of the archaea-like methanogenesis genes in the bacterial domain.
Materials and Methods
Sequences
Amino acid sequences of the following taxa were included in the phylogenetic analyses. Archaea: Aeropyrum pernix K1, Archaeoglobus fulgidus DSM 4304, Halobacterium sp. strain NRC-1, Methanocaldococcus jannaschii DSM2661, Methanococcus maripaludis, Methanosarcina acetivorans C2A, Methanopyrus kandleri AV19, Methanosarcina barkeri Fusaro, Methanosarcina mazei Goel, Methanothermobacter marburgensis, Methanothermobacter thermautotrophicus, Methanothermobacter wolfeii, Methanothermus fervidus, Pyrococcus abyssi GE5, Pyrococcus furiosus DSM 3638, Pyrococcus horikoshii OT3, Sulfolobus solfataricus P2, Sulfolobus tokodaii 7. Bacteria: Burkholderia fungorum LB400, Gemmata obscuriglobus UQM2246, Hyphomicrobium methylovorum, Methylobacillus flagellatus KT, Methylobacterium extorquens AM1, Methylobacterium organophilum, Methylococcus capsulatus Bath, Methylococcus thermophilus, Methylomicrobium album, Methylomonas rubra, Methylophilus methylotrophus, Methylosinus trichosporium, Pirellula sp. strain 1 (“Rhodopirellula baltica”), Xanthobacter autotrophicus. Sequence accession numbers are provided in the phylogenetic trees in Fig. 3.
In general, protein sequences were retrieved from databases of the International Nucleotide Sequence Database Collaboration (GenBank/EMBL/DDBJ). The genome of “R. baltica” has been fully sequenced and annotated recently within the framework of the REGX project (http://www.regx.de/) (Glöckner et al. 2003). For the unfinished genomes of G. obscuriglobus UQM2246 and M. capsulatus Bath, preliminary sequence data from The Institute for Genomic Research (http://www.tigr.org/), and for those of B. fungorum LB400 and M. barkeri, preliminary sequence data from The DOE Joint Genome Institute (JGI; http://www.jgi.doe.gov/tempweb/JGI_microbial/html/index.html) were accessible through the BLAST database of unfinished microbial genome sequences at the National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov/sutils/genom_table.cgi). Amino acid sequences encoded by the genes under study from methanogenic archaea, methylotrophic proteobacteria, and “R. baltica” were used to identify regions encoding similar proteins in the unfinished genomes by BLAST similarity searches (default settings). The nucleotide sequences of these regions of the unfinished genomes were translated in all six reading frames and the appropriate amino acid sequences were extracted. In Fig. 3, the regions of the DNA sequence contigs that were translated to yield the respective amino acid sequences are indicated.
All publicly available sequences, including paralogs, of FmdA, FmdB, FmdC, Ftr, Mch, Fae, and β-RFAP-synthase were used in the analyses. In the case of FmdC, MTH106 (“tungsten formylmethanofuran dehydrogenase, subunit C homolog”; TrEMBL:O26209), MTH192 (“tungsten formylmethanofuran dehydrogenase, subunit C homolog”; TrEMBL:O26294), and MJ1350 (“hypothetical protein, belongs to the fwdC/fmdC family”; SwissProt: YD50_METJA) were not considered for the analyses since they have been classified in COG0070 (glutamate synthase domain 3) rather than in COG2218 (FwdC) (http://www.ncbi.nlm.nih.gov/COG/).
In general, full-length sequences were used. Exceptions are indicated in Fig. 3 and summarized in the following: (i) For the Mch protein of some organisms, only partial sequence information was available. (ii) FmdC sequences from M. thermautotrophicus, M. marburgensis, and M. wolfeii exhibit an FwdD-like domain in their C terminus and were consequently truncated C terminally according to the domain boundary given in COG2218 (FwdC) for the M. thermautotrophicus FmdC sequence. (iii) In some Archaea, open reading frames (ORFs) of unknown function occur which exhibit sequence similarity to 3-hexulose 6-phosphate synthase (Humps; EC 4.1.2.—) in their C-terminal domain and similarity to Fae in their N-terminal domain (Vorholt et al. 2000). The Fae-like sequence portions of these products of larger archaeal ORFs were included in the analyses. (iv) In five cases, sequences have been shortened at their N terminus after inspection of initial multiple alignments because of likely incorrect gene start prediction: “R. baltica” RB9834 (FmdA), M. acetivorans MA0833 (FwdA), B. fungorum (FmdB and Ftr), and M. thermautotrophicus MTH1474 (Humps-related protein).
Throughout the text, gene names are noted in lowercase. Gene product names begin with a capital letter.
Alignments
Amino acid sequence alignments were constructed with ClustalW 1.8 (Thompson et al. 1994) using the Gonnet250 substitution matrix (Gonnet et al. 1994). ClustalW’s initial pairwise alignments were done in slow/accurate mode. A gap open penalty of 10 and a gap extension penalty of 0.1 (pairwise alignments)/0.2 (multiple alignments) was employed. Where possible, ClustalW alignments were checked with appropriate seed alignments of the Pfam database (Pfam, release 11; http://www.sapger.ac.uk/Software/Pfam/index.shtml): PF02289 for Mch, PF01493 for FmdC, and PF01913 and PF02741 for Ftr.
Phylogenetic Analysis
Length heterogeneity at the beginning and end of sequences was masked prior to phylogenetic analyses, keeping positions 6–598 (96% of original alignment positions) for FmdA, positions 26–493 (93%) for FmdB, positions 36–342 (85%) for FmdC, positions 21–350 (93%) for Ftr, positions 25–364 (88%) for Mch, positions 11–183 (91%) for Fae, and positions 27–374 (89%) for β-RFAP-synthase. No indel gap positions were removed. Phylogenetic trees were reconstructed using the maximum likelihood method implemented in MrBayes v3 (http://morphbank.ebc.uu.se/mrbayes/) (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003). MrBayes has been shown to be one of the most accurate likelihood programs available (Williams and Moret 2003). It performs Bayesian inference estimations to assess phylogeny and is sufficiently fast to allow branch support evaluation by posterior probabilities based on a large set of sampled trees. Phylogenetic analyses with MrBayes were carried out using the Jones amino acid substitution model, four Markov chains, and an approximated gamma distribution of evolutionary rates with five categories. For each set of aligned sequences, 5000 trees were sampled, of which the first 300 were discarded as “burn-in” of the Markov chains. The remaining 4700 trees were used to construct a majority rule consensus tree, showing posterior clade probabilities and a topology based on mean branch lengths. Trees were visualized with ARB (http://www.arb-home.de/).
Additionally, maximum likelihood and maximum parsimony trees were calculated using the programs ProML (Jones/Taylor/Thornton model of amino acid substitution, one category of sites, constant rate of change among sites, 10 jumbles of species input order, global rearrangements) and ProtPars (default settings) from the phylogenetic inference package PHYLIP v.3.6a4 (http://evolution.genetics.washington.edu/phylip.html) with 100 bootstrap samples. Extended majority rule consensus trees were constructed with the program Consense from PHYLIP v.3.6a4.
Results
Genes
The genome of “R. baltica” harbors four genes encoding polypeptides with considerable sequence similarity to H4MPT-dependent enzymes/enzyme subunits from methanogenic and sulfate-reducing archaea (41–66% average sequence similarity) as well as from methylotrophic proteobacteria (29–68% average sequence similarity): the genes encoding subunits A (FmdA) and C (FmdC) of formylmethanofuran dehydrogenase (Fmd), formylmethanofuran:H4MPT-formyltransferase (Ftr), and methenyl-H4MPT cyclohydrolase (Mch). Inspection of the unfinished genome sequence of G. obscuriglobus UQM2246 revealed that in this organism the four genes are also present. Moreover, G. obscuriglobus possesses a gene encoding a protein with high similarity to the catalytic subunit B of archaeal Fmd which could not be detected in “R. baltica.” In addition and also in contrast to “R. baltica,” an ftr paralog (ftr-2; Ftr-1 and Ftr-2 share 38% amino acid identity) is present in G. obscuriglobus. Both the “R. baltica” and the G. obscuriglobus genomes, respectively, contain two genes, fae-1 and fae-2, encoding proteins with high sequence similarity to the formaldehyde-activating enzyme from methylotrophic proteobacteria and to putative Fae-like gene products from Methanopyrus kandleri and members of the genus Methanosarcina. Fae-1 and Fae-2 share 28% (G. obscuriglobus) and 31% amino acid identity (“R. baltica”), respectively.
Besides archaea-like genes encoding Fae and H4MPT-dependent enzymes, the genomes of the two planctomycetes also harbor a gene encoding a protein with high similarity (52% average) to the novel bifunctional H4MPT/H4F-methylene dehydrogenase MtdA of methylotrophic proteobacteria. In contrast to methylotrophs, however, a gene for the strictly H4MPT-dependent methylene-dehydrogenase MtdB is not present in “R. baltica” and could also not be found in G. obscuriglobus, although this is a preliminary result since genome sequence data are not yet complete for this organism.
In Methylobacterium extorquens, fae and the genes encoding the H4MPT-dependent enzymes have been found in a cluster with several other genes whose precise function has not been elucidated so far (orf5, orfY, orf7, orf9 and orf17 [Chistoserdova et al. 1998]). However, from mutational studies on M. extorquens it is known that they play an important role during growth on C1 compounds. Some of them may be involved in the biosynthesis of cofactors necessary for methylotrophy (orf7, orf9, and orf17) (Chistoserdova et al. 2003). Recently, the product of one of the genes present in the M. extorquens cluster, orf4 (Chistoserdova et al. 1998), has been characterized as β-RFAP (β-ribofuranosyl-aminobenzene-5′-phosphate)-synthase (Scott and Rasche 2002). This enzyme catalyzes the first reaction distinguishing the methanopterin biosynthesis pathway from that of folate biosynthesis.
We found that both “R. baltica” and G. obscuriglobus harbor a gene encoding a protein with high sequence similarity to β-RFAP synthase (41% average). The two planctomycete strains also seem to have genes encoding polypeptides similar to the gene products of orf5, orf7, and orfY, albeit this similarity is less pronounced than in the case of the other proteins mentioned above. Orf9- and Orf17-like gene products could only be detected in “R. baltica” and not in G. obscuriglobus at this stage of the genome sequencing effort. Their sequence similarity to methylotrophic counterparts is also limited.
In summary (Table 1), both planctomycete genomes encode proteins with high sequence similarity to enzymes required for a methylotrophic pathway (Chistoserdova et al. 1998; Vorholt 2002) that produces energy from C1-substrates and/or detoxifies the toxic intermediate formaldehyde generated during growth on these substrates. Furthermore, the two members of the Planctomycetes under study here also seem to possess at least one of the genes (and probably others) encoding proteins for the biosynthesis of the cofactor methanopterin on which the activity of the enzymes mentioned above depends.
Genomic Arrangement of the Genes
Figure 2 shows the genomic context of the genes encoding H4MPT enzymes in “R. baltica” and G. obscuriglobus in comparison to representatives of proteobacterial methylotrophs for which sequence data of large genomic fragments were available. In contrast to what is known about the location of the genes in methanogenic and sulfate-reducing archaea, in the alphaproteobacterium M. extorquens, the genes encoding the proteins that are involved in the detoxifying formaldehyde oxidation pathway cluster on the genome (Chistoserdova et al. 1998). When we inspected preliminary whole-genome sequence data of other methylotrophic proteobacteria for the arrangement of the genes of interest, we found that this clustering is by and large conserved, with some exceptions where disruptions occur (Fig. 2). The gene order within these cluster(s) on methylotroph genomes is similar, albeit not entirely conserved.

Genomic arrangement of genes involved in H4MPT-dependent C1-transfer: comparison of methylotrophic proteobacteria and Planctomycetes. Enzyme abbreviations are as given in Table 1, RPS: β-RFAP synthase. Boxed arrows (dotted lines): gene or “gene module” present in the genome; arrangement of genes or modules relative to each other and on the genome unknown (due to incomplete genome sequence data). Shaded areas: inversion of “gene module.” M. extorquens: GenBank accession numbers AF032114 and L27235; position of the mtdA gene from Vorholt et al. (1999). Arrows, representing the genes in their coding direction, are not drawn to scale. Physical distances between the genes of less than 2 kb are not indicated by spaces between the arrows.
In contrast, counterparts of these archaea-like genes in “R. baltica” are scattered more widely over the genome and their gene order is quite different from that seen in methylotrophs. For G. obscuriglobus at this stage of the sequencing effort the precise genomic arrangement of the genes cannot be deduced. However, it is already obvious that G. obscuriglobus shares some “gene modules” with “R. baltica” and methylotrophs, respectively (Fig. 2). Regarding chromosomal gene order of the archaea-like genes in Bacteria, two observations are remarkable, which are considered in more detail in the discussion: (i) only in “R. baltica” and G. obscuriglobus are the fae (fae-1) and mtdA genes adjacent to each other; and (ii) the genomic arrangement of the genes coding for Ftr and the different subunits of Fmd is identical in all known bacterial genomes containing these archaea-like genes (Fig. 2; solid-line box). Of the two ftr genes in G. obscuriglobus, only one (ftr-1) is found in a genomic context of genes encoding H4MPT-dependent enzymes: In keeping with what has been found for methylotrophic proteobacteria and “R. baltica,” it is located upstream of fmdC. The genomic context of ftr-2 in G. obscuriglobus is not related to genes encoding H4MPT-dependent enzymes (data not shown).
Phylogenetic Analysis of the Proteins Encoded by the Archaea-like Genes
Phylogenetic analyses in this study are mainly based on maximum likelihood reconstruction methods (MrBayes, ProML) since these are known to be least prone to reconstruction artifacts such as long branch attraction. In addition, maximum parsimony (ProtPars) analysis was performed. Figure 3 summarizes the results of tree reconstructions, showing the consensus topologies resulting from the MrBayes analyses. ProML and ProtPars topologies are not displayed. In the following, wherever ProML and/or ProtPars topologies disagree with MrBayes’ topology, alternative branching patterns are described and branch support of the respective method is given in parentheses.

Unrooted phylogenetic trees for FmdA (FwdA; FhcA), FmdB (FwdB or FwuB; FhcB), FmdC (FwdC; FhcC), Ftr (FfsA or FhcD), Mch, Fae, and β-RFAP synthase, constructed with MrBayes (maximum likelihood). Details of the phylogenetic reconstruction are given under Materials and Methods. Branch support is generally shown as posterior clade probability by MrBayes (based on 4700 sampled trees) and coded in shaded circles as indicated. Confidence values for planctomycete branchings are shown in the order “posterior clade probability” by MrBayes/bootstrap value by ProML (100 sets)/bootstrap value by ProtPars (100 sets). Dashes indicate that the respective treeing method yielded a different topology than MrBayes, which is described under Results. Bars represent estimated sequence divergence. Full species names are given under Materials and Methods. Parentheses after species names provide the sequence accession number (generally: sequence information accessible through http://www.ncbi.nlm.nih.gov/; sp: SwissProt, http://www.expasy.org/), the GenBank “locus_tag” (e.g., MA1710), where applicable, as well as—in the case of incomplete genome sequence data—the contig number together with the translated region of the nucleotide sequence and—in the case of sequences that were used only in part—the amino acid (aa) range of the respective sequence (see also Materials and Methods). partial: full sequence information was not present in the public databases. FwdA(B,C): subunit of tungsten Fmd, FwuB: subunit B of tungsten Fmd, selenocysteine containing. FmdA(B,C): subunit of molybdenum Fmd. FhcA(B,C,D): subunit of formylhydrolase complex. Humps, Hps-2: hexulose-6-phosphate synthase (D-arabino 3-hexulose-6-phosphate formaldehyde lyase). Only the N-terminal, Fae-like domains of these products of larger archaeal open reading frames (see Vorholt et al. 2000) were included in the analysis and used as an outgroup in the Fae tree.
Tree Topologies in the Bacterial Clade
In the Mch tree as well as in the β-RFAP synthase tree, the planctomycete sequences group together at a position between the bacterial and the archaeal sequence clusters. In the case of the components of the Fhc complex (FmdB, FmdA, Ftr, and FmdC), only the sequences of G. obscuriglobus show this deep-branching position, whereas the FmdA, FmdC, and Ftr sequences of “R. baltica” fall within the bacterial clade (FmdB is absent in this organism). The Ftr-2 sequence of G. obscuriglobus clusters with a set of archaeal Ftr-like sequences some distance from the other archaeal Ftr sequences. The Fae tree is remarkable in that it does not show the clear separation of archaeal and bacterial sequences seen in the other trees (the affiliation of Ftr-2 from G. obscuriglobus with archaeal paralogs being the single exception). Instead, Fae sequences from methylotrophic proteobacteria and Planctomycetes as well as methanogenic archaea (Methanosarcina and Methanopyrus) are interspersed. The planctomycete Fae-1 paralogs are more closely related to each other than they are to the Fae-2 paralogs, and vice versa, suggesting a fae gene duplication in the Planctomycetes that preceded planctomycete speciation.
As shown in Fig. 3, all of the G. obscuriglobus branches as well as the branches of planctomycete clusters are supported with more than 0.95 MrBayes clade probability. Furthermore, all “R. baltica” branches within the bacterial cluster are supported with clade probability values greater than 0.75. The analyses with ProML and ProtPars support the planctomycete branches of MrBayes’ topology with typically more (only in some cases on the Ftr and Fae trees with less) than 70% bootstrap. However, not in all cases are ProML and ProtPars consensus tree topologies congruent with that of the MrBayes consensus tree. Discrepancies in the bacterial cluster affect predominantly the “R. baltica” position: (i) In the FmdA tree, according to ProML (64) and ProtPars (100), “R. baltica” branches in a cluster with a B. fungorum/M. capsulatus group (ProML, 68; ProtPars, 90); (ii) in the FmdC tree, according to ProtPars, “R. baltica” branches (36) after M. extorquens and before B. fungorum (39) and M. capsulatus (94); (iii) in the Ftr tree, ProML places “R. baltica” (87) in a cluster with a B. fungorum/M. capsulatus group (50); and (iv) in the β-RFAP synthase tree, ProtPars does not support the planctomycete cluster of the MrBayes topology but rather exhibits a topology in which “R. baltica” branches (32) after G. obscuriglobus (58) at the deepest position within the bacterial cluster.
Aside from the varying branching positions of the “R. baltica” sequences, the topologies of the bacterial clusters in the trees in Fig. 3 are consistent with those of the currently accepted 16S rRNA tree for FmdA, FmdB, Ftr, and, in principle, Fae (although archaeal sequences are interspersed with the bacterial sequences in this tree): (((Alphaproteobacteria, (Betaproteobacteria, Gammaproteobacteria)), Planctomycetes)) (Garrity 2001). In contrast, the FmdC and β-RFAP synthase trees deviate from this 16S rRNA tree topology within the bacterial cluster, as does the Mch tree (Fig. 3). In the latter, only Gammaproteobacteria appear monophyletic, whereas some members of the Alpha- and Betaproteobacteria show unusual affiliations (X. autotrophicus with Gammaproteobacteria, albeit with relatively low branch support, some Betaproteobacteria with Alphaproteobacteria, and B. fungorum some distance to the other Betaproteobacteria closer to the base of the bacterial cluster).
Tree Topologies in the Archaeal Clade
Regarding the branching pattern in the archaeal clusters of the MrBayes consensus trees shown in Fig. 3, the FmdA, FmdB, FmdC, and Ftr trees, show a by-and-large comparable topology which does not contradict the currently accepted 16S rRNA species tree topology (Garrity 2001). In Archaea, an ftr-gene duplication seems to have occurred and a divergent evolution of the paralogs is obvious from their branching position some distance to the remainder of the archaeal sequences in the trees as well as from the long branches.
Extensive archaeal gene duplication, especially in the genus Methanosarcina, is seen for fmdA, fmdB, and fmdC. A common theme of the FmdA, FmdB, and FmdC trees shown in Fig. 3 is the occurrence of Fwd and Fmd clusters as well as the presence of Methanosarcina–A. fulgidus Fwd clusters, which branch separately and at positions closer to the base of the archaeal clade than the Methanothermobacter/Methanocaldococcus/Methanopyrus Fwd clusters. The relative positions of these Fwd, Fmd, and Methanosarcinal–A. fulgidus Fwd clusters to each other vary among FmdA, FmdB, and FmdC trees.
The topology in the archaeal clade of the Mch tree shown in Fig. 3 is essentially comparable to that of the Fmd-subunit trees and the Ftr tree, except that sequences from A. fulgidus and the genus Methanosarcina do not form a cluster but branch next to each other in the maximum likelihood trees, and a halobacterial sequence branches between the Methanothermobacter/Methanocaldococcus/Methanopyrus cluster and A. fulgidus. The archaeal branching pattern in the β-RFAP synthase tree in Fig. 3 is noteworthy in that it shows obvious contradictions to the currently accepted species phylogeny based on 16S rRNA (Garrity 2001): sequences from anaerobic methanogenic Euryarchaeota (Methanocaldococcus) cluster with those from aerobic sulfur-oxidizing Crenarchaeota (Sulfolobus), and sequences from anaerobic hyperthermophilic Euryarchaeota (Pyrococcus) cluster with those from aerobic hyperthermophilic Crenarchaeota.
Conserved “Signatures” of Amino Acids Putatively Involved in Mch Catalysis
Detailed analysis of the 3D structure of the M. kandleri Mch combined with earlier observations on the characterized bifunctional human H4F-dependent enzyme (Allaire et al. 1998) has led to the notion that two residues might be potentially involved in the catalytic function of Mch: lysine94 and tyrosine190 (Grabarse et al. 1999). Inspection of the alignment of Mch sequences available to date (Fig. 4) reveals that at the two positions that are homologous to lysine94 and tyrosine190 of the M. kandleri enzyme, identical residues (lysine and tyrosine, respectively) are present in methanogenic archaea except for members of the genus Methanosarcina. The archaeal sulfate-reducer and halophile sequences exhibit lysine and phenylalanine at the respective positions, whereas in all bacterial Mch sequences a tyrosine and a histidine residue are strictly conserved. Interestingly, at these positions of presumed functional importance, members of the genus Methanosarcina show the same “signature” as all Bacteria harboring these archaea-like genes (for which complete sequence information for Mch is available).

Conserved “signatures” of amino acid residues potentially involved in Mch catalysis. Residues shown in bold may be involved in the catalytic activity of Mch (as discussed by Grabarse et al. 1999). Highlighting shows that all bacterial species exhibit the same “signature” as members of the genus Methanosarcina.
Discussion
Surprisingly, two members of the independent bacterial phylum of the Planctomycetes, “R. baltica” and G. obscuriglobus, were found to harbor archaea-like genes encoding proteins similar to H4MPT-dependent enzymes. Thus, after their relatively recent and unexpected detection in methylotrophic proteobacteria (Chistoserdova et al. 1998), these genes have now been discovered in a second phylum of the bacterial domain, a finding that raises two interesting questions: (i) Are these genes functional in planctomycete species, and if so, which metabolic role do they play? and (ii) What were the evolutionary processes leading to the observed patchy occurrence of the pathway in which these archaea-like genes participate?
For methylotrophic proteobacteria, it is known that the archaea-like genes together with special bacterial genes constitute a pathway that is essential during growth on C1 compounds (Chistoserdova et al. 1998). It has been demonstrated in the alphaproteobacterium Methylobacterium extorquens AM1, a well-studied methylotrophic model organism, that this pathway accounts for the efficient detoxification of formaldehyde generated in large amounts under these conditions (Marx et al. 2003). The role of the archaea-like and bacteria-like genes found in Planctomycetes remains to be elucidated. Members of this phylum do not have a record of methylotrophic metabolism, and preliminary physiological studies on “R. baltica” do not show a capability of this organism to grow on common C1-substrates (Schlesner and Gade, personal communication). Moreover, primary oxidation systems for C1-substrates that might generate formaldehyde as an intermediate could not be detected in the genome of “R. baltica” (Glöckner et al. 2003). Interestingly, however, a normalized codon usage analysis (Karlin and Mrazek 2000) of the “R. baltica” genes predicts fae-1 to be highly expressed (Lombardot, unpublished data). This distinct codon adaptation of fae-1 indicates special metabolic constraints imposed on the Fae-1 protein in “R. baltica.” Moreover, a comprehensive survey of the “R. baltica” proteome revealed that fae-1, mtdA, and mch are expressed (Gade et al., personal communication). Thus, there is evidence that at least some of these genes are of physiological relevance to “R. baltica.”
In the first place, however, the unexpected detection of the archaea-like genes in Planctomycetes revives the discussion on the origin of these genes in the bacterial domain that was stirred with their discovery in methylotrophic proteobacteria. Figure 5 schematically summarizes different evolutionary scenarios that may be imagined to account for the observed isolated occurrence of the archaea-like genes in members of the Proteobacteria and Planctomycetes. We consider the genes encoding H4MPT-dependent enzymes as originally archaeal, since they are involved in the single dissimilatory pathway currently known, e.g., in methanogenic archaea, whereas they seem to be dispensable under certain conditions for methylotrophic proteobacteria (Chistoserdova et al. 2003; Marx et al. 2003; Vorholt 2002). Consequently, evolutionary pathways in Fig. 5 are oriented exclusively in the direction from Archaea to Bacteria.

Possible evolutionary scenarios leading to the observed distribution of originally archaeal genes in the bacterial domain. Straight arrows indicate interdomain and curved arrows intradomain LGT events. Dotted lines symbolize events of gene loss; thick lines mean gene retention over evolutionary time. Thick dashed lines indicate retention of the genes only in certain groups of the indicated phylum/domain. Planctomycetes are not shown as the deepest-branching bacterial phylum since their phylogenetic position is still debated (Brochier and Philippe 2002; Di Giulio 2003; Jenkins and Fuerst 2001, and references therein) and a deepest branching of the Planctomycetes is not supported by a most recent analysis from our group (Teeling et al. 2004).
Figure 5, Panel 1: The Archaeal Genes Entered the Bacterial Kingdom in Two Independent Events of Lateral Transfer
This evolutionary scenario seems quite plausible, since (i) unlike the scenario illustrated in panel 4, it would not have to postulate the rather improbable occurrence of massive parallel gene loss to explain the isolated existence of the archaea-like genes in only two independent bacterial phyla, and (ii) it has been suggested earlier that exchange of genetic material between the archaeal and the bacterial domain takes place to a remarkable extent (Deppenmeier et al. 2002; Nelson et al. 1999).
Despite the general plausibility of the evolutionary scenario depicted in Fig. 5, panel 1, the observation of a conserved gene order “findA–ftr–findC” in Proteobacteria and Planctomycetes (Fig. 2) would argue against two independent events of lateral interdomain transfer. In both members of the Planctomycetes, and in methylotrophic proteobacteria, the ftr gene is located adjacent to a gene(s) encoding a subunit(s) of the Fmd enzyme, although the presence of the conserved gene arrangement in G. obscuriglobus can be approved only in part at this time (gene order ftr–fmdC). In contrast, the archaeal counterparts of the bacterial genes encoding Fmd subunits and Ftr are localized a great distance from each other on the genomes of extant archaea (data not shown). It seems highly unlikely that the observed bacterial arrangement should have arisen twice in two independent bacterial phyla by independent LGT events from archaeal genomes on which the ftr genes are separated widely from the Fmd subunit genes. In “R. baltica,” conservation of this gene order could be the result of LGT from Proteobacteria (see discussion of panel 2), and as such its existence does not count as an argument against the hypothesis of dual LGT between domains. Nevertheless, a valid argument against this hypothesis persists in the evidence for conservation of the gene order also in G. obscuriglobus, whose corresponding sequences underwent separate evolution from proteobacterial sequences (Fig. 3).
We consider the discussion of the conserved order of fmdA–ftr–fmdC as particularly relevant, since this arrangement mirrors a physical interaction of the gene products (Fmd subunits and Ftr) in methylotrophic Proteobacteria, which does not exist in Archaea: In methylotrophic proteobacteria the encoded proteins form a functional complex and have been proven to catalyze a different biochemical reaction compared to the archaeal proteins—transfer of the formyl group to an MFR analogue and hydrolysis to formate compared to transfer and dehydrogenation to carbon dioxide (Pomper et al. 2002). Thus, the rearrangement of genes into the conserved order findA-ftr-fmdC found in Bacteria seems to have accompanied a functional adaptation of the gene products of archaea-like genes in the bacterial domain which reflects different metabolic needs due to different environmental conditions (e.g., oxic versus anoxic habitats).
An additional argument against two independent events of LGT between Archaea and Bacteria arises from the fact that all bacterial Mch sequences, including those from Planctomycetes, show a conserved signature of the residues putatively involved in Mch catalysis (Fig. 4). This signature is identical with the residue pattern found in members of a single archaeal genus, Methanosarcina, and differs from the signature(s) shown by the other archaeal sequences.
Figure 5, Panel 2: Proteobacteria Were the Primary Recipients of the Archaeal Genes. Planctomycetes—and Probably, although as yet undetected, Other Bacterial Phyla—Picked up the Archaeal Genes from Proteobacteria by Lateral Transfer
Proteobacteria would lend themselves to being likely primary recipients of the archaeal genes in the light of two hypotheses on the origin of the eukaryotic cell (Martin and Muller 1998; Moreira and Lopez-Garcia 1998), which both propose the existence of an intimate symbiotic association of representatives of Proteobacteria with an archaeal methanogen. Such a close association would have facilitated the transfer of even large chunks of genetic material, thus enabling a concerted transfer of pathway components also in the case of their potential dispersed localization on the archaeal donor genome. Then, in the progenitor of Proteobacteria, these archaeal genes evolved further and were subsequently distributed to other bacterial lineages like the Planctomycetes.
The apparent closer relationship of FmdA, FmdC, and Ftr sequences from “R. baltica” to methylotroph sequences than to those of G. obscuriglobus (Fig. 3) could be taken as an indication for such a LGT, albeit there is no proof for this potential transfer being actually directed from Proteobacteria to “R. baltica”: Neither are the genes in this organism conspicuous with respect to their G+C content, nor did a normalized codon usage analysis of the “R. baltica” genes (Karlin and Mrazek 2000) classify any of the archaea-like genes as “putative alien” (PA) (Lombardot, unpublished data). Thus, there is no evidence for lateral transfer of the genes of interest to the “R. baltica” genome, at least not for a recent event.
Although scenario 2 (curved arrow) may explain the history of fmdA, ftr, and fmdC in “R. baltica,” it seems unlikely that the evolutionary route leading to the distribution of the archaeal genes in the bacterial domain can be described by this scenario in general: The majority of the protein sequences encoded by the archaea-like genes in Planctomycetes branch some distance to the methylotroph sequences at the base of the bacterial clade. This suggests a separate evolution of the genes in the planctomycete lineage and in methylotrophic proteobacteria, which would be inconsistent with the general hypothesis of their lateral transfer from Proteobacteria to Planctomycetes, and vice versa (see panel 3, path b).
Figure 5, Panel 3: Planctomycetes Were the Primary Recipients of the Archaeal Genes. From There, the Genes Were Either Passed on Vertically to Other Bacterial Lineages (a) or Transferred Laterally to Proteobacteria (b)
Provided that Planctomycetes constitute a deep-branching bacterial phylum, which has been suggested by recent phylogenetic analyses (Brochier and Philippe 2002; Di Giulio 2003) and some earlier analyses (Jenkins and Fuerst 2001 and references therein), they would represent possible candidates for the primary uptake of the genes from an ancient archaeal donor. The assumption of further vertical inheritance of the genes (path a in scenario 3) would have to invoke rather improbable multiple events of independent gene loss to account for the observed isolated occurrence of the archaea-like genes in the bacterial domain. Most recent findings of our group, however, place Planctomycetes in relationship to Chlamydia and do not show evidence for a deep-branching position (see discussion of panel 4 and Teeling et al. [2004]). In this light, path a becomes much more likely. Provided that the archaea-like genes and the bacterial genes forming the pathway depicted in Fig. 1 were always part of the same metabolic module, some facts arguing in favor of scenario 3a (and against scenario 2) are implied in the absence of mtdB in Planctomycetes and in the adjacent localization of mtdA and fae-1 in Planctomycetes. In methylotrophic proteobacteria, mtdB most likely arose from a duplication of the mtdA gene, and MtdB was subsequently adapted to function specifically with H4MPT in the effective oxidative detoxification of formaldehyde generated under certain growth conditions (Hagemeier et al. 2000). Planctomycetes seem to have a more ancient version of the pathway found in methylotrophic proteobacteria: In the absence of MtdB, they would have been dependent on MtdA catalyzing the step of the formaldehyde oxidation pathway that follows the Fae-catalyzed reaction (Fig. 1). Thus, a concerted expression and regulation of the two genes would have been advantageous for Planctomycetes and is suggested by the operon-like organization observed exclusively in members of this phylum (Fig. 2). An alternative scenario, the loss of mtdB due to different metabolic constraints in Planctomycetes after lateral import of the pathway from methylotrophic proteobacteria, and concomitant or subsequent serendipitous clustering of mtdA and fae-1, seems very unlikely.
As already discussed for scenario 2 (curved arrow), path b Fig. 5, panel 3, as the general evolutionary route of the archaeal genes in the bacterial domain would be inconsistent with the observed separate evolution of most of the genes in Planctomycetes and Proteobacteria concluded from their separate branching positions in the phylogenetic trees (Fig. 3).
Figure 5, Panel 4: The Common Ancestor of Bacteria and Archaea Was Already Equipped with the Genes
The discovery of the archaea-like genes in at least two of the four cultured genera of the Planctomycetes together with the recently suggested deepest-branching position of this phylum in the Bacteria (Brochier and Philippe 2002) immediately brings to mind the possibility that the genes of interest may have already been present in the common ancestor of Bacteria and Archaea (scenario 4 in Fig. 5). This hypothesis would not suffer from the dilemma of how to explain how a functional pathway in Bacteria can emerge from lateral transfer of several genes that are putatively scattered widely on the genome of the archaeal donor. On the other hand, however, countless independent events of traceless gene loss would have to be assumed on the bacterial and on the archaeal side, which seems rather unlikely. In addition, a strong selective pressure for retention of the genes in anaerobic archaeal methanogens and sulfate reducers as well as in aerobic methylotrophic proteobacteria and Planctomycetes would have to be postulated. Although such a strong selective force is obvious for methanogens, as they rely on methanogenesis as the sole energy-producing process, the physiological relevance of the pathway involving the archaea-like genes varies in methylotrophs (Chistoserdova et al. 2003; Marx et al. 2003; Vorholt 2002), and its functional significance in Planctomycetes is altogether unknown. Moreover, a most recent reevaluation of the phylogenetic position of the Planctomycetes, based on a large data set of concatenated ribosomal proteins and DNA-directed RNA-polymerase subunits as well as on genome trees of “R. baltica,” does not show evidence for a deepest branching of the Planctomycetes but rather reaffirms their earlier proposed relationship to Chlamydia (Teeling et al. 2004). In the light of these arguments, we consider the presence of the genes of interest in the common ancestor of Bacteria and Archaea combined with multiple differential loss as the explanation for the observed patchy distribution of these archaea-like genes in the bacterial domain as rather unlikely.
Synopsis
The picture of the evolution of the genes involved in archaeal and bacterial H4MPT-dependent C1-transfer that arises from the data presented here seems to be most consistent with the scenario shown in Fig. 6: A common ancestor of Planctomycetes and Proteobacteria received the archaeal genes by lateral transfer from an archaeal donor that might have been a representative of Methanosarcina, since all members of this genus show the “bacterial” signature of putative functional amino acids in the Mch protein (Fig. 4). In the bacterial domain, the acquired genes subsequently evolved according to distinct environmental and metabolic constraints, which is reflected by rearrangements of gene order (fmdA–ftr–fmdC) and concomitant functional divergence (Pomper et al. 2002), by gene recruitment (mtdA) as well as by gene duplication (ftr, fae, mtdB) and subsequent functional specialization (for MtdB and probably also for Ftr and Fae paralogs). In the course of evolution, some of the genes were lost from planctomycete genomes (fmdB in “R. baltica”) and some may have been replaced by orthologous genes from proteobacterial lineages (fmdA, fmdC, and ftr in “R. baltica”).

Synopsis: most probable evolutionary scenario leading to the observed distribution of originally archaeal genes in the bacterial domain, based on the data presented here. Symbols are as in Fig.5. The thin dotted arrow symbolizes partial loss of the genes in the genus Pirellula.
That LGT played a significant role in the evolution of the genes involved in archaeal and bacterial H4MPT-dependent C1-transfer is strongly suggested by the observed clustered arrangement of the genes in methylotrophic proteobacteria vs. their dispersed localization in methanogenic and sulfate-reducing archaea, since LGT has been proposed to be a major driving force for the formation and maintenance of operons (Lawrence 1999; Lawrence and Roth 1996). We assume that initially only one event of interdomain LGT has occurred (discussion of Fig. 5, panel 1; Fig. 6). However, bacterial Fae may have been transferred secondarily to the archaeal domain (not indicated in Fig. 6) according to the branching position of Methanosarcina sequences among bacterial sequences in the Fae tree (Fig. 3).
In addition, the inconsistencies with the 16S rRNA species tree (Garrity 2001) observed in the bacterial clusters of the FmdC, Mch, and β-RFAP synthase trees may indicate repeated events of LGT within the bacterial domain (not shown in Fig. 6), as may the varying branching positions of the “R. baltica” sequences in the FmdA, FmdC, and Ftr trees. However, it is difficult to assess whether the instability of the “R. baltica” branching positions—and also the general differences in bacterial branching patterns among the FmdA/FmdB/Ftr/Fae trees (α,(β,γ)), the FmdC tree ((α,β),γ), and the Mch/β-RFAP synthase trees ((α,γ),β)—rather results from the scarce bacterial sequence information and the imbalanced species representation between the Mch tree and the rest of the trees or reliably indicates independent lateral transfer of pathway fractions. Separate dissemination in the bacterial domain of pathway chunks containing the genes for Mch and β-RFAP synthase seems at least possible considering the inversion of the gene modules observed between methylotrophic proteobacteria (Fig. 2, shaded areas).
In Archaea, the gene encoding β-RFAP synthase has apparently been exchanged laterally between cren- and euryarchaeota, and the branching position of the halobacterial Mch sequence next to that of A. fulgidus in the phylogenetic tree also suggests an event of LGT. Halobacterium sp. strain NRC-1 could have received its mch gene laterally from the archaeal sulfate reducer, which would be corroborated by the identical signature of potentially functional amino acids in the two sequences (Fig. 4). Alternatively, considering its long branch, the halobacterial Mch sequence might be a highly diverged single remnant of the whole pathway once present in this organism, since Halobacteriales share a common root with certain methanogens (Garrity 2001).
With respect to the potential archaeal donor of the genes under study here, it is tempting to speculate that it belongs to Methanosarcina, even though conservation of two potentially catalytic amino acids in the Mch protein may be only a weak hint about its identity: Some representatives of Methanosarcina are found in syntrophic relationships with Bacteria in certain anaerobic environments and may represent a possible gate for LGT between Bacteria and Archaea. The analysis of the complete genome of M. mazei has yielded strong evidence for such transfer events between domains (Deppenmeier et al. 2002).
Considerations
Overall, some critical points have to be kept in mind while interpreting the data presented here. (i) The above combination of evidence available for single genes (mch) or gene groups (fmdA/fmdC/ftr, archaea-like genes, bacterial genes) to assess the history of the whole pathway assumes an initially coherent dissemination of these pathway components, which cannot be proven. (ii) By the methods of phylogenetic reconstruction used in this study it is not possible to exclude that the apparent deep branching of the planctomycete sequences has been caused by artifacts of phylogenetic reconstruction such as long branch attraction (LBA), although, of all treeing methods commonly used, maximum likelihood is known to be least prone to this effect. LBA might be an explanation for the observed association of the Ftr-2 paralog from G. obscuriglobus with a group of highly diverged archaeal Ftr-like sequences and, likewise, for the observed clustering of the highly diverged planctomycete Fae-2-paralogs with the Fae sequence from M. kandleri. (iii) The separate branching of the majority of planctomycete sequences at the base of the bacterial cluster was interpreted to indicate the presence of the genes in a common ancestor of Proteobacteria and Planctomycetes but not very likely in the last common ancestor of Bacteria and Archaea. This conclusion is based on our recent reevaluation of the phylogenetic branching position of Planctomycetes (Teeling et al. 2004) but it should be emphasized that the phylogenetic position of the Planctomycetes is still a matter of discussion (Brochier and Philippe 2002; Di Giulio 2003; Jenkins and Fuerst 2001).
Conclusions
In summary, the data presented here allow us to propose some constraints on the processes involved in the evolution of archaea-like C1-transfer genes in Bacteria. However, they do not solve the dilemma of their origin in the bacterial domain: None of the scenarios depicted in Fig. 5 can be supported unequivocally, and for the summary shown in Fig. 6 several assumptions in the interpretation of the data have to be made, whose validity cannot be proven. Further work will be needed for a clearer assessment of the processes involved in the evolution of these genes. For instance, the inclusion of so far missing sequence information for all the organisms known to harbor the archaea-like genes, combined with the application of elaborate phylogenetic methods based on the selection of alignment positions with particular evolutionary rates, will enhance the discriminatory power of the phylogenetic analyses. Further insights may also be gained by putting the sequence alignments under new scrutiny as knowledge on functionally important sites of the proteins encoded by the genes becomes available from biochemical and structural research. Additional information generated by these efforts might change the picture drawn here of the evolutionary history of the archaea-like C1-transfer genes, as might the potential discovery of the archaea-like genes in bacterial lineages other than Proteobacteria and Planctomycetes.
References
M Allaire YG Li RE MacKenzie M Cygler (1998) ArticleTitleThe 3-D structure of a folate-dependent dehydrogenase/cyclohydrolase bifunctional enzyme at 1.5 angstrom resolution Structure 6 173–182
C Brochier H Philippe (2002) ArticleTitlePhylogeny—A non-hyperthermophilic ancestor for Bacteria Nature 417 244–244
L Chistoserdova S-W Chen A Lapidus ME Lidstrom (2003) ArticleTitleMethylotrophy in Methylobacterium extorquens AM1 from genomic point of view J Bacteriol 185 2980–2987 Occurrence Handle1:CAS:528:DC%2BD3sXjs1aqu7s%3D Occurrence Handle12730156
L Chistoserdova JA Vorholt RK Thauer ME Lidstrom (1998) ArticleTitleC-1 transfer enzymes and coenzymes linking methylotrophic bacteria and methanogenic archaea Science 281 99–102 Occurrence Handle10.1126/science.281.5373.99 Occurrence Handle1:CAS:528:DyaK1cXksVOmsrw%3D Occurrence Handle9651254
U Deppenmeier A Johann T Hartsch R Merkl RA Schmitz R Martinez-Arias A Henne A Wiezer S Baumer C Jacobi H Bruggemann T Lienard A Christmann M Bomeke S Steckel A Bhattacharyya A Lykidis R Overbeek HP Klenk RP Gunsalus HJ Fritz G Gottschalk (2002) ArticleTitleThe genome of Methanosarcina mazei: Evidence for lateral gene transfer between Bacteria and Archaea J Mol Microbiol Biotechnol 4 453–461 Occurrence Handle1:CAS:528:DC%2BD38XlsVGks7Y%3D Occurrence Handle12125824
M Di Giulio (2003) ArticleTitleThe ancestor of the Bacteria domain was a hyperthermophile J Theor Biol 224 277–283
JA Fuerst (1995) ArticleTitleThe Planctomycetes—Emerging models for microbial ecology, evolution and cell biology Microbiology-UK 141 1493–1506
G Garrity (2001) Bergey’s manual of systematic bacteriology Springer New York
F Glöckner M Kube M Bauer H Teeling T Lombardot W Ludwig D Gade A Beck K Borzym K Heitmann R Rabus H Schlesner R Amann R Reinhardt (2003) ArticleTitleComplete genome sequence of the marine planctomycete Pirellula sp strain 1. Proc Nat Acad Sci USA 100 8298–8303
GH Gonnet MA Cohen SA Benner (1994) ArticleTitleAnalysis of amino acid substitution during divergent evolution: the 400 by 400 dipeptide substitution matrix Biochem Biophys Res Commun 199 489–496
W Grabarse M Vaupel JA Vorholt S Shima RK Thauer A Wittershagen G Bourenkov HD Bartunik U Ermler (1999) ArticleTitleThe crystal structure of methenyltetrahydromethanopterin cyclohydrolase from the hyperthermophilic archaeon Methanopyrus kandleri Structure 7 1257–1268
CH Hagemeier L Chistoserdova ME Lidstrom RK Thauer JA Vorholt (2000) ArticleTitleCharacterization of a second methylene tetrahydromethanopterin dehydrogenase from Methylobacterium extorquens AM1 Eur J Biochem 267 3762–3769 Occurrence Handle10.1046/j.1432-1327.2000.01413.x Occurrence Handle1:CAS:528:DC%2BD3cXksVKgtbg%3D Occurrence Handle10848995
JP Huelsenbeck F Ronquist (2001) ArticleTitleMRBAYES: Bayesian inference of phylogenetic trees Bioinformatics 17 754–755 Occurrence Handle10.1093/bioinformatics/17.8.754 Occurrence Handle1:STN:280:DC%2BD3MvotV2isw%3D%3D Occurrence Handle11524383
IntegratedGenomics (GOLD: Genomes online database) Genome sequencing of Gemmata sp. str. Wal-1
C Jenkins JA Fuerst (2001) ArticleTitlePhylogenetic analysis of evolutionary relationships of the planctomycete division of the domain bacteria based on amino acid sequences of elongation factor Tu J Mol Evol 52 405–418
S Karlin J Mrazek (2000) ArticleTitlePredicted highly expressed genes of diverse prokaryotic genomes J Bacteriol 182 5238–5250
H-P Klenk A Clayton Rebecca J-F Tomb O White E Nelson Karen A Ketchum Karen J Dodson Robert M Gwinn K Hickey Erin D Peterson Jeremy L Richardson Delwood R Kerlavage Anthony E Graham David C Kyrpides Nikos D Fleischmann Robert J Quackenbush H Lee Norman G Sutton Granger S Gill F Kirkness Ewen A Dougherty Brian K McKenny D Adams Mark B Loftus S Peterson I Reich Claudia K McNeil Leslie H Badger Jonathan A Glodek L Zhou R Overbeek D Gocayne Jeannine F Weidman Janice L McDonald T Utterback D Cotton Matthew T Spriggs P Artiach P Kaine Brian M Sykes Sean W Sadow Paul A Garland Stacey M Mason Tanya J Olsen Gary M Fraser Clare O Smith Hamilton R Woese Carl JC Venter (1997) ArticleTitleThe complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus Nature 390 364–370 Occurrence Handle10.1038/37052 Occurrence Handle9389475
E Konig H Schlesner P Hirsch (1984) ArticleTitleCell-wall studies on budding bacteria of the Planctomyces Pasteuria group and on a Prosthecomicrobium sp. Arch Microbiol 138 200–205
J Lawrence (1999) ArticleTitleSelfish operons: The evolutionary impact of gene clustering in prokaryotes and eukaryotes Curr Opin Genet Dev 9 642–648
JG Lawrence JR Roth (1996) ArticleTitleSelfish operons: Horizontal transfer may drive the evolution of gene clusters Genetics 143 1843–1860 Occurrence Handle1:CAS:528:DyaK28XlvVSqt78%3D Occurrence Handle8844169
W Liesack H Konig H Schlesner P Hirsch (1986) ArticleTitleChemical composition of the peptidoglycan-free cell envelopes of budding bacteria of the Pirella Planctomyces group Arch Microbiol 145 361–366
MR Lindsay RI Webb JA Fuerst (1997) ArticleTitlePirellulosomes: A new type of membrane-bounded cell compartment in planctomycete bacteria of the genus Pirellula Microbiology-UK 143 739–748
W Martin M Muller (1998) ArticleTitleThe hydrogen hypothesis for the first eukaryote Nature 392 37–41 Occurrence Handle10.1038/32096 Occurrence Handle1:CAS:528:DyaK1cXhvVOht74%3D Occurrence Handle9510246
CJ Marx L Chistoserdova ME Lidstrom (2003) ArticleTitleFormaldehyde-detoxifying role of the tetrahydromethanopterin-linked pathway Methylobacterium extorquens AM1 J Bacteriol 185 760–768
D Moreira P Lopez-Garcia (1998) ArticleTitleSymbiosis between methanogenic archaea and delta-proteobacteria as the origin of eukaryotes: The syntrophic hypothesis J Mol Evol 47 517–530
A Neef R Amann H Schlesner KH Schleifer (1998) ArticleTitleMonitoring a widespread bacterial group: In situ detection of Planctomycetes with 16S rRNA-targeted probes Microbiology-UK 144 3257–3266
KE Nelson RA Clayton SR Gill ML Gwinn RJ Dodson DH Haft EK Hickey LD Peterson WC Nelson KA Ketchum L McDonald TR Utterback JA Malek KD Linher MM Garrett AM Stewart MD Cotton MS Pratt CA Phillips D Richardson J Heidelberg GG Sutton RD Fleischmann JA Eisen O White SL Salzberg HO Smith JC Venter CM Fraser (1999) ArticleTitleEvidence for lateral gene transfer between Archaea and Bacteria from genome sequence of Thermotoga maritima Nature 399 323–329 Occurrence Handle10.1038/20601 Occurrence Handle1:CAS:528:DyaK1MXjs1WnsLo%3D Occurrence Handle10360571
BK Pomper O Saurel A Milon JA Vorholt (2002) ArticleTitleGeneration of formate by the formyltransferase/hydrolase complex (Fhc) from Methylobacterium extorquens AMI FEBS Lett 523 133–137
BK Pomper JA Vorholt (2001) ArticleTitleCharacterization of the formyltransferase from Methylobacterium extorquens AM1 Eur J Biochem 268 4769–4775
BK Pomper JA Vorholt L Chistoserdova ME Lidstrom RK Thauer (1999) ArticleTitleA methenyl tetrahydromethanopterin cyclohydrolase and a methenyl tetrahydrofolate cyclohydrolase in Methylobacterium extorquens AM1 Eur J Biochem 261 475–480 Occurrence Handle10.1046/j.1432-1327.1999.00291.x Occurrence Handle1:CAS:528:DyaK1MXislGqtr8%3D Occurrence Handle10215859
F Ronquist JP Huelsenbeck (2003) ArticleTitleMrBayes 3: Bayesian phylogenetic inference under mixed models Bioinformatics 19 1572–1574 Occurrence Handle10.1093/bioinformatics/btg180 Occurrence Handle1:CAS:528:DC%2BD3sXntlKms7k%3D Occurrence Handle12912839
H Schlesner C Rendsmann BJ Tindall D Gade R Rabus S Pfeiffer P Hirsch (2004) ArticleTitleTaxonomic heterogeneity within the Planctomycetales as derived by DNA/DNA-hybridization, description of Rhodopirellula baltica gen. nov., sp. nov., transfer of Pirellula marina the genus Blastopirellula gen. nov. as Blastopirellula marina comb, nov., and an emended description of the genus Pirellula Int J Syst Evol Microbiol . .
JW Scott ME Rasche (2002) ArticleTitlePurification, overproduction, and partial characterization of beta-RFAP synthase, a key enzyme in the methanopterin biosynthesis pathway J Bacteriol 184 4442–4448 Occurrence Handle10.1128/JB.184.16.4442-4448.2002 Occurrence Handle1:CAS:528:DC%2BD38XlvV2gsrk%3D Occurrence Handle12142414
M Strous JA Fuerst EHM Kramer S Logemann G Muyzer KT van de Pas-Schoonen R Webb JG Kuenen MSM Jetten (1999) ArticleTitleMissing lithotroph identified as new planctomycete Nature 400 446–449 Occurrence Handle10.1038/22749 Occurrence Handle1:CAS:528:DyaK1MXltVGnt7c%3D Occurrence Handle10440372
H Teeling T Lombardot M Bauer L Ludwig FO Glöckner (2004) ArticleTitleReevaluation of the phylogenetic position of the Planctomycetes by means of concatenated ribosomal protein sequences, DNA-directed RNA polymerase subunit sequences and whole genome trees Int J Syst Evol Microbiol 54 791–801
RK Thauer (1998) ArticleTitleBiochemistry of methanogenesis: A tribute to Marjory Stephenson Microbiology-UK 144 2377–2406
JD Thompson DG Higgins TJ Gibson (1994) ArticleTitleClustal-W—Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice Nucleic Acids Res 22 4673–4680 Occurrence Handle1:CAS:528:DyaK2MXitlSgu74%3D Occurrence Handle7984417
JA Vorholt (2002) ArticleTitleCofactor-dependent pathways of formaldehyde oxidation in methylotrophic bacteria Arch Microbiol 178 239–249 Occurrence Handle10.1007/s00203-002-0450-2 Occurrence Handle1:CAS:528:DC%2BD38XovValt78%3D Occurrence Handle12209256
JA Vorholt RK Thauer (2002) Molybdenum and tungsten enzymes in C1 metabolism Molybdenum and tungsten: Their roles in Biological processes Marcel Dekker New York 571–619
JA Vorholt L Chistoserdova ME Lidstrom RK Thauer (1998) ArticleTitleThe NADP-dependent methylene tetrahydromethanopterin dehydrogenase in Methylobacterium extorquens AM1 J Bacteriol 180 5351–5356 Occurrence Handle1:CAS:528:DyaK1cXmvVWitLs%3D Occurrence Handle9765566
JA Vorholt L Chistoserdova SM Stolyar RK Thauer ME Lidstrom (1999) ArticleTitleDistribution of tetrahydromethanopterin-dependent enzymes in methylotrophic bacteria and phylogeny of methenyl tetrahydromethanopterin cyclohydrolases J Bacteriol 181 5750–5757 Occurrence Handle1:CAS:528:DyaK1MXmtFKitLw%3D Occurrence Handle10482517
JA Vorholt CJ Marx ME Lidstrom RK Thauer (2000) ArticleTitleNovel formaldehyde-activating enzyme in Methylobacterium extorquens AM1 required for growth on methanol J Bacteriol 182 6645–6650 Occurrence Handle10.1128/JB.182.23.6645-6650.2000 Occurrence Handle1:CAS:528:DC%2BD3MXitVCnu78%3D Occurrence Handle11073907
J Wang C Jenkins RI Webb JA Fuerst (2002) ArticleTitleIsolation of Gemmata-like and Isosphaera-like planctomycete bacteria from soil and freshwater Appl Environ Microbiol 68 417–422 Occurrence Handle10.1128/AEM.68.1.417-422.2002 Occurrence Handle1:CAS:528:DC%2BD38Xjt1Wluw%3D%3D Occurrence Handle11772655
Ward N, Butler MK, Smith RL, Fuerst JA (2003) Genome sequencing of Gemmate obscuriglobus UQM2246 http://www.tigr.org.
Williams TL, Moret BME (2003) An investigation of phylogenetic likelihood methods. Proceedings of the 3rd IEEE Symposium on Bioinformatics and Bioengeneering (BIBE: 03), pp 79–86
www.anammox.com. Joint environmental genomics program: Genome sequencing of Candidatus “Kuenenia stuttgartiensis” Genoscope/Technical University of Munich/University of Nijmegen
Acknowledgments
Preliminary sequence data were obtained from The Institute for Genomic Research (http://www.tigr.org). Sequencing of Gemmata obscuriglobus UQM2246 and sequencing of Methylococcus capsulatus Bath were accomplished with support from DOE. The Gemmata obscuriglobus UQM2246 genome sequencing project is a collaboration between The Institute of Genomic Research and the group of John A. Fuerst at the University of Queensland, Australia. Preliminary sequence data were also obtained from the DOE Joint Genome Institute (http://www.jgi.doe.gov/). Sequencing of Burkholderia fungorum LB400 was accomplished with support from DOE. This work was supported by the Max Planck Society and the German Federal Office of Research and Education.
Author information
Authors and Affiliations
Corresponding author
Additional information
Reviewing Editor: Dr. W. Ford Doolittle
Rights and permissions
About this article
Cite this article
Bauer, M., Lombardot, T., Teeling, H. et al. Archaea-Like Genes for C1-Transfer Enzymes in Planctomycetes: Phylogenetic Implications of Their Unexpected Presence in This Phylum. J Mol Evol 59, 571–586 (2004). https://doi.org/10.1007/s00239-004-2643-6
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-2643-6