
Abstract
Several human neurodegenerative disorders are caused by the expansion of polymorphic trinucleotide repeat regions. Many of these loci are functional short tandem repeats (STRs) located in brain-expressed genes, and their study is thus relevant from both a medical and an evolutionary point of view. The aims of our study are to infer the comparative pattern of variation and evolution of this set of loci in order to show species-specific features in this group of STRs and on their potential for expansion (therefore, an insight into evolutionary medicine) and to unravel whether any human-specific feature may be identified in brain-expressed genes involved in human disease. We analyzed the variability of the normal range of seven expanding STR CAG/CTG loci (SCA1, SCA2, SCA3-MJD, SCA6, SCA8, SCA12, and DRPLA) and two nonexpanding polymorphic CAG loci (KCNN3 and NCOA3) in humans, chimpanzees, gorillas, and orangutans. The study showed a general conservation of the repetitive tract and of the polymorphism in the four species and high heterogeneity among loci distributions. Humans present slightly larger alleles than the rest of species but a more relevant difference appears in variability levels: Humans are the species with the largest variance, although only for the expanding loci, suggesting a relationship between variability levels and expansion potential. The sequence analysis shows high levels of sequence conservation among species, a lack of correspondence between interruption patterns and variability levels, and signs of conservative selective pressure for some of the STR loci. Only two loci (SCA1 and SCA8) show a human specific distribution, with larger alleles than the rest of species. This could account, at the same time, for a human-specific trait and a predisposition to disease through expansion.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The field of comparative genetics has undergone a recent boom due to its power as a tool for the understanding of the evolutionary and functional factors shaping a given genome region and for the search of the genetic basis of species uniqueness. Despite the large effort made to obtain comparative genetic information, little is still known about similarities and differences between the human genome and that of our closest phylogenetic relatives, the apes. In recent years, several human-specific genetic traits have been detected by a comparative analysis of human and primate species, mostly from the analysis of genetic regions in chimpanzees (for reviews, see Gagneux and Varki 2001; Hacia 2001). Comparative information has mainly been obtained for noncoding regions, and such data have been the key to the understanding of hominoid phylogeny (Chen and Li 2001; see Ruvolo 1997 for a previous review). Comparative analyses of functional regions usually aim to understand the functional constraints on a particular genetic region, including purifying, positive, and balancing selection. In some cases, analysis may help to explain the appearance of a new genetic variant in a particular species, such as lysozyme enzymes in primates (Messier and Stewart 1997) or in the FOXP2 gene in humans (Lai et al. 2001; Enard et al. 2002).
We focused our study on the comparative analysis of a special group of genetic elements: functional CAG/CTG repetitive tracts. These are functional CAG/CTG short tandem repeats (STRs), mostly coding for polyglutamine tracts; they are found in genes that are highly expressed in the brain and their expansion in repeat number causes neurodegenerative disorders. Many of the genes in this study cause spinocerebellar ataxia (SCA) and are named, accordingly, SCAn loci, where n denotes a locus defining number.
These type of loci share the mutation dynamics of the rest of STRs of the genome, that is, they mutate by adding or subtracting one (or rarely more than one) repeat unit, but differ by expanding into abnormally long alleles that produce ataxia, dystrophy or similar diseases. This group of genetic diseases has not been detected in nonhuman species, a fact that could just be attributed to the greater knowledge of human disease but might also reflect a unique human pathogenic trend. Beyond the mere description, the comparison of the patterns of variability in the normal range of humans and apes can allow the testing of hypotheses on the causes of expansion and disease. This study can therefore be viewed in the context of evolutionary medicine, where the comprehension of the natural history of a disease may lie in the particular characteristics (either of the locus or of the species) of disease predisposition.
The variability of expanding loci has been studied in humans in order to determine population-specific disease risk factors and to understand the evolutionary forces shaping this variability (Watkins et al. 1995; Jodice et al. 1997; Andrés et al. 2003). On the other hand, interspecific comparative studies on these loci have focused on allele length comparison between humans and other primate species and have dealt mostly with a few species used as references in a single locus approach. The special interest in allele length differences among species is based on the observation that long alleles have an increased mutation rate and higher probability of very long leaps (Webster et al. 2002) and the possibility of expansion into the pathogenic range (as shown [Fu et al. 1991; Nolin et al. 2003] for fragile X).
We have analyzed, in four species (human, chimpanzee, gorilla, and orangutan), nine STR loci including those present on the SCA1 (spinocerebellar ataxia 1 locus), SCA2, SCA3 (or Machado–Joseph disease locus), SCA6, SCA8, SCA12, DRPLA (dentatorubral–pallidoluysian atrophy), KCNN3 (potassium intermediate/small conductance calcium-activated channel, subfamily N, member 3), and NCOA3 (nuclear receptor coactivator 3) genes (Table 1). They all are functional, have different genomic locations and functions, and share a high central nervous system expression and the presence of a CAG/CTG repetitive tract. Seven are expanding disease-related loci, while the remaining two are coding but do not seem to expand into pathogenic alleles. Variation at KCNN3, other than expansion, has been proposed as being associated with mental diseases and ataxia (Dror et al. 1999; Figueroa et al. 2001), although as a predisposing factor rather than as a single direct cause.
A strong correlation between expansion and variability in the normal range has been demonstrated by the observation that expanding STRs are the most variable group of STRs of the human genome (Chakraborty et al. 1997; Jodice et al. 1997; Deka et al. 1999). As these expansions have not been detected in apes, our aim is to determine whether these loci show similar levels of variability in apes to those observed in humans (and therefore diversity and expansion potential would not be directly related) or whether the high levels of polymorphism are exclusive to the species for which the expanding disease has been detected.
We have also determined whether shared factors among loci (such as the existence of a coding poly [CAG] tract) or locus-specific ones (such as differential mutation patterns or selective events) led to the diversity of observed allele distribution of expanding loci.
Ascertainment bias may be a relevant problem when trying to infer general patterns from a group of loci selected in one of the species or populations compared. Nevertheless, this problem does not affect our study, as we are interested in determining the evolution of this specific group of loci (where the observed tendencies are clear in terms of expansion and disease) but do not try to generalize our observations to infer traits for the rest of STRs, which would produce a strong ascertainment bias.
Materials and Methods
Allele Typing
Twenty common chimpanzees (Pan troglodytes subspecies troglodytes [one individual] and verus), 13 gorillas (Gorilla gorilla subspecies gorilla), and 4 to 6 orangutans (Pongo pygmaeus subspecies abelii [two individuals] or unknown) were typed for the number of repeats at the nine loci shown in Table 1. Data for SCA1 and SCA3 in chimpanzees (16 individuals) were obtained from the literature (Limprasert et al. 1996, 1997). In order to avoid sampling a single primate population, we obtained the samples from very different sources: Coriell Cell Repositories (USA), European Collection Cell Cultures (UK), Barcelona Zoo (Spain), Kumamoto Primate Park (Japan), Institute of Zoology, London (UK), and Dr. Takafumi Ishida, Tokyo (Japan). Although it is not possible to assume that this sample set is representative of genetic variation of all apes, care was taken to try to obtain a diverse sample set, whose heterogeneous origin reduces the limitations of studying a small population sample. Subspecies of ape specimens were determined by amplification by PCR of both hypervariable regions of the mtDNA D-loop region and direct sequencing with internal primers. Subspecies identification may not be crucial for our study, however, as variation at nuclear loci does not cluster by subspecies, at least for chimpanzees (Kaessmann et al. 1999).
All DNA samples were amplified for the region containing the repetitive element by PCR, with primers and conditions previously described for human analysis (Table A1). In the KCNN3 gene, which contains two CAG repetitive segments, the most variable repetitive tract (nucleotides 513 to 569 in NCB1 Reference Sequence NM_002249.3) was amplified in a fragment that did not include the other repetitive tract. Lengths of the amplified fragments were subsequently typed with GeneScan software version 3.7 (Applied Biosystems) after electrophoresis on 6% denaturing gel performed on an ABI Prism377 automatic sequencer (Applied Biosystems). A human sample of known genotype, previously sequenced and typed, was used as a standard size control for all GeneScan runs.
At least one individual per species and locus was directly sequenced to verify the correspondence between amplified fragment length and number of repeats. Sequences were determined using BigDyes sequencing kits, versions 2.0 and 3.0 (Applied Biosystems), on automatic sequencers ABIPrism 377 and 3100 (Applied Biosystems). The only exception is DRPLA, for which ape sequences from GenBank were used (accession number AJ133270 [Pan paniscus], AJ133271 [Gorilla gorilla], AJ133272 [Pongo pygmaeus]). Sequences were assembled and analyzed with the SeqmanII program (Lasergene 1999 package; DNASTAR, Inc.). Homology between human and mouse sequences was obtained by BLAST, additional alignment of sequences (Seqman II and Clustal programs), and manual determination of the conserved regions.
Allele Frequencies and Statistical Analysis
The correspondence between allele size and number of repeats for every species allowed the estimation of allele frequencies by direct allele counting. Human distributions for healthy individuals result from pooling African, European, Indian, and East Asian samples for all loci; the distributions were obtained from the literature or the typing of healthy individuals when necessary (Andrés et al. 2003). As they come from normal individuals, we do not expect to find a significant proportion of premutated alleles (those with an intermediate allele length, i.e., between normal and pathogenic, and which have high expansion probability). Allele frequencies were averaged across populations without weighing by sample size, as the number of chromosomes varied greatly among populations. All subsequent analyses were performed with the pooled human distribution.
The different parameters of allele size distributions for every locus and species were determined. Mean, standard deviation, variance, and variation coefficient of repeat number were calculated with the SPSS statistical package. Expected heterozygosity was calculated with the Arlequin 2.000 package (Schneider et al. 2000). In order to analyze the possibility that variance was influenced by differences in sample size, we computed it on an increasing sample size, for every locus and species, with a program that takes a pseudosample of 8–40 chromosomes (the range in size between our smallest and our largest ape samples) from the original distribution and calculates its variance. After 1000 random extractions for each pseudosample size, the average variance for every pseudosample size is plotted in a graph that shows how variance relates to pseudosample size.
To determine whether variances observed in species with a smaller sample size could be obtained from a hypothetical human sample of eight chromosomes (our smallest sample size, that of orangutans), we performed a second resampling experiment. In this case, a pseudosample of eight chromosomes was obtained for every human distribution and its variance was calculated; after 10,000 sample extractions, we obtained a distribution of variances from pseudosamples of eight human chromosomes. To determine the significance of our results, the variance was compared to the 95% confidence interval of the pseudosample variance distribution.
The four species were compared in terms of mean and variance of repeat number. Two tests were performed for mean repeat number with permutation tests: the comparison between the four species for every individual locus (considering every species as a different category) and the comparison between humans and the rest of the species for every locus (considering “apes” a category, which included all ape species, and comparing it with the “human” category). The permutation test was performed as follows: Individual chromosomes were randomly shuffled between classes (species or species groups), maintaining the original sample sizes. For every permutated data set, the difference in the average number of repeats between the two classes was computed. This process was repeated 1000 times. The test is significant if the probability of obtaining a difference in average repeat number in the permutations as large as in the observed data set, in a one-tailed test, is <0.05 for the human–ape comparison or <0.0056 (after Bonferroni correction) for the four-species comparison.
Variance comparison among the four species, for every locus, was performed with Scheffé–Box (log ANOVA) test for homogeneity of variances (Sokal and Rohlf 1995, p. 397); as this test is not available in statistical packages, an ad hoc program has been written and is available on request (oscar.lao@upf.edu). In order to obtain a single overall significance value for all loci, the individual Scheffé–Box p values for every locus were combined with Fisher’s test for probability combination (Sokal and Rohlf 1995, p. 795). When a species showed a single allele for a given locus the test was performed using the rest of the species.
Results
Data on nine CAG repeat loci, whose characteristics are shown in Table 1, were obtained for humans (of African, European, Indian, and East Asian origin), for 20 chimpanzees, 13 gorillas, and 4 to 6 orangutans; all chromosomes analyzed were in the “normal,” nonexpanded range. Allele size distributions are plotted by locus in Fig. 1, and statistical parameters of the distributions and expected heterozygosity are shown in Table 2. The amplified fragment was sequenced in at least one individual for species and locus; sequences of all repetitive regions are shown in Fig. 2, and their accession numbers are available in Table A2, including compared mouse loci. Comparative locus analysis was carried out on different levels, searching for general species trends and for locus-specific trends.

Graphical representations of individual allele size distributions and average length and variability parameters for species and loci. 1.1–1.9 Allele length distributions in the four species. The distributions are the result of typing, for repeat number, chimpanzee (in red), gorilla (blue), and orangutan (yellow) chromosomes, and pooling human distributions from African, European, Indian, and East Asian origin (black). 1.10 Mean allele size for species and locus. 1.11 Variance of repeat number for species and locus.

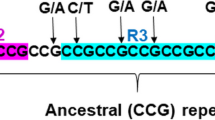
Sequence of the repetitive region for the expanding loci in the four species. The line over the sequences marks the STR sequence region, and the total length of the overlined sequence region is shown. Repetitive segments are indicated without detailing the exact number of repeats, and segments containing six or more repeat units, with higher probabilities of slippage events than shorter segments, aremarked in bold face.
General species-specific trends can be detected by comparing the parameters of the distribution for every species over all loci (Table 3). Humans show a higher mean number of repeats than the other species, reaching statistical significance (permutation test humans vs. apes, p < 0.001; see Materials and Methods for the groups considered), showing that a trend exists in allele length among the different species. Nevertheless, the trend is not followed by all loci, as discussed below.
Variance and coefficient of variation of repeat number show a decreasing trend from humans to orangutans, as shown in Table 3. Statistically significant differences in variance exist between species, with all individual loci showing significant differences among species (p < 0.05) and the combined p value also being statistically significant. Interestingly, for the seven expanding loci humans presented the largest variance.
The small sample size in the ape species may bias the estimation of the dispersion parameters. As stated in Materials and Methods, the samples for nonhuman species are of very heterogeneous origin, which reduces the possibility of underestimating variance by sampling from a single, localized population or inbred zoo collection. Furthermore, to explore to what extent human variance is larger than that of the rest of the species as a consequence of its larger sample size, we performed two independent tests.
First, we studied whether variance increased with sample size by a permutation test on pseudosamples of 8–40 chromosomes for the four species in every locus. Results showed that reduction of sample size does not lead to a reduction of variance (data not shown) and that the resampling average value remains within the range of the values obtained from the original distribution. Therefore, as expected, a reduced sample does not determine lower variance value. A second resampling test was performed, in which we tested whether variances similar to those obtained for apes could be obtained from pseusosamples of eight human chromosomes from our human distributions (see Materials and Methods). Of the seven loci with larger variance in humans than in the rest of the species, four loci (SCA3, SCA8, SCA12, and DRPLA) showed large and statistically significant differences (p < 0.05) between humans and orangutans (the species with the lowest sample size), and two of them (SCA3 and DRPLA) showed significantly larger variances in humans than in any other species. This analysis suggests that beyond some sample size influence, our results cannot be exclusively explained for differences in sample size, and variance divergence for this group of loci seems to be a species characteristic.
Heterozygosity is high and very similar between humans and chimpanzees and low in gorillas and orangutans (Table 3). This observation is due to the large influence of SCA2 and NCOA3, with higher heterozygosities in chimpanzees than in humans; without these two loci, mean heterozygosity is higher in humans than in chimpanzees.
In order to determine which loci are mostly responsible for the species trends, mean repeat number (Fig. 1.10) and variance of repeat number (Fig. 1.11) were compared for each species and locus. Humans do not always show larger allele sizes at all loci or at all expanding loci, and only three (SCA1, SCA8, and NCOA3) show statistically significant larger mean allele length in humans than in any other species. On the other hand, variance is higher in humans than in the rest of species for all expanding loci but not for the two nonexpanding loci. Nonetheless, the low number of nonexpanding loci analyzed prevents us from generalizing this interesting difference in variance.
The individual comparison among loci distributions shows a strong heterogeneity in allele size distribution (Fig. 1), and sequences of the STR alleles over the four species (Fig. 2) illustrate that repeat regions are very complex and that different loci and lineages show heterogeneous amounts and patterns of divergence. The sequence distribution comparison clearly shows a lack of general patterns (in STR sequence and interrupting complexity) that could explain the observed allele size and variability of expanding loci.
Interestingly, when comparing human and mouse, only very short tracts were detected for almost all mouse loci, from the complete absence of the tract (in SCA6, SCA8, and SCA12 the repetitive region could not be detected) to very short CAG/CAA repetitive regions (four CAG repeats in SCA1, two in SCA2, five in SCA3, four in DRPLA, and one in NCOA3). Only KCNN3 keeps the repetitive tract over phylogenetically distant species, as a repetitive CAG/CAA region exists in the mouse sequence, with the interrupting codons CAA (Gln) and TCG (Ser), which are absent in primates.
Discussion
Our study shows that the “flexible conservation” that exists in human functional CAG/CTG tracts (high polymorphism in number of repeats combined with sequence conservation) is also found in apes. The conservation of the variable tracts in all species strongly supports the presence of the repeat regions in the common ancestor of humans and apes in all nine loci studied, and the presence of polymorphism in almost all species suggests the existence of ancestral polymorphism.
Previous studies comparing human and mouse CAG/CAA tracts found a relationship between interrupting levels and conservation between very distant species (Albà et al. 1999), suggesting that older tracts would be more frequently interrupted by non-CAG codons. We failed to find this relationship in our set of repetitive tracts, as the most conserved locus (KCNN3, with a long repetitive region in both humans and mice) is totally uninterrupted in primates, and many tracts showing low conservation between distant species are profoundly interrupted in primates.
Species-Specific Characteristics
The combined analysis of all loci points to species-specific trends. In allele length comparison, larger alleles in humans than in other species were reported for nonfunctional STRs (Rubinsztein et al. 1995a; Crouau-Roy et al. 1996; Cooper et al. 1998), a controversial conclusion (Ellegren et al. 1995, 1997). Previous studies found longer alleles in humans than in other species for SCA1, SCA3, AR, and HD CAG repeats and in the FA (GAA) locus (Rubinsztein et al. 1995b; Djian et al. 1996; Limprasert et al. 1997; Choong et al. 1998; Gonzalez-Cabo et al. 1999; Justice et al. 2001). This suggests a general increase in the number of repeats from monkeys to apes and humans for expanding loci (although similar allele length was found by Limprasert et al. [1996] in the SCA3 locus). On the other hand, a study on functional nonexpanding CAG STR shows shorter alleles in humans than in apes (Saleem et al. 2001). Therefore, a clear picture of the human specific characteristics in functional STRs had not emerged beyond single locus comparisons.
The data presented in this paper for nine functional trinucleotides show significantly higher number of repeats in humans, a trend that is unique to this lineage and shows that, beyond locus heterogeneity, a specificity in allele length exists in this set of STRs. The trend is not present in all loci and is mainly (but not exclusively) due to SCA1, SCA8, and NCOA3.
In our set of samples and loci, we found differences in variance among the four species, with the highest values in humans. In the genomic regions studied so far, DNA sequence diversity is lower in humans than in chimpanzees and other apes (Crouau-Roy et al. 1996; Kaessmann et al. 2001; Noda et al. 2001), possibly due to a demographic bottleneck in the human lineage (Jorde et al. 2000). Surprisingly, the present results show that humans are more diverse (measured as variance of allele distribution) than any of the other species studied for an ample set of expanding functional trinucleotide tandem repeats. No demographic factor (which would affect the whole genome) or ascertainment bias effect for the selection of the STRs in humans could explain this finding, which is not a general STR trend, but it is exclusive of functional STRs that can expand and produce disease. This is not found in noncoding STRs (and previous results [Crouau-Roy et al. 1996; Garza et al. 1995; Wise et al. 1997] are not concordant) or in other similar STRs (such as the nonexpanding KCNN3 and NCOA3, analyzed in this study).
The possible sampling error in apes has been reduced by choosing individuals of different origins as much as possible (see Materials and Methods), and the effects of different sample size for the different species have been proven to be small through resampling procedures (see Results). Moreover, variability levels may be investigated to test whether our primate samples present lower variability levels than humans at other genetic loci. Seventeen of the chimpanzees typed in this study were previously analyzed for 16S rRNA (Noda et al. 2001); the subset of individuals analyzed in both studies show variability levels (π = 0.0014 ± 0.0003) comparable with those existing for the 16S rRNA of the whole human species (π = 0.0016 ± 0.0004) (Ingman et al. 2000). These results show that a part of our sample of chimpanzees is as variable as humans at a global scale, and thus the expected diversity of chimpanzees in CAG repeats should be higher than humans. Therefore, our results are not the consequence of a small sample size or the selected sample of individuals, and locus-specific factors (related to CAG loci) acting in different ways in different species are needed to explain the observation that the loci that can expand in humans are more variable in humans than in any other species.
Human expanding STRs have previously been shown to be more variable than other di-, tri-, or tetranucleotides in the human genome (Chakraborty et al. 1997; Jodice et al. 1997; Deka et al. 1999), suggesting a relationship among variability, expansion, and disease. Moreover, our results show that these loci are more variable in humans than in any ape, suggesting that this pattern could be related to a human specifically capacity for expansion. The relationship suggests that loci with increased variance may be more likely to expand to pathogenic alleles, and thus lead to disease.
Different scenarios would be compatible with the increase in variance in one species: the first is a high mutation rate in the absence of strong selective constraints, which would increase variability of the STR (Di Rienzo et al. 1998), leading to new alleles that, if long enough, would increase slippage probability to expanded alleles. Humans do not have mean larger alleles, and therefore allele length differences do not seem to explain the observation. Nevertheless, we observed a statistically significant relationship between the longest allele and variance, even correcting for influence of mean allele size (r = 0.6552, p < 0.0005). The observed relationship between variance and longest allele can be explained for the nature of variance calculation (as alleles far away from the mean allele size will strongly influence variance). Thus normal but long alleles might affect the STR dynamics: as mutation rate increases with allele size, very long alleles can contribute to the high mutation rate and high variability levels. But this factor, although important for the production of pathogenic alleles, would have a minor effect on the overall variance, as they are rare in the population, and its increased mutation rate will probably not be large enough to explain the huge differences observed in variance for the whole distribution. When computing variances without the very long alleles (eliminating the five longer alleles in a range of less than 40 repeats), variance decreases only 20% on average, far from the differences with other species, showing that very long alleles are not the main responsible alleles for the large human variances in expanding loci. No other factors seem to exist to explain differences in mutation rates.
A second possibility, given the functionality of the loci, is the existence of some kind of balancing selection that, as in the case of other human genes like HLA (Hedrick and Thomson 1983; Hughes and Nei 1988) or CCR5 (Bamshad et al. 2002), would maintain the high diversity by favoring the existence of different alleles in the population. This is a truly speculative explanation, as no external evidence exists for the presence of balancing selection acting over these loci, but the fact that they are functional require consideration of a nonneutral explanation for our findings.
A detailed knowledge both of the mutation rate and pattern for each locus and of the functional behavior of the different alleles may be necessary to interpret these findings and to infer whether mutation or selection are the main causes of the increase of variability in humans.
Locus-Specific Characteristics
Differences in a single functional locus among species may influence its function, and these differences are especially interesting as these genes are expressed in brain and their variation outside the normal range has pathogenic effects. These characteristics (functionality and variability) make poly(CAG) regions attractive candidates for having brain-related functions, especially in the human lineage, which shows many brain-specific phenotypic traits compared to the rest of species.
The analysis of each locus demonstrates the existence of a strong evolutionary heterogeneity, showing that specific evolutionary forces have been acting on each gene, governing its diversification and evolution. No important differences in allele length exist between humans and apes, the only exception being SCA1 and SCA8, the two candidate loci to present functional differences between humans and the other species if the length of the repetitive tract influenced protein function. This possibility has been suggested for SCA1 (Yue et al. 2001) and SCA8 (see Andrés et al. 2003 for a detailed explanation of SCA8 interspecific differences). The variability levels are also highly heterogeneous among loci, ranging from loci with similar diversity in all species (as in SCA6 and SCA12) to those with extreme differences (SCA3 and DRPLA).
As in allele length distributions, there is a strong heterogeneity in the repetitive sequence conservation among loci for the different species: in contrast to the total conservation of the CAG tract in some loci (SCA6, SCA12, and KCNN3), others show important sequence differences among species, both in the presence of interruptions and in the composition of the repetitive segment (as SCA2, SCA3, SCA8, or DRPLA), which results in differences in repeat number. Nevertheless, the complexity in sequence interruption patterns could not be related to the amount of allele size diversity; and any simple pattern of STR mutation depending on the repetitive sequence that could be inferred from the analysis of one functional STRs locus does not seem to apply to others. Heterogeneity is the main rule.
The comparative study of seven expanding and two nonexpanding brain-expressed functional STRs has shown the overall maintenance of polymorphism with a higher mean in humans for some loci and a larger amount of variability in humans for expanding loci. Comparative studies focused on the search of species specificities on functional regions often do not deal with intraspecific variability, trying to find out fixed changes among species. Here we suggest that differences in genetic variability in important brain-expressed genes should also be considered. Species variability levels on phenotypic traits can also be a species-unique characteristic, and the study of diversity in specific genes with important gene function may give a clue toward understanding the species-specific evolution of these genes.
References
MM Albà MF Santibanez-Koref JM Hancock (1999) ArticleTitleConservation of polyglutamine tract size between mice and humans depends on codon interruption Mol Biol Evol 16 1641–1644 Occurrence Handle10555295
AM Andres O Lao M Soldevila F Calafell J Bertranpetit (2003) ArticleTitleDynamics of CAG repeat loci revealed by the analysis of their variability Hum Mutat 21 61–70 Occurrence Handle10.1002/humu.10151 Occurrence Handle1:CAS:528:DC%2BD3sXot1alsA%3D%3D Occurrence Handle12497632
MJ Bamshad S Mummidi E González SS Ahuja DM Dunn WS Watkins S Wooding AC Stone LB Jorde RB Weiss SK Ahuja (2002) ArticleTitleA strong signature of balancing selection in the 5′ cis-regulatory region of CCR5 Proc Natl Acad Sci USA 99 10539–10544 Occurrence Handle10.1073/pnas.162046399 Occurrence Handle1:CAS:528:DC%2BD38Xmt1Chs7k%3D Occurrence Handle12149450
R Chakraborty M Kimmel DN Stivers LJ Davison R Deka (1997) ArticleTitleRelative mutation rates at di-, tri-, and tetranucleotide microsatellite loci Proc Natl Acad Sci USA 94 1041–1046 Occurrence Handle10.1073/pnas.94.3.1041 Occurrence Handle1:CAS:528:DyaK2sXhtVyht70%3D Occurrence Handle9023379
FC Chen WH Li (2001) ArticleTitleGenomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees Am J Hum Genet 68 444–456 Occurrence Handle10.1086/318206 Occurrence Handle1:CAS:528:DC%2BD3MXhtl2lsLg%3D Occurrence Handle11170892
CS Choong JA Kemppainen EM Wilson (1998) ArticleTitleEvolution of the primate androgen receptor: a structural basis for disease J Mol Evol 47 334–342 Occurrence Handle1:CAS:528:DyaK1cXmt1anurs%3D Occurrence Handle9732460
G Cooper DC Rubinsztein W Amos (1998) ArticleTitleAscertainment bias cannot entirely account for human microsatellites being longer than their chimpanzee homologues Hum Mol Genet 7 1425–1429 Occurrence Handle10.1093/hmg/7.9.1425 Occurrence Handle1:CAS:528:DyaK1cXmtVSltrw%3D Occurrence Handle9700197
B Crouau-Roy S Service M Slatkin N Freimer (1996b) ArticleTitleA fine-scale comparison of the human and chimpanzee genomes: linkage, linkage disequilibrium and sequence analysis Hum Mol Genet 5 1131–1137 Occurrence Handle10.1093/hmg/5.8.1131 Occurrence Handle1:CAS:528:DyaK28XkslSrurc%3D
R Deka S Guangyun D Smelser Y Zhong M Kimmel R Chakraborty (1999) ArticleTitleRate and directionality of mutations and effects of allele size constraints at anonymous, gene-associated, and disease-causing trinucleotide loci Mol Biol Evol 16 1166–1177 Occurrence Handle1:CAS:528:DyaK1MXmtVWntb8%3D Occurrence Handle10486972
A Di Rienzo P Donnelly C Toomajian B Sisk A Hill ML Petzl-Erler GK Haines DH Barch (1998) ArticleTitleHeterogeneity of microsatellite mutations within and between loci, and implications for human demographic histories Genetics 148 1269–1284 Occurrence Handle1:CAS:528:DyaK1cXks1eitLo%3D Occurrence Handle9539441
P Djian JM Hancock HS Chana (1996) ArticleTitleCodon repeats in genes associated with human diseases: fewer repeats in the genes of nonhuman primates and nucleotide substitutions concentrated at the sites of reiteration Proc Natl Acad Sci USA 93 417–421 Occurrence Handle10.1073/pnas.93.1.417 Occurrence Handle1:CAS:528:DyaK28XjslylsA%3D%3D Occurrence Handle8552651
V Dror E Shamir S Ghanshani R Kimhi M Swartz Y Barak R Weizman L Avivi T Litmanovitch E Fantino K Kalman EG Jones KG Chandy JJ Gargus GA Gutman R Navon (1999) ArticleTitlehKCa3/KCNN3 potassium channel gene: Association of longer CAG repeats with schizophrenia in Israeli Ashkenazi Jews, expression in human tissues and localization to chromosome 1q21 Mol Psychiatry 4 254–260 Occurrence Handle10.1038/sj.mp.4000508 Occurrence Handle1:CAS:528:DC%2BD3cXlvVWktrs%3D Occurrence Handle10395215
H Ellegren S Moore N Robinson K Byrne W Ward BC Sheldon (1997) ArticleTitleMicrosatellite evolution—A reciprocal study of repeat lengths at homologous loci in cattle and sheep Mol Biol Evol 14 854–860 Occurrence Handle1:CAS:528:DyaK2sXltVeit70%3D Occurrence Handle9254923
H Ellegren CR Primmer BC Sheldon (1995) ArticleTitleMicrosatellite ‘evolution’: directionality or bias? Nat Genet 11 360–362 Occurrence Handle10.1038/ng1295-360 Occurrence Handle1:CAS:528:DyaK2MXpvVCks7g%3D Occurrence Handle7493011
W Enard M Przeworski SE Fisher CS Lai V Wiebe T Kitano AP Monaco S Pääbo (2002) ArticleTitleMolecular evolution of FOXP2, a gene involved in speech and language Nature 418 869–872 Occurrence Handle10.1038/nature01025 Occurrence Handle1:CAS:528:DC%2BD38XmtlWltbY%3D Occurrence Handle12192408
KP Figueroa P Chan L Schols C Tanner O Riess SL Perlman DH Geschwind SM Pulst (2001) ArticleTitleAssociation of moderate polyglutamine tract expansions in the slow calcium-activated potassium channel type 3 with ataxia Arch Neurol 58 1649–1653 Occurrence Handle10.1001/archneur.58.10.1649 Occurrence Handle1:STN:280:DC%2BD3MrntVakuw%3D%3D Occurrence Handle11594924
YH Fu DP Kuhl A Pizzuti M Pieretti JS Sutcliffe S Richards AJ Verkerk JJ Holden RG Fenwick SuffixJr. ST Warren BA Oostra DL Nelson CT Caskey (1991) ArticleTitleVariation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox Cell 67 1047–1058 Occurrence Handle10.1016/0092-8674(91)90283-5 Occurrence Handle1:CAS:528:DyaK38XhtVekurk%3D Occurrence Handle1760838
P Gagneux A Varki (2001) ArticleTitleGenetic differences between humans and great apes Mol Phylogenet Evol 18 2–13 Occurrence Handle10.1006/mpev.2000.0799 Occurrence Handle1:CAS:528:DC%2BD3MXmt1WktQ%3D%3D Occurrence Handle11161737
JC Garza M Slatkin NB Freimer (1995) ArticleTitleMicrosatellite allele frequencies in humans and chimpanzees, with implications for constraints on allele size Mol Biol Evol 12 594–603 Occurrence Handle1:CAS:528:DyaK2MXmsVOqs7o%3D Occurrence Handle7659015
P González-Cabo MI Sanchez J Canizares JM Blanca R Martínez-Arias M Castro ParticleDe J Bertranpetit F Palau MD Molto R Frutos Particlede (1999) ArticleTitleIncipient GAA repeats in the primate Friedreich ataxia homologous genes Mol Biol Evol 16 880–883 Occurrence Handle10368965
JG Hacia (2001) ArticleTitleGenome of the apes Trends Genet 17 637–645 Occurrence Handle10.1016/S0168-9525(01)02494-5 Occurrence Handle1:CAS:528:DC%2BD3MXnslemur4%3D Occurrence Handle11672864
PW Hedrick G Thomson (1983) ArticleTitleEvidence for balancing selection at HLA Genetics 104 449–456 Occurrence Handle1:STN:280:BiyB1cfltVQ%3D Occurrence Handle6884768
AL Hughes M Nei (1988) ArticleTitlePattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection Nature 335 167–170 Occurrence Handle10.1038/335167a0 Occurrence Handle1:CAS:528:DyaL1cXlvVCktrY%3D Occurrence Handle3412472
M Ingman H Kaessmann S Pääbo U Gyllensten (2000) ArticleTitleMitochondrial genome variation and the origin of modern humans Nature 408 708–713 Occurrence Handle10.1038/35047064 Occurrence Handle1:CAS:528:DC%2BD3cXptVCrsr4%3D Occurrence Handle11130070
C Jodice B Giovannone V Calabresi M Bellocchi L Terrenato A Novelletto (1997a) ArticleTitlePopulation variation analysis at nine loci containing expressed trinucleotide repeats Ann Hum Genet 61 IssueIDPt 5 425–438 Occurrence Handle10.1017/S0003480097006489
LB Jorde WS Watkins MJ Bamshad ME Dixon CE Ricker MT Seielstad MA Batzer (2000) ArticleTitleThe distribution of human genetic diversity: a comparison of mitochondrial, autosomal, and Y-chromosome data Am J Hum Genet 66 979–988 Occurrence Handle10.1086/302825 Occurrence Handle1:CAS:528:DC%2BD3cXisFShsbk%3D Occurrence Handle10712212
CM Justice Z Den SV Nguyen M Stoneking PL Deininger MA Batzer BJ Keats (2001) ArticleTitlePhylogenetic analysis of the Friedreich ataxia GAA trinucleotide repeat J Mol Evol 52 232–238 Occurrence Handle1:CAS:528:DC%2BD3MXislOksrY%3D Occurrence Handle11428460
H Kaessmann V Wiebe S Pääbo (1999) ArticleTitleExtensive nuclear DNA sequence diversity among chimpanzees Science 286 1159–1162 Occurrence Handle10.1126/science.286.5442.1159 Occurrence Handle1:CAS:528:DyaK1MXnt1KrsrY%3D Occurrence Handle10550054
H Kaessmann V Wiebe G Weiss S Pääbo (2001) ArticleTitleGreat ape DNA sequences reveal a reduced diversity and an expansion in humans Nat Genet 27 155–156 Occurrence Handle10.1038/84773 Occurrence Handle1:CAS:528:DC%2BD3MXhtFGktL0%3D Occurrence Handle11175781
Y Kawaguchi T Okamoto M Taniwaki M Aizawa M Inoue S Katayama H Kawakami S Nakamura M Nishimura I Akiguchi (1994) ArticleTitleCAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1 Nat Genet 8 221–228 Occurrence Handle10.1038/ng1194-221 Occurrence Handle1:CAS:528:DyaK2MXitVOgtLs%3D Occurrence Handle7874163
MD Koob ML Moseley LJ Schut KA Benzow TD Bird JW Day LP Ranum (1999) ArticleTitleAn untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8) Nat Genet 21 379–384 Occurrence Handle10.1038/7710 Occurrence Handle1:CAS:528:DyaK1MXitFKrs7s%3D Occurrence Handle10192387
CS Lai SE Fisher JA Hurst F Vargha-Khadem AP Monaco (2001) ArticleTitleA forkhead-domain gene is mutated in a severe speech and language disorder Nature 413 519–523 Occurrence Handle10.1038/35097076 Occurrence Handle1:STN:280:DC%2BD3MrjvFegsw%3D%3D Occurrence Handle11586359
P Limprasert N Nouri RA Heyman C Nopparatana M Kamonsilp PL Deininger BJ Keats (1996) ArticleTitleAnalysis of CAG repeat of the Machado-Joseph gene in human, chimpanzee and monkey populations: A variant nucleotide is associated with the number of CAG repeats Hum Mol Genet 5 207–213 Occurrence Handle10.1093/hmg/5.2.207 Occurrence Handle1:CAS:528:DyaK28XptVCmsQ%3D%3D Occurrence Handle8824876
P Limprasert N Nouri C Nopparatana PL Deininger BJ Keats (1997) ArticleTitleComparative studies of the CAG repeats in the spinocerebellar ataxia type 1 (SCA1) gene Am J Med Genet 74 488–493 Occurrence Handle10.1002/(SICI)1096-8628(19970919)74:5<488::AID-AJMG6>3.0.CO;2-K Occurrence Handle1:STN:280:DyaK1c%2FgtVantg%3D%3D Occurrence Handle9342197
W Messier CB Stewart (1997) ArticleTitleEpisodic adaptive evolution of primate lysozymes Nature 385 151–154 Occurrence Handle10.1038/385151a0 Occurrence Handle1:CAS:528:DyaK2sXks1Sjug%3D%3D Occurrence Handle8990116
S Nagafuchi H Yanagisawa K Sato T Shirayama E Ohsaki M Bundo T Takeda K Tadokoro I Kondo N Murayama (1994) ArticleTitleDentatorubral and pallidoluysian atrophy expansion of an unstable CAG trinucleotide on chromosome 12p Nat Genet 6 14–18 Occurrence Handle10.1038/ng0194-14 Occurrence Handle1:CAS:528:DyaK2cXhtlWgtrw%3D Occurrence Handle8136826
R Noda CG Kim O Takenaka RE Ferrell T Tanoue I Hayasaka S Ueda T Ishida N Saitou (2001) ArticleTitleMitochondrial 16S rRNA sequence diversity of hominoids J Hered 92 490–496 Occurrence Handle10.1093/jhered/92.6.490 Occurrence Handle1:CAS:528:DC%2BD38XjslCkt74%3D Occurrence Handle11948216
SL Nolin WT Brown A Glicksman GE Jr. Houck AD Gargano A Sullivan V Biancalana K Brondum-Nielsen H Hjalgrim E Holinski-Feder F Kooy J Longshore J Macpherson JL Mandel G Matthijs F Rousseau P Steinbach ML Vaisanen H Koskull ParticleVon SL Sherman (2003) ArticleTitleExpansion of the fragile X CGG repeat in females with premutation or intermediate alleles Am J Hum Genet 72 454–464 Occurrence Handle10.1086/367713 Occurrence Handle1:CAS:528:DC%2BD3sXht12rtLw%3D Occurrence Handle12529854
HT Orr MY Chung S Banfi TJ Kwiatkowski SuffixJr. A Servadio AL Beaudet AE McCall LA Duvick LP Ranum HY Zoghbi (1993) ArticleTitleExpansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1 Nat Genet 4 221–226 Occurrence Handle10.1038/ng0793-221 Occurrence Handle1:CAS:528:DyaK3sXmsVOisLk%3D Occurrence Handle8358429
SM Pulst A Nechiporuk T Nechiporuk S Gispert XN Chen I Lopes-Cendes S Pearlman S Starkman G Orozco-Diaz A Lunkes P DeJong GA Rouleau G Auburger JR Korenberg C Figueroa S Sahba (1996) ArticleTitleModerate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2 Nat Genet 14 269–276 Occurrence Handle10.1038/ng1196-269 Occurrence Handle1:CAS:528:DyaK28XntVClsb8%3D Occurrence Handle8896555
DC Rubinsztein W Amos J Leggo S Goodburn S Jain SH Li RL Margolis CA Ross MA Ferguson-Smith (1995a) ArticleTitleMicrosatellite evolution—Evidence for directionality and variation in rate between species Nat Genet 10 337–343 Occurrence Handle10.1038/ng0795-337 Occurrence Handle1:CAS:528:DyaK2MXms1Kjurw%3D
DC Rubinsztein J Leggo GA Coetzee RA Irvine M Buckley MA Ferguson-Smith (1995b) ArticleTitleSequence variation and size ranges of CAG repeats in the Machado-Joseph disease, spinocerebellar ataxia type 1 and androgen receptor genes Hum Mol Genet 4 1585–1590 Occurrence Handle1:CAS:528:DyaK2MXnvVensb8%3D
M Ruvolo (1997) ArticleTitleMolecular phylogeny of the hominoids: Inferences from multiple independent DNA sequence data sets Mol Biol Evol 14 248–265 Occurrence Handle1:CAS:528:DyaK2sXhs12jtbs%3D Occurrence Handle9066793
Q Saleem A Anand S Jain SK Brahmachari (2001) ArticleTitleThe polyglutamine motif is highly conserved at the Clock locus in various organisms and is not polymorphic in humans Hum Genet 109 136–142 Occurrence Handle10.1007/s004390100550 Occurrence Handle1:CAS:528:DC%2BD3MXmtVWitL4%3D Occurrence Handle11511917
S Schneider D Roessli L Excoffier (2000) A software for population genetics data analysis Genetics and Biometry Laboratory, University of Geneva Switzerland
RR Sokal FJ Rohlf (1995) Biometry WH Freeman New York
WS Watkins M Bamshad LB Jorde (1995) ArticleTitlePopulation genetics of trinucleotide repeat polymorphisms Hum Mol Genet 4 1485–1491 Occurrence Handle1:CAS:528:DyaK2MXnvVens7Y%3D Occurrence Handle8541830
MT Webster NG Smith H Ellegren (2002) ArticleTitleMicrosatellite evolution inferred from human-chimpanzee genomic sequence alignments Proc Natl Acad Sci USA 99 8748–8753 Occurrence Handle10.1073/pnas.122067599 Occurrence Handle1:CAS:528:DC%2BD38XltF2htr4%3D Occurrence Handle12070344
CA Wise M Sraml DC Rubinsztein S Easteal (1997) ArticleTitleComparative nuclear and mitochondrial genome diversity in humans and chimpanzees Mol Biol Evol 14 707–716 Occurrence Handle1:CAS:528:DyaK2sXksVahs7g%3D Occurrence Handle9214743
S Yue HG Serra HY Zoghbi HT Orr (2001) ArticleTitleThe spinocerebellar ataxia type 1 protein, ataxin-1, has RNA-binding activity that is inversely affected by the length of its polyglutamine tract Hum Mol Genet 10 25–30 Occurrence Handle10.1093/hmg/10.1.25 Occurrence Handle1:CAS:528:DC%2BD3MXovVKqtA%3D%3D Occurrence Handle11136710
O Zhuchenko J Bailey P Bonnen T Ashizawa DW Stockton C Amos WB Dobyns SH Subramony HY Zoghbi CC Lee (1997) ArticleTitleAutosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel Nat Genet 15 62–69 Occurrence Handle10.1038/ng0197-62 Occurrence Handle1:CAS:528:DyaK2sXjsFSgtw%3D%3D Occurrence Handle8988170
Acknowledgments
We thank Monica Vallés for her technical support and Anna Pérez-Lezaun for her technical help and feedback. The authors would like to especially thank Arcadi Navarro for reading the manuscript and for his help with statistical comparisons, Michael Greenacre for statistical help, Lynn B. Jorde, Pascal Gagneux, and Mar Albà for reading an early version of the manuscript, and Lynda Vigilant, Pascal Gagneux, and Anne C. Stone for helpful advice on primate subspecies determination. Tanzanian human samples were kindly supplied by Dr. Clara Menéndez from the Unitat d’Epidemiologia i Bioestadística (Hospital Clínic, Barcelona). Primate samples were supplied by the Barcelona Zoo (under the agreement of the Primate DNA Bank with the Pompeu Fabra University), Ikuo Hayasaka, and Dr. Takafumi Ishida of the University of Tokyo (Japan) or obtained from the Institute of Zoology (London), Coriell Cell Repositories (CCR), and European Collection of Cell Cultures (ECACC). This study was supported by the Dirección General de Investigación (Spanish Goverment), Grants SAF 2001-0772 to J.B. and BOS2001-0794 to F.C., and Grants-in-Aids to Priority Area from MEXT, Japan, to N.S. A.M.A. was financially supported by a fellowship from Generalitat de Catalunya, 2000FI 00686.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Andrés, A.M., Soldevila, M., Lao, O. et al. Comparative Genetics of Functional Trinucleotide Tandem Repeats in Humans and Apes. J Mol Evol 59, 329–339 (2004). https://doi.org/10.1007/s00239-004-2628-5
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-2628-5