
Abstract
There are three telencephalic commissures which are paleocortical (the anterior commissure), archicortical (the hippocampal commissure), and neocortical. In non-placental mammals, the neocortical commissural fibers cross the midline together with the anterior and possibly the hippocampal commissure, across the lamina reuniens (joining plate) in the upper part of the lamina terminalis. In placental mammals, a phylogenetically new feature emerged, which is the corpus callosum: it results from an interhemispheric fusion line with specialized groups of mildline glial cells channeling the commissural axons through the interhemispheric meninges toward the contralateral hemispheres. This concerns the frontal lobe mainly however: commissural fibers from the temporo-occipital neocortex still use the anterior commissure to cross, and the posterior occipito-parietal fibers use the hippocampal commissure, forming the splenium in the process. The anterior callosum and the splenium fuse secondarily to form the complete commissural plate. Given the complexity of the processes involved, commissural ageneses are many and usually associated with other diverse defects. They may be due to a failure of the white matter to develop or to the commissural neurons to form or to migrate, to a global failure of the midline crossing processes or to a selective failure of commissuration affecting specific commissural sites (anterior or hippocampal commissures, anterior callosum), or specific sets of commissural axons (paleocortical, hippocampal, neocortical commissural axons). Severe hemispheric dysplasia may prevent the axons from reaching the midline on one or both sides. Besides the intrinsically neural defects, midline meningeal factors may prevent the commissuration as well (interhemispheric cysts or lipoma). As a consequence, commissural agenesis is a malformative feature, not a malformation by itself. Good knowledge of the modern embryological data may allow for a good understanding of a specific pattern in a given individual patient, paving the way for better clinical correlation and genetic counseling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
In 1968, Rakic and Yakovlev published a cardinal article on the development of the cerebral commissures and septum pellucidum which until now probably has remained the fundamental and most often quoted (sometimes erroneously) article on the subject. In this article, they reviewed the previous relevant literature and reported on their own study of 113 brains including 58 brains of embryos and fetuses aged 4–20 weeks, 40 brains of fetuses aged 20–43 weeks, four brains of infants/toddlers aged 1.5–24 months, and 15 brains of adults aged 28–78 years [1]. This helped to settle a then long-standing controversy regarding the mode of development of the corpus callosum, septum pellucidum, and cavum septi pellucidi between two opposed schools of thought. Simply stated, a first group of embryologists (Mihalkovics, His and mostly Zuckerkandl) [quoted in 1] assumed that the callosal fibers crossed the midline through an area of secondary interhemispheric fusion and that the cavum septi pellucidi was the portion of the interhemispheric fissure that became enclosed by the development of the surrounding corpus callosum (Mihalkovics, His, Zuckerkandl, Déjerine) [1, 2]. A second group (Elliot Smith, Johnston and above all Hochstetter) [quoted in 1] developed the view that all the fibers crossed through the same thickened upper portion of the pre-existing lamina terminalis, the so-called commissural plate, and that the cavum resulted from a secondary mechanical cleavage of this commissural plate (Hochstetter) [quoted in 1]. This second view became the most popular at that time, but in their report and from the study of their own specimens, Rakic and Yakovlev strongly made the point that it was the first model of development (Mihalkovics, His, Zuckerkandl) that was correct. They studied the development of the three commissures and stressed a few very important points [1]:
-
The midline crossing bed of the developing corpus callosum (dorsal interhemispheric fusion line, weeks 12–13) is clearly separate in space and time from the lamina reuniens (“joining plate”) through which the anterior commissure (ventral lamina reuniens, week 10) and the hippocampal commissure (dorsal lamina reuniens, week 11) cross.
-
The cavum septi pellucidi is a pocket of the anterior interhemispheric fissure that is secondarily isolated by the developing corpus callosum.
-
The corpus callosum grows according to the expansion of the hemispheres: its frontal segment prenatally, the splenium mostly post-natally. As a consequence, its apparent backward progression is due to the splenium being pushed dorsally by the anterior growth of the frontal lobe and of the associated callosal rostrum, genu, and body.
More recent further embryological studies in mice models as well as in humans have refined but essentially confirmed those findings [3–15].
Anatomy of the great forebrain commissures
The great interhemispheric forebrain commissures are cortico-cortical bundles of white matter connecting the cortex of one hemisphere with the other, mostly in a symmetrical, homotopic fashion. There are three main telencephalic commissures: the anterior commissure, the hippocampal commissure (also called commissure of the fornix or psalterium Davidi [or David’s lyre] in the older literature), and the corpus callosum. These three commissures are readily seen on the midsagittal plane on MR imaging. Together with the third ventricle, optic chiasm, pituitary, brainstem and vermis, they form the prominent structures of the midline. Their morphology (hypoplasia, hyperplasia, agenesis, dysgenesis, even atrophy) necessarily somehow reflects the development of the brain. Their agenesis, complete or partial, is one of the most commonly observed features in the malformations of the brain and is a part of many syndromes. A good understanding of their development is therefore likely to provide a better apprehension of the pathogenetic processes leading to the disorder in a given patient. This development involves complex mechanisms of neuronal migration and cellular and chemical axonal guidance (common to the white matter in general) in addition to midline crossing (like decussating fibers). Therefore, as can be expected, their agenesis/dysgenesis is rarely isolated: although the term “agenesis of corpus callosum” (callosal agenesis) is almost universally used, most clinical cases associate defects of the hippocampal commissure in addition to those of the corpus callosum, often an agenetic or hypoplastic anterior commissure, and typically other white matter, or grey and white matter, derangements. With modern imaging techniques, more attention is paid to individual structures within the white matter, with obvious consequences on the clinical understanding and the genetic counseling.
Summary of comparative anatomy
Commissures are bundles of white matter that connect homologous structures on both sides of the central nervous system (CNS; e.g., corpus callosum). Decussations, by contrast, are bundles of white matter that connect different structures on both sides of the central nervous system (e.g., pyramidal decussation). Although it is obviously not proved with certainty, it is generally assumed that cross-wiring in the central nervous system results from the physics of vision: the retinal image being inverted by the lens, the chiasmatic decussation restores the continuity of the image in the topographically organized occipital cortex; tactile crossing follows to allow easy integration of the sensory inputs, and motor crossing to allow prompt motor response on the appropriate side [Ramon y Cajal, quoted by 16]. As a step further, commissures are needed for bilateral integration and better body coordination.
While the anterior and hippocampal commissures are common to all vertebrates, a corpus callosum is found in placental mammals only [17–21] and results from a complex evolutionary history. Very primitive vertebrates are mostly olfactory and would have a paleopallium only (olfactory brain, “good and bad”) [20]; accordingly, they would have a paleocortical commissure which is the anterior commissure. More advanced vertebrates have developed an archipallium (hippocampus, memory) immediately superimposed to the paleopallium and together with it a hippocampal commissure [20]. Both commissures cross in a very simple way where the two hemispheres are in continuity at the level of the upper lamina terminalis (lamina reuniens of His). Further advanced species have developed a neopallium (neocortex, mostly sensory and motor integration) in addition to the paleopallium and the archipallium [20]. Surprisingly, the corresponding new commissuration developed along different paths in different animal subclasses. In monotremes and marsupials, the neocortical commissural fibers travel through the anterior commissure together with the paleopallial fibers, via the internal capsule (some fibers may travel with the hippocampal commissure also) [21]. On the contrary, in a remarkable process of phylogenetic innovation [4, 17, 18, 21], placental mammals have developed an independent corpus callosum that crosses the midline separately from the anterior and the hippocampal commissures [17, 21]. This new evolutionary process involves the development of a phylogenetically new structure, the transient midline glial “sling” (or “zipper”) that bridges the interhemispheric fissure across the primitive meninge to facilitate the crossing of the neocortical fibers. Not all neocortical commissural fibers follow this path however: most of those from the lateral and inferior temporo-occipital neocortex still travel with the anterior commissure in placental mammals, similar to the marsupial pattern; those from the posteromedial neocortex travel together with the hippocampal commissure and form the splenium. This probably is because they are not submitted to the same constraints of distance as the neocortical fibers of the rest of the hemispheres [21].
Embryologically, the anterior commissure and the hippocampal commissure cross ventrally and dorsally, respectively, through the lamina reuniens, which is located in direct continuity with both hemispheres (“telencephalon impar” of Yakovlev [22]) in front of the anterior insertion of the tela choroidea of the third ventricle. However, in advanced mammals and especially in primates and humans, the enormous development of the mostly associative frontal lobes displaces the hippocampus (parieto-temporal in location in rodents) toward the medial temporal lobes and the corresponding hippocampal commissure from the lamina terminalis to over the posterior third ventricle, stretching the fornix and isolating the velum interpositum in the process [20]. In humans, the hippocampal commissure connects the subicular and parahippocampal cortices and corresponds to the dorsal hippocampal commissure of non-primate mammals. In these, a ventral hippocampal commissure also exists which connects the cornua ammonis and crosses in front of the dorsal hippocampal commissure, between the anterior segments of the fornix. In primates, this ventral hippocampal commissure is markedly reduced in size and connects parts of the hippocampal heads only; it is at best vestigial in humans [23–25].
The anterior commissure
The paleopallial anterior commissure is phylogenetically the oldest of the great forebrain commissures. It extends from one hemisphere to the other in the depth of the anterior portion of the basal ganglia and between the amygdalae, above and behind the septal nuclei. It contains approximately 3.5 million fibers in humans [26], while its diameter is never greater than 6 mm (personal data). In rhesus monkeys and probably in humans alike, it is made of the apposition of paleopallial and neocortical components [27]. The paleopallial component forms the basal telencephalic commissure; it crosses the midline in front of the neocortical component, with a well-defined glial plane in between [27]. The basal telencephalic commissure connects the olfactory bulbs and the paleopallial structures: septal area (medial septal nucleus – or subcallosal gyrus – and lateral septal nucleus), amygdalae, and overlying entorhinal cortices [26]. The neocortical component connects some orbitofrontal and insular neocortex [26], as well as most of the anterior, lateral, and inferior temporo-occipital neocortex [28]. In the monkey, the paleopallial basal telencephalic bundle consists mostly of small unmyelinated fibers, while the more prominent neocortical bundle contains the small myelinated commissural fibers of the temporo-occipital associative neocortex [27].
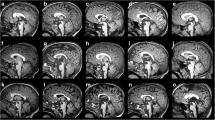
On MR imaging (Fig. 1a–c), the anterior commissure can easily be identified on the axial plane (between the capsular genua) and coronal plane (arching between the amygdalae). On the midsagittal plane, it crosses the midline at the upper end of the lamina terminalis, in front of the interventricular foramen of Monro, within the bifurcation of the fornix where it divides into its pre-commissural (septal) and post-commissural (mammillary) tracts; seen on the axial plane, the anterior commissure on the midline forms the letter π with the post-commissural fibers.

Imaging of the telencephalic commissures. a Midline sagittal T1WI. All commissures originate in front of and above (ventral and rostral to) the foramen of Monro, from where they span the frontoparietal lobes and overhang the posterior third ventricle. The anterior commissure sits at the top of the lamina terminalis; it is joined to the anterior callosum (lamina rostralis, genu, body); the isthmus is marked with a slight narrowing at the level where the fornix abuts the callosum; it contains fibers from the rolandic area; therefore, the anterior callosum contains frontal fibers only; more posteriorly, the splenium contains (clockwise) parietal, medial occipital, and medial temporal fibers associated with the subicular–parahippocampal fibers; the septal area is immediately below the lamina rostralis; the septum pellucidum above it is also limited by the callosal body and the fornix: septum pellucidum and anterior callosum go together. b Coronal view of the anterior commissure stretched between the anterior mesial temporal lobes (olfactory structures); it contains not only the paleocortical fibers but also the neocortical lateral temporo-occipital fibers. c Axial view of the AC, between the septal area anteriorly and the postcommissural and hippocampo-mammillary tracts posteriorly (forming the Greek letter π). d Coronal view of the hippocampal commissure as a transverse velum attached to the fornical crura laterally and to the undersurface of the posterior callosum medially. e Axial T1WI. The HC appears as a division of the septum pellucidum in front of the splenium; the space it limits is the velum interpositum
The hippocampal commissure
Until quite recently, the hippocampal commissure was thought to be only residual in humans [29, 30]. However, electrophysiological recordings in epileptic patients [23, 31], clinical correlation in patients with global (i.e., involving the hippocampal commissure) or anterior (i.e., sparing it) callosotomy [32], and anatomical studies in primates mostly [23–25] suggested that it was really functional. In their detailed anatomical and electrophysiological study, Gloor et al. demonstrated that, like in primates, the (dorsal) hippocampal commissure in humans connects the presubiculum, entorhinal, and parahippocampal cortices, but not the hippocampus proper [23], and found that the ventral hippocampal commissure (that would connect the Ammon’s horns) was at most vestigial in humans [23].
The hippocampal commissure is part of the fornix. The hippocampal fibers of the alveus (the white matter of the hippocampal cortex) gather into the fimbria and form the crus of the fornix. Upon reaching the septum pellucidum on the midline, the fornical columns join each other to form the body of the fornix. There, they course in the lower margin of the pellucidal leaves until they reach the superior-anterior edge of the foramen of Monro, where each column divides into a pre-commissural, hippocampo-septal tract that contains the fibers of the hippocampus proper and connects with the lateral septal nucleus and a post-commissural hippocampo-mammillary tract that contains the fibers from the subicular area and reaches the mammillary body. While 80% of the fibers (hippocampal and subicular/parahippocampal) form this ipsilateral longitudinal fornix, about 20% cross the midline between the fornical crura and form the transverse hippocampal commissure, a triangular transverse structure stretched between the fornical crura with an anterior vertex behind the septum pellucidum and fornical body, a posterior base anterior to the forceps major, and a midline attachment to the undersurface of the callosal splenium. It corresponds to the dorsal hippocampal commissure of the non-primate mammals and connects the subicular and parahippocampal cortices of either side. In monkeys, the hippocampal commissure consists of small myelinated associative fibers; it is clearly separated from the callosal splenium by a glial plane [27]. It should be mentioned that, in addition to the hippocampal commissure, a small number of hippocampal fibers decussate anteriorly to join the contralateral septal area (hippocampal decussation) [25].
On MR imaging (Fig. 1a, d, e), the hippocampal commissure appears in coronal cuts as a transverse layer of white matter attached to the corpus callosum along the midline while laterally its wings are attached to the fornical columns; it forms the roof of the cistern of the velum interpositum. In rare cases, a persistent space between the commissure and the corpus callosum forms the cavum Vergae. In axial cuts, the lateral wings of the hippocampal commissure can only be identified in the form of thin white matter layers diverging posteriorly from the septum pellucidum on either side of the cistern of the velum interpositum. On a sagittal midline cut, the commissure cannot be seen as it is fused with the corpus callosum. Its location, however, can be identified behind the callosal isthmus where the fornix joins the undersurface of the corpus callosum and in the concavity of the splenium. When the ventricles are dilated, especially in case of hydrocephalus, the anatomy is changed: the two wings become verticalized; this is easy to recognize on coronal cuts, but on the midline sagittal the fornix appears lowered and detached from the corpus callosum.
The corpus callosum
The corpus callosum holds its name from its compactness. It is the most prominent forebrain commissure in advanced mammals, spanning much of the frontal and parietal lobes from the anterior commissure anteriorly to the hippocampal commissure posteriorly (Fig. 1a). The callosal commissural neurons are located predominantly in intermediate cortical layers [13]. Given its large size, the human corpus callosum is anatomically subdivided in segments.
From a functional and developmental anatomical point of view, the isthmus is a pivotal segment (Fig. 2). It is located where the columns of the fornix join each other on the undersurface of the corpus callosum on the midline, between the septum pellucidum anteriorly (ventrally) and the hippocampal commissure posteriorly (dorsally). It contains the commissural fibers of the perirolandic area: motor strip, somato-sensory strip, and primary auditory cortex (all primary cortical areas). It divides the corpus callosum into a prominent anterior frontal associative segment that carries the commissural fibers of the frontal associative cortex and a smaller posterior splenial segment that carries the commissural fibers of the primary visual (calcarine) cortex as well as the more associative posterior parietal and medial occipito-temporal cortices. The anterior, frontal callosal segment is related to the septum pellucidum and more remotely to the fornical body, while the splenial segment is related to the hippocampal commissure. As will be detailed in the next paragraph on embryology, this subdivision of the corpus callosum reflects its development, and explains the malformations, better than the classic anatomy.

Wallerian degeneration of the perirolandic commissural fibers. a In this child who presented with a hypoxic-ischemic injury at term, there is a bilateral band of encephalomalacia in the depth of the central sulcus and in the underlying white matter. b The corpus callosum accordingly presents a notch in the location of the isthmus, reflecting the fact that it contains fibers from the central (perirolandic) cortex
The classic, more descriptive callosal segmentation includes, in a clockwise order (brain looking left), the lamina rostralis, the genu, the body, the isthmus, and the splenium.
-
The semi-horizontal rostrum/lamina rostralis (beak) extends anteriorly from the anterior commissure to the posterior inferior aspect of the genu. Although commonly assumed to be the last callosal segment to develop, it is already present in the 14-week fetus [33]. It borders the septal area, or subcallosal gyrus, superiorly and the septum pellucidum anterior-inferiorly and closes the fetal cavum septi pellucidi in the fetus (post-natally a virtual space). The fibers it contains have not been specifically studied but are likely to connect the fronto-basal cortex [34, 35].
-
The genu (knee) is a thickened part of the corpus callosum, so named because of the abrupt change in orientation it marks between the lamina rostralis and the callosal body. It forms the anterior limit of the septum pellucidum. It is made of the commissural fibers of the whole anterior frontal lobe which are collectively called the forceps minor: they connect the prefrontal cortex and the anterior cingulate area [35]. The fibers of the ventro-medial prefrontal cortex are in the ventral genu while the fibers of the dorso-lateral prefrontal cortex are in the dorsal genu [34]. A “MAC line” (for mammillary body–anterior commissure–corpus callosum) has been defined to evaluate the anterior development of the corpus callosum during evolution: the genu falls behind that line in the rat, rabbit, or cat and in front of it in the dog, primate, and human [36].
-
The callosal body is the horizontal portion that extends from the genu to the point where the fornix abuts the undersurface of the corpus callosum. It borders the septum pellucidum superiorly. Laterally, the fibers of the callosal body form the roofs of the lateral ventricular bodies, running between the cingular bundle superiorly and the occipito-frontal fascicle inferiorly and across the anterior radiations of the thalamus (all these form the latero-ventricular crossroad). They connect the precentral cortex (premotor area, supplementary motor area), the adjacent portion of the insula, and the overlying cingulate gyrus mostly [34, 35].
-
The isthmus usually appears as a mild focal narrowing found where the fornix joins the corpus callosum. It carries the commissural fibers of the pre- and post-central gyri (motor and somatoisensory strips) [34, 35] and of the primary auditory area [21, 37].
-
The splenium (spleen) is the thickest portion of the corpus callosum. It protrudes in the ambient cistern and overhangs the tectal plate, while the vein of Galen sweeps around it. Its morphology is extremely variable, from rounded to flat. It should be located above or just at the line drawn along the third ventricular floor [38]; not uncommonly, it drops below this line in cases of idiopathic developmental delay [38]. The splenial fibers form the forceps major and participate in the tapetum, or sagittal stratum, in the lateral wall of the ventricular atrium. They can be subdivided in three groups: the superior group contains the commissural fibers from the posterior parietal cortex; the posterior group, the commissural fibers of the medial occipital cortex; the inferior group, the commissural fibers of the medial temporal cortex [21, 34, 35]. By its undersurface, the splenium is attached to the hippocampal commissure.
-
The upper surface of the corpus callosum is lined with the indusium griseum (gray velum). This thin midline layer of assumedly “vestigial” cortex appears to be the supracallosal continuation of the hippocampal gyrus dentatus and posterior pericallosal fasciola cinerea. The physiological role of the indusium griseum is unknown. However, it contains a glial component which has the important developmental role of directing the pioneering callosal fibers when they start crossing the midline at about weeks 12–13.
Intuitively, for a long time, it has been felt that because most of the callosal fibers are homotopic, their topography would reflect the cortical organization, if only because of specific disconnection syndromes developed after injury to the corpus callosum or callosotomy [reviewed in 39]. However, precisely documented studies are surprisingly scarce, essentially because of methodological difficulties [35, 37, 40, 41]. The problem is compounded by the size of the fibers, their density, and how many fibers each cortical area sends in the corpus callosum, all of which will affect their segmental representation in the corpus callosum. Today’s MR imaging provides a unique opportunity to study the callosal topography in health and disease, using lesional topography (Fig. 2) and/or diffusion tensor imaging [35]. Depending on whether the callosal fibers connect primary sensory–motor or associative areas, they are large and heavily myelinated or small and poorly myelinated, respectively [21, 37]. The highest density of large fibers (3–5 μm) is in the isthmus (motor, somatosensory, auditory cortex) and in the posterior splenium (visual cortex), while the highest density of thin fibers (< 0.4 μm) is in the genu and anterior splenium (high-order prefrontal and temporo-parietal associative areas). The largest callosal fibers in humans correspond to the primary auditory cortex [37].
The septum pellucidum
Abbie [17] and Rakic and Yakovlev [1] strongly emphasized that it was impossible to understand the development of the commissures without understanding the nature of the septum pellucidum. These structures are anatomically, functionally, and developmentally closely related, and the septum pellucidum must be included in any description of the great forebrain commissures. If the septum pellucidum is a simple structure to look at (Fig. 1a, b, e), its detailed anatomy is still somewhat mysterious. Above all, it is often confused with the septal area (or septum) which is the paleopallial portion of the mediobasal frontal cortex. The septal area is made of gray matter that forms two nuclei, a medial septal nucleus (or subcallosal gyrus) and a lateral septal nucleus, both associated with the diagonal band of Broca; the septal area is located below the lamina rostralis and in front of the lamina terminalis and hypothalamus. The septum pellucidum contains no gray matter; it is located above the lamina rostralis, closing the medial aspect of and separating the lateral ventricles. Although classical anatomy textbooks (e.g., Déjerine [2, 42]) mention that its medial aspect is lined with a vestigial cortex, this is not confirmed by the few reported histological studies [43–45]. In addition, myelinated white matter fibers are seen histologically (and radiologically), but the origin and target of those fibers are unclear. Finally, while Rakic and Yakovlev [1, p. 58] (Fig. 8) demonstrated convincingly that the cavum found in fetuses would represent part of the interhemispheric fissure isolated by the growth of the corpus callosum, no meningeal element is reported to be associated with it.
In any case, the septum pellucidum clearly does not contain organized gray matter and is not simply a membrane. It is made of two sheaths of white matter apposed to each other along the midline and has been shown to carry fibers in rats [46], monkeys, [47] and humans [2, 42]. Within its lower (choroidal) edge, it contains the columns of the fornix with hippocampo-septal and hippocampo-mammillary fibers. The fibers contained into the septum pellucidum itself have been described as limbic fibers, namely, the “peduncle of the septum pellucidum” (pédoncule du septum lucidum), the “olfactory fascicle of the fornix” (faisceau olfactif du trigone), and the “olfactory fascicle of cornu Ammonis of Zuckerkandl” (faisceau olfactif de la corne d’Ammon de Zuckerkandl) [2, 42], referred to as the septocingulate perforating pathway [46] that connects the medial septum and diagonal band of Broca to the superior cingulate cortex by intersecting (hence perforating) the corpus callosum [46] and as a bundle of cingulothalamic perforant fibers [47].
In the hindbrain, the posterior medullary velum is a lamina of white matter running along and interposed between the flocculo-nodular lobe and the tela choroidea of the fourth ventricle [48]. In a similar way, a lamina of white matter runs along and is interposed between the limbic lobe and the tela choroidea, closing the lateral ventricles medially and thus forming a medial telencephalic medullary velum [49]. It carries limbic fibers and forms the fimbria along the temporal horns, the crus of the fornix and the hippocampal commisssure around the thalamus at the atrium, and the body of the fornix and the septum pellucidum between the ventricular bodies and frontal horns. The septum pellucidum may be considered an extension of the fornix that contains direct fibers between the cingulate gyrus and the septal nuclei, just like the fornix contains direct fibers from the parahippocampal gyrus to the septal nuclei [46]. It must be noted, however, that this limbic connection pattern alone cannot explain why septal defects or ageneses are commonly found in cases of lateral hemispheric schizencephaly.
The usual position of the two pellucidal sheaths along the midline is a secondary, relatively late event. In the fetus or premature infant, a space is found between the pellucidal leaves, tela choroidea, and corpus callosum, the well-known cavum septi pellucidi, with its occasional posterior recess between the splenium and hippocampal commissure, the cavum Vergae [50] (not to be confused with a cavum veli interpositi, located below the hippocampal commissure). It progressively closes from the back to the front [50] typically before the third post-natal month [51], leaving a tiny triangular residual cavity behind the callosal genu only, but may persist post-natally. Persistent cava usually communicate with the rest of the cerebrospinal fluid (CSF) spaces and are variably considered as normal or abnormal depending on the clinical context (including functional or psychiatric disorders) [51, 52]. They may not communicate with the CSF spaces and become cystic, either diffusely or separately [53]. The lateral, ventricular surface of the septum pellucidum is lined with ependyma, with a subependymal plate that contains primitive neural cells like the remainder of the lateral ventricular wall and may be the origin of various glial and neurono-glial tumors [54–56]. The core of the pellucidal leaves contains small myelinated fibers [44, 57] that are in keeping with associative commissural fibers. The medial surface of the pellucidal laminae (when not fused together) presents without gray matter and without an epithelium (especially no meningeal tissue, even vestigial); it is histologically rather poorly characterized: from the few histological reports of fetal/neonatal or persistent cava or cysts, it appears that it is made of glia in neonates [43–45, 50], which may become covered in part with ependyma-looking cells [43, 44] as the subject matures [43]. The pellucidal laminae may be completely fused and undistinguishable from each other or be grossly fused but still separated by a layer of loose tissue, or form a cavum, reportedly in above 50%, 25%, and 15–25% (or much less), respectively [43, 51].
From the MR imaging point of view (Fig. 1a, b, e), the gross anatomic description of the septum pellucidum is well known. It has the shape of an irregular triangle. The body of the corpus callosum forms its upper edge, the lamina rostralis forms its anterior-inferior edge (and separates it from the septal area), and the body of the fornix forms its posterior-inferior edge. Its anterior vertex is at the genu; its posterior vertex is where the fornix joins the corpus callosum; its inferior vertex is at the anterior commissure. It is typically seen as a thin single lamina between the lateral ventricles. The septal subependymal veins are coursing on the lateral aspect of the septum pellucidum from the anterior corpus callosum toward the internal cerebral veins, which they join (typically) at the foramina of Monro.
Summary of the commissural organization
The telencephalic commissures form an almost continuous structure, very consistently organized throughout. Starting clockwise from the upper end of the lamina terminalis (in front of the tela choroidea of the third ventricle), they include the anterior commissure, the lamina rostralis, genu, and body of the corpus callosum; the isthmus; the splenium with the underlying hippocampal commissure, the body of the fornix closing the loop toward the anterior commissure. The septum pellucidum is circumscribed by the anterior callosum and the body of the fornix. The paleopallial commissural fibers cross the midline through the anterior commissure together with the neocortical fibers of the lateral and inferior surfaces of the temporo-occipital lobes. Unlike in lower mammals, there are no commissural fibers between the cornua ammonis in humans: the hippocampal commissure carries commissural fibers of the subiculum and parahippocampal neocortex. The whole anterior callosum (in front of the isthmus) contains associative frontal commissural fibers. The isthmus contains mostly primary motor, somatosensory, and auditory commissural fibers. The splenium contains a mixture of primary visual and associative temporo-occipital and parietal commissural fibers (in this regard, the splenium may be considered as forming a single segment with the hippocampal commissure that carries parahippocampal fibers). This organization correlates well with the embryological development, as will appear in the next paragraph.
Embryology
The cerebral anatomical terminology has been defined for the mature brain and is not always adapted to the still evolving embryological and early fetal structures. To clearly understand the descriptions of the literature, some review of the brain morphology in its early stages and correlation with the evolving anatomy are recommended. The terminology used here refers to the various articles addressing the embryology of the forebrain midline used in this review [1, 3–15, 46], as well as the Atlas of Central Nervous System Development of SA Bayer and J Altman, specifically, the two volumes covering the late first trimester and the second trimester [58, 59]. The lamina reuniens corresponds to the future septal area [1, 58], while the “primordium hippocampi” [1] corresponds to the fornix [58], that is, to the early form of the medial telencephalic medullary velum. The anterior portion of this medullary velum, between the anterior and the hippocampal commissure, represents the future septum pellucidum. The corticoseptal boundary is the boundary between the medial hemispheric cortex and the septum pellucidum.
During week 4, the neural tube closes, the closing anterior neuropore (about day 25) being located at the level of what will be the lamina terminalis (anterior wall of the 3rd ventricle). During week 6 the hemispheric vesicles begin to expand on either side of the lamina terminalis, so that the lamina terminalis forms the continuity between them (telencephalon impar of Yakovlev [22]). At this stage and until the early fetal weeks, the forebrain is completely embedded in the solid meninx primitiva. During week 7, the diencephalic roof and adjacent parts of the hemispheric vesicles undergo the changes that transform them into the tela choroidea; this is completed by week 8.
During week 8, the portion of the lamina terminalis immediately adjacent to the tela choroidea on the midline becomes thicker (Fig. 3a); because of its continuity between the lateral vesicles, it is called the lamina reuniens, (for “joining plate”) (of His) [1]: it forms the shortest pathway between the hemispheres (Fig. 4a). This lamina reuniens forms the primordium of the septal area, with a medial septal nucleus and a lateral septal nucleus already present [58]. The early fibers of the anterior commissure appear laterally at week 8, get closer to the midline at week 9, and the pioneer fibers cross at week 9 [58] or week 10 [1] in the lamina reuniens [1, 58] (Figs. 3b and 4a). The progression of these fibers is facilitated by “commissural” cellular glial tunnels that guide them along their path [4, 14] (this cellular structure persists as late as week 14 [14]). At this stage and until week 10, there is no evidence of hippocampal commissure or corpus callosum. Yet, the early fibers that connect the septal area anteriorly to the ipsilateral hippocampus (still dorsal to the thalamus [58]) on the medial edge of the hemispheres along the tela choroidea form the primordium of the fornix as early as week 8 [58] or week 9 [1] (Fig. 3b). This ribbon of white matter closes the medial aspect of the ventricle along the tela choroidea and forms a medial telencephalic medullary velum (Fig. 4a).


Commissural embryology, medial aspect of the forebrain. a W7. Thickening of the upper end of the lamina terminalis (LT) forming the lamina reuniens (LR) or “joining plate”. b W9. The fibers of the anterior commissure (AC) have started crossing in the ventral part of the LR; hippocampo-septal fibers are developing along the medial aspect of the hemisphere, forming the early fornix (FO). c W11. Some of the fibers of the fornix cross the midline in the dorsal part of the LR, just anterior to the tela choroidea, and form the hippocampal commissure (HC). d W12. Deepening of the interhemispheric fissure (sulcus medianus telencephali medii or SMTM) divides the LR; the hemispheric portion of the LR forms the early septum pellucidum; glial cells migrate through the corticoseptal boundary, which is the margin between the cortex and the septum pellucidum, and form an interhemispheric glial sling. e Early W13. Pioneer cingulate fibers cross the midline via the glial sling and soon joined by other neocortical fibers; they all form the anterior corpus callosum along the corticoseptal boundary; other neocortical fibers use the AC and the HC to cross, so that at this stage there are three neocortical commissural beds. f Late W13–14. The addition of further neocortical fibers results in the anterior corpus callosum (associated with the glial sling) fusing with the splenium (associated with the HC) to form a complete corpus callosum (CC) and with the AC to form a complete commissural plate; further growth of the callosal plate will push the splenium and HC dorsally (black arrow), so that together they override the third ventricle, stretching the fornix and septum pellucidum and forming the roof of the cistern of the velum interpositum

Commissural embryology, coronal. a W9–10. The LR is divided by the deepening of the SMTM, the lateral portions of which form the medial wall of the lateral ventricles (medullary velum); the AC crosses in the medial portion of the LR; the junction between the cortex and the medullary velum is the corticoseptal boundary. b W11. At the corticoseptal boundary, three specialized glial structures develop: the glial sling (attractant) forms a bridge between the hemispheres to channel the callosal fibers across the midline, together with the indusium griseum glia above and the glial wedge below (both repellents). c W13. The cingulate fibers first, then the fibers from the rest of the frontal lobes commissurate along the glial sling and between the glia of the indusium griseum and glial wedge; were the sling missing, they would turn into the medullary velum to form the Probst’s bundles; the space enclosed by the developing corpus callosum forms the cavum septi pellucidi
The fornix expands during weeks 9–11 [58]. By weeks 10–11, some of the fornical fibers are noted to exchange across the midline just in front of the attachment of the tela choroidea, where they form the early hippocampal commissure (Fig. 3c). The crossing occurs dorsal and rostral to the anterior commissure and the septal area, either through the lamina reuniens itself [1] or at its surface, under the meningeal lining [8]. The segment of the fornix located between the septal nuclei and the hippocampal commissure represents the early septum pellucidum (Fig. 3d). At week 11, no corpus callosum is seen yet; the anterior commissure is well apparent, ventral to the interventricular foramina [58, plate 6A]; more rostrally within the septal area-lamina reuniens, the hippocampal commissure can be recognized ([1] Figs. 5b and 16c) ([58] plate 4B). Still more dorsally ([58] plates 6A–12A), the fornix connects the septal nuclei to the hippocampus; it is interposed between the medial edge of the limbic cortex and the tela choroidea as a true limbic medullary velum.

Complete commissural agenesis. a Midline sagittal T1WI. The AC is tiny (containing fibers of the olfactory cortex only?); there is no CC or HC; the medial hemispheric sulci radiate toward the cerebral hilum; the third ventricular roof is expanded as it is not maintained by the commissural plate. b Coronal T2WI. The lateral ventricles are widely separated, containing the heavily myelinated bundles of Probst (rerouted commissural fibers); the cingulate cortex is rolled out over the thalami; the internal cerebral veins are separated by the high-riding tela choroidea, and the free edge of the falx is low; the lumen of the temporal horns is abnormal in shape and extends deep into the parahippocampal gyrus (temporal portion of the cingular bundle missing). c Axial T2WI. Gross posterior ventricular dilatation forming the so-called colpocephaly
As mentioned before, the corpus callosum is a new phylogenetic acquisition of the placental mammals that develops through a process of interhemispheric midline fusion with groups of specialized midline glial guiding the callosal fibers to the other side. At about 10 weeks, the interhemispheric midline is still filled with the meninx primitiva [1]. Deepening of the interhemispheric fissure results in a clefting of the dorsal lamina reunions and in the formation of a sulcus named sulcus medianus telencephali medii (SMTM) [1] (Fig. 4a). The two lips of this sulcus come in close approximation at the edge of the cortical plate [1] now designated as the genetically determined corticoseptal boundary [6, 9, 11, 13, 60] which is where a clear demarcation is seen between the cortex and the fornical fibers (medial medullary velum) (Figs. 3d and 4a). Glial cells (and seemingly also neuronal cells, at least in mice [12]) migrate medially from the subventricular zone toward the midline and invade the primitive meninge along the corticoseptal boundary to form a “glial sling” [3–6, 10, 12, 14, 15] or “midline zipper glia” [9, 11, 13] (“massa commissuralis” of Rakic and Yakovlev) [1] (Figs. 3d and 4b); this process begins at about week 12 [1]. This interhemispheric glial bridge provides a support for the first pioneer axons to cross at about weeks 12–13 [1] (Fig. 4c), before disappearing shortly after [1]. The glial sling is needed for the callosal crossing: if it is severed in mouse embryos, the callosal fibers do not cross and instead travel parallel to the midline in the future septum pellucidum [3], but they resume crossing if the sling is restored [61]. The glial sling is independent anatomically and developmentally from both the anterior and the hippocampal commissures: it is located rostral and dorsal to them [1, 4]; in genetically acallosal mice, both anterior and hippocampal commissures develop normally [4]; surgical disruption of the sling in mouse embryo results in callosal agenesis without altering the other commissures [2, 4].
In addition to the glial sling, two other glial structures are needed to convey the pioneer callosal fibers toward the glial sling and the midline: the indusium griseum glia and the glial wedge. The indusium griseum glia is located just above the corticoseptal boundary; the glial wedge is located at the dorsomedial aspect of the lateral ventricle [9–11, 13] and is part of the radial glia scaffold [13] (Fig. 4b). It should be mentioned that, posteriorly, the glial wedge is located between the hippocampal commissure and the sling, so that the callosum at this stage is clearly separate from the hippocampal commissure [9]. Indusium griseum glia and glial wedge act as repellents (chemorepulsive molecule slit-2) and channel the incoming pioneer axons across the corticoseptal boundary toward the glial sling and the midline (Fig. 4c). There, the pioneer callosal axons follow the sling and cross the midline without penetrating the sling or invading the septum pellucidum [3, 4] The first pioneer axons to cross are cingulate axons from the adjacent cingulate cortex [7, 10]: they are eventually followed by further neocortical axons which fasciculate along them. This complex process occurs within a brief period of time of about a week (week 13) [1]; while the first axons begin to cross, the sling is still forming ventrally and dorsally.
There are three separate commissural sites at this stage, all in the close vicinity of the foramen of Monro: the anterior commissure within the ventral lamina reuniens, the hippocampal commissure within the dorsal lamina reuniens, and rostral to the lamina reuniens and separately from from it by the furure septum pellucidum, the glial sling and the pioneer callosal axons crossing the interhemispheric fissure at the corticoseptal (Fig. 3d, e). Later-arriving neocortical commissural axons eventually cross according to the cortical area they originate from: axons from the lateral and inferior temporo-occipital neocortex fasciculate along the anterior commissure; those from the anterior hemispheric neocortex, along the pioneer callosal fibers and glial sling; those from posterior neocortex, along the hippocampal commissure (Fig. 3e). These two latter segments grow by addition of more neocortical fibers and eventually meet to form a single corpus callosum (Fig. 3f). Thus, the corpus callosum forms by fusion of two separate segments, not as a single structure [8], and this explains some “unusual” callosal malformations. The fusion between the anterior, sling-derived callosum (frontal associative and possibly primary sensory motor) with the hippocampal commissure-associated splenium (parieto-temporo-occipital) may be hypothesized to occur just anterior to the hippocampal commissure, but this has to be established.
A week later or so, the corpus callosum is short but essentially complete and stretches from the anterior commissure to the hippocampal commissure. From that moment, it grows by addition of fibers that fasciculate along pre-existing fibers as the cortex expands and the connectivity develops until well after birth. It was classically postulated that the splenium would form after the genu and the body and that the rostrum would form last: it is quite clear now that the hippocampal commissure comes first, then the anterior callosum, then the whole commissural plate from the lamina rostralis to the splenium. The corpus callosum (lamina rostralis, genu, and body fused with the splenium) is complete and easily recognized by week 14 [33, 36] or week 15 [15], the splenium becoming prominent by weeks 18–19 [15]. It was also classically postulated that the corpus callosum would develop from the front to the back: yet, as early as 1968, Rakic and Yakovlev convincingly demonstrated that prenatally it was the anterior part of the corpus callosum that developed most [1]. Actually, because of the disproportionate expansion of the frontal lobes in humans, and the related anterior accumulation of commissural fibers, the hippocampal commissure with its associated splenium, although initially located in the lamina reuniens in front of the tela choroidea of the third ventricle, is shifted posteriorly (dorsally) over the tela choroidea (isolating the velum interpositum and stretching the fornical columns) until it projects above the posterior third ventricle and the tectal plate, while the hippocampal commissure becomes overlaid by the splenium. This front-to-back translation of the splenium results from the back-to-front expansion of the frontal lobes and the corresponding accumulation of fibers in the anterior callosal segment and not from a front-to-back progression of the corpus callosum. This is an important point to consider when trying to explain why a corpus callosum is hypogenetic in a given patient.
Although the shape of the corpus callosum is essentially final by week 20, its sagittal cross-sectional area is only 5% of what it will be in a mature brain [65], roughly in proportion with the volume of the brain at this stage (about 80 g). The corpus callosum enlarges together with the connectivity and the tangential growth of the cortex: in utero and in the early maturation it grows by addition of fibers; later pruning of the callosal fibers is compensated by the development of the myelin, which becomes significant post-natally at about 6 months at the splenium and at about 8 months in the genu on T2 imaging. The myelination is said to proceed from posterior to anterior: this reflects the fact that myelination of the primary cortical areas (somatosensory, motor, auditory, visual) connected through the isthmus and splenium antecedes the myelination of the body, genu, and rostrum related to the more anterior associative areas.
The septum pellucidum forms in close association with the anterior corpus callosum. Its lower margin contains the early fibers (week 10) of the segment of the fornix located between the septal area and the hippocampal commissure (week 11). The fibers that connect the medial septal nucleus with the ipsilateral cingulate cortex develop in the pellucidal leaves themselves at the same time as the pioneer callosal fiber cross the midline in the rat: they intersect the glial sling and the corpus callosum along the way [46]. Because the septum pellucidum evolves from the banks of the sulcus mediani telencephali medii, it becomes circumscribed by the lamina rostralis, genu, and body of the corpus callosum that develop along the corticoseptal boundary (Figs. 3f and 4c). The interhemispheric space isolated between the pellucidal leaves becomes the cavum septi pellucidi [1, 2] (with or without the posterior extension of the cavum Vergae). This space was initially thought to be a meningeal space [1, 2], but some more recent evidence suggests that it is the space that is left after the disappearance of the glial sling [6]: this would explain why no meningeal epithelium is found on the medial aspect of the pellucidal leaves in premature infants with cavum septi pellucidi [43]. The cavum is not truly apparent before 20 weeks [60] and usually disappears within 3 months post-natally, its lumen becoming progressively effaced from the back to the front [50]. There is no convincing explanation given for this effacement.
Commissural axons reach the midline only after being guided by cellular influences (cellular tunnels, midline glia), short-range guidance factors (attractants and repellents), long-range guidance factors (attractants and repellents), midline crossing factors (short range and long range), fasciculation factors ( “pull together” and “ push together”), and defasciculation factors. It is beyond the scope of this article and beyond the expertise of the author to provide a detailed description of the multiple genes, transcription factors, receptors, etc. involved in such complex processes; the interested readers are invited to refer to the relevant literature [3–15, 46, 62–68].
Abnormal commissures may be expected in a large number of genetic defects, familial or not, identified or not, syndromic or not [for review see 13]. As a consequence, “agenesis of corpus callosum” should not be considered a specific entity: it is one anatomical feature of many different diseases.
Classic commisural agenesis
The classic, or pure, commissural agenesis is a well-known constellation of features, the most prominent one being the absence of apparent interhemispheric connection. Given the normal prominence of the commissures, this absence results in a characteristic deformity of the brain (Fig. 5).
First of all, in the classic commissural agenesis, the fibers of the corpus callosum are not agenetic, but rather heterotopic. Unable to cross when reaching the corticoseptal boundary, the fibers instead make a right angle and travel into the medullary velum. Gathering there, the fibers form a parasagittal bundle that encroaches medially upon the lateral ventricular lumen, giving it a crescentic appearance and therefore giving the appearance of a bull’s head to the section of the lateral and third ventricles on the coronal cuts (Fig. 5b). This bundle is called the bundle of Probst in the literature, from M Probst who described it in 1901 [69]; it had already been described and recognized to be made of re-routed callosal fibers by Onufrowicz in 1887 [71] and by Sachs in 1892 [72]. This bundle is easily identified because of its dense myelination of T1 and T2 images, on coronal and on axial planes. In addition to the corpus callosum, the hippocampal commissure is missing also in the classic total commissural agenesis (Fig. 5a). The hippocampal commissural fibers may be re-routed as well, either in the lower edge of the medial telencephalic medullary velum together the ipsilateral fibers of the fornix or in the bundle of Probst; the accumulation of the callosal and fornical fibers explains the thick appearance of the velum. In about 50% of the cases, the anterior commissure is either absent or too thin to be recognized, or apparent but hypoplastic [49]; this could be due to the absence of its neocortical component (Fig. 5a). It is classically mentioned that it may be enlarged, as if compensating for the missing corpus callosum. This was specifically observed not to occur in a large series of cases [49, 70], and in the cases where it was mentioned it could actually have been confused with a dislocated hippocampal commissure or a small residual genu. This difference is important to make: if no commissural fiber can cross, it means that there is a global defect of the midline crossing independent from local signals; if only neocortical fibers are concerned, it is a specifically neocortical defect; if some neocortical fibers at least are re-routed across another commissural site, it means that the defect is local and concerns the glial sling only and that the commissural fibers would be free to “choose” their course.
Because of the lack of commissure, the midline anatomy is changed. The interhemispheric fissure is wide with an upward bulge of the tela choroidea of the third ventricle, and the free margin of the falx is low (Fig. 5b). Not being joined together by the commissures, the lateral ventricles are separate, away from the midline and parallel to each other, without the usual image of a thin midline septum pellucidum. This is explained by the addition of the widened interhemispheric fissure, the rolling out of the limbic/cingulate cortex above the medullary velum, and the thickness of the bundles of Probst on the medial aspect of the lateral ventricles. On a sagittal cut of the medial aspect of the hemisphere, the midline sulci converge toward the third ventricle (the hemispheric hilum), as apparently the cingulate gyrus cannot form properly in the absence of a corpus callosum (Fig. 5a).
The lateral ventricular shape also is abnormal. Besides the crescentic appearance of the frontal horns and bodies, this is especially striking at the level of the temporal horns. On a coronal cut, the ventricular lumen extends into the parahippocampal gyrus, under the rounded hippocampus itself, so that the section of the horn has the shape of a horizontal Y (Fig. 5b). This can be explained only by the lack of the temporal portion of the cingulum (or cingular bundle) [48, 73]. This white matter fascicle runs along the midline into the entorhinal, parahippocampal, retrosplenial, and cingulate gyri. In monkeys and presumably in humans, it consists of three main components originating from (1) the anterior and dorsal thalamus, (2) the cingulate gyrus, and (3) the associative cortex [74]. The temporal segment of the cingulum corresponds to the fibers that connect the presubiculum and parahippocampal gyrus to the posterior cingulate gyrus and parietal associative cortex [74]. Whether the abnormalities of the cingulum extend further toward the anterior frontal regions is not known; it could help to explain why the cingulate cortex does not form a clearly defined gyrus in commissural agenesis. Finally, the ventricular morphology is also remarkable for the colpocephaly, which is a broad expansion of the atrium and occipital horn (Fig. 5c). A common explanation given is the lack of the forceps major, and it has been mentioned that the commissural fibers of the posterior part of the hemisphere would not become part of the bundles of Probst and might lack entirely in commissural agenesis [75]. We could not find any report in the literature to confirm this point; it would imply that the posterior fibers are “programmed” to fasciculate with the hippocampal commissural fibers but not to pierce the corticoseptal boundary to follow the interhemispheric glial sling. The other fiber tracts surrounding the posterior part of the ventricle are the optic radiations, the occipito-frontal and the occipito-temporal fasciculi, the cingulate and parietal fibers converging toward the temporal segment of the cingulum, and the mostly associative visual parieto-temporo-occipital short fascicles [42]: anyone of these may be missing, in part or globally. This white matter defect is associated with shallowness of the interhemispheric occipital sulci. Systematic DTI studies of these fascicles could be extremely informative, as the identification of the white matter defects could help to explain the neurocognitive disorders observed in the patients.
Not all complete commissural agenesis present the whole set of features described above: it is probably highly significant to demonstrate that an apparently common complete commissural agenesis presents with or without an agenesis of the anterior commissure, or with or without the bundles of Probst, as this can point to such or such defect along the cascade of events that lead to the abnormal-versus-normal appearance of the brain. Also, white matter progression, fasciculation and midline crossing occur throughout the central nervous system, and it is therefore important to examine the whole intracranial CNS at least, and possibly also the spinal cord, to identify a global CNS involvement and whether the supratentorial abnormalities are isolated, part of a constellation of neural defects, or part of a global syndrome with associated extraneural defects. Again, commissural (or callosal) agenesis is not a disease entity but a feature that may be a part of many different malformations. The specific characters of the disorder must be identified if one wants to focus the search for a causal mechanism.
Partial commisural agenesis (commisural hypogenesis)
It has long been assumed that the corpus callosum developing from front to back, the partial commissural ageneses, most commonly apparently posterior, would be lesser forms of agenesis and would represent later-occurring (implicit: lesser) disorders. It was also assumed that the rostrum/lamina rostralis would develop last. However, modern embryological data in man indicates that the corpus callosum develops within a very short time during week 13 and that at week 14 it is virtually complete, though still short [1, 15, 36], and that from then on it grows anteriorly mostly, pushing the splenium dorsalward. Therefore, when looking at any instance of incomplete commissuration, attention should be paid separately to all constitutive segments of the commissures (anterior commissure, hippocampal commissure with associated splenium, anterior callosal segment, and septum pellucidum), as well as to their fusion areas (anterior callosum with anterior commissure, anterior callosum with splenium, splenium with hippocampal commissure) and to their expansion in thickness as well as dorso-ventrally. It quickly appears from such an analysis of the malformations based on the modern embryological understanding that partial ageneses are not different degrees of a continuum but rather different malformations, each with a specific developmental disorder and, potentially, a specific genetic defect.
In the most common and typical form of partial commissural agenesis, the commissural plate is short but essentially complete: relatively prominent genu, short body with small septum pellucidum; the splenium is clearly demarcated and its anatomical relationship with the fornix and hippocampal commissure look normal, suggesting that the hippocampal commissure is normally developed (Fig. 6). The anterior cuts through the completed commissural segment have a normal anatomical appearance (Fig. 6b), while the posterior coronal cut behind it is typical for agenesis with well apparent bundles of Probst (Fig. 6c). While classically this would have been read as an agenesis of the posterior part of the commissures (splenium and hippocampal commissure), the understanding now is that, both the splenium and the hippocampal commissure being well defined, the defect results from a poor dorsoventral expansion, that is, a defect of late addition of neocortical commissural fibers to an otherwise normally induced and complete commissural plate. The insufficient number of neocortical callosal fibers crossing could correlate with the small size of the anterior commissure (neocortical component incomplete?). Whether all fibers that constitute the bundle of Probst posteriorly cross or not is unknown; DTI would help in determining this point.

Partial commissural agenesis, common form. a Midline sagittal T1. The commissural plate is short but virtually complete; the anterior segment is attached to a proportionately small septum pellucidum, the posterior fornix and HC are normally attached to the splenium, also proportionate with the whole commissural plate: it may be assumed that both the HC and glial sling formed normally, but that the addition of neocortical fibers was not sufficient to produce the normal dorso-ventral expansion of the commissural plate; this may explain the small AC as well. b Coronal T2WI, anterior. Normal appearance. c Coronal T2WI, posterior. The appearance is similar to a usual agenesis, but the bundles of Probst may be less prominent
The nature of extreme types of partial commissural agenesis is more difficult to ascertain, but embryology suggests answers. They typically present with a rudiment of what is commonly assumed to be a genu because of its anterior location (Fig. 7a). The anterior commissure is present ventral to the foramen of Monro, with the “callosal” rudiment slightly more dorsal and rostral (Fig. 7a, b). The medial aspect of the brain presents with radiating sulci; the coronal cuts show typical bundles of Probst posteriorly (Fig. 7c), with the commissure interposed anteriorly (Fig. 7b). The important point to consider is that the commissural rudiment is located in the rostro-dorsal portion of the lamina terminalis just anterior to the tela choroidea, and this corresponds embryologically to the lamina reuniens (see Fig. 3a, b). Therefore, it is in the expected location of a hippocampal commissure that would not have been displaced dorsally by the callosal growth, and the malformation may therefore be considered a complete callosal agenesis with the anterior and hippocampal commissures being present. It is not impossible that this hippocampal commissure would carry some callosal (neocortical) fibers. These callosal fibers could come from the posterior part of the hemispheres via the bundles of Probst to form a true splenium in an anterior location or, alternatively, they could be selected by the anterior location of the hippocampal commissural bed and originate from the anterior part of the hemisphere (Fig. 8). Modern MR imaging with DTI might help in answering that question. An anteriorly located hippocampal commissure can be differentiated from a rudimentary callosum by the fact that it is located in the ventricular roof, anterior to or apparently in the tela choroidea (Fig. 9), while the anterior callosum would be associated with the septum pellucidum it circumscribes (Figs. 10 and 11).

Extreme partial commissural agenesis. a Midline sagittal T1WI. There is a commissural rudiment located in the third ventricular roof just anterior to the foramen of Monro, close to the AC; this would commonly be identified as a callosal “genu” because of its location according to a complete callosum; however, it is located where the HC should be in the early stage of development before the CC develops and pushes the HC dorsally (compare with Fig. 3c): therefore, it is likely a HC with complete callosal agenesis. b Anterior coronal T2WI. The commissure between the septal leaves. c Posterior coronal T2WI. Classical appearance of commissural agenesis

Possible configurations of a non-shifted HC. a The hippocampal commissure is not shifted dorsally because the callosum failed to form: it is located anterior to the tela choroidea but contains no callosal fibers. b Posterior callosal fibers being programmed to cross with the HC might contribute to it. c Inversely, the posterior fibers might be unable to cross because the HC is not in the appropriate location, but the anterior callosal fibers might be able to do it

Extreme partial commissural agenesis. a Midline sagittal T1WI. The commissural rudiment is smaller and located more posteriorly than in Fig. 7 but still in the roof of the third ventricle. b Coronal T2WI. The commissure connects the fornices, not the bundles of Probst, and therefore is a HC

Hypoplastic callosum, absent HC. Midline sagittal T2WI. The commissural rudiment is separated from the roof of the ventricle by a small but proportionate septum pellucidum; its appearance is that of a callosal genu with a rudimentary lamina rostralis and no appreciable splenium. The HC is likely agenetic or extremely hypoplastic

Hypoplastic callosum. a Midline sagittal T1WI. The appearance is similar to that of Fig. 7 and could suggest that it is a HC. b Axial FLAIR. A septum pellucidum is present, although hypoplastic, located centrally in relation to the commissure: the commissural rudiment therefore is a hypoplastic callosum, not a HC
Inversely, the corpus callosum may be present, but not the hippocampal commissure. In such a case, the anterior corpus callosum is well defined as it is associated with a well-defined fornix and septum pellucidum (Fig. 12a, b); posteriorly, however, the fornix does not join the posterior callosum, and no hippocampal commissure is seen (so assumedly no splenium) and no bundle of Probst (Fig. 12c).

Agenesis of the hippocampal commissure. a The anterior CC is mildly hypoplastic but present; together with the fornix it circumscribes the septum pellucidum; however, the fornix does not join the callosal undersurface and there is no well-defined splenium. b Intermediate coronal T2WI. Normal appearance of the callosum, septum pellucidum, and fornix but abnormal appearance of the parahippocampal gyrus (defect of the temporal portion of the cingular bundle). c Posterior T2WI. No splenium, no HC, no bundle of Probst
A particular pattern of partial callosal agenesis is the segmental callosal agenesis, in which the most posterior, dorsal part of the anterior callosum did not fuse with the splenium and the attached hippocampal commissure (Figs. 13 and 14). This feature was classically explained as resulting from a destructive process; however, no evidence of hemispheric cleft or encephalomalacia is found. In addition, such a pattern has been reported in two siblings [49]. On sagittal images, the callosum appears to be made of two separate segments, the ventral one bordering the septum pellucidum with a rostrum that extends to the anterior commissure and the dorsal one attached to the fornix (Figs. 13a and 14). On coronal images, the ventral segment appears to be a true callosal genu and anterior body (Fig. 13b), the intermediate segment is a thin lamina of hippocampal commissure (Fig. 13c), and the posterior segment appears as a true splenium (Fig. 13d).

Segmental agenesis of the corpus callosum, with associated hypoplasia. a Midline sagittal T1WI. The CC is globally hypoplastic; with a tiny rostrum, genu, and anterior body, very thin posterior body and small splenium; the septum pellucidum is not apparent on this cut. b Anterior coronal T2WI. The septum pellucidum forms a cavum, with its leaves on either side of the midline (therefore missed in a). c Intermediate coronal T2WI. The intermediate segment of the commissural plate is made of a HC, and the posterior callosal body is missing. d Posterior coronal T2WI. The splenium is present with the HC

Segmental agenesis of the corpus callosum. Frontonasal dysplasia. Midline sagittal T2WI. The anterior callosum is fairly well developed, with a well-defined septum pellucidum, but it does not join the splenium posteriorly. The continuity of the splenium and HC with the fornix is well demonstrated
A posterior splenial tapering is fairly common also and is assumed to result from a poor commissuration of the posterior hemispheric neocortical commissural fibers, in spite of the fact that the hippocampal commissure is normally present; the anterior callosum may be normal or hypoplastic (Fig. 15). Finally, the commissural plate may be complete, but diffusely hypoplastic. While it is obviously malformative when the remainder of the white matter is normal (Fig. 16), it is difficult to tell whether it is destructive or developmental when it is associated with a significant ventriculomegaly (Fig. 17).

Posterior tapering of the corpus callosum. Midline sagittal T1WI. The CC, septum pellucidum, AC, fornix, and HC are well defined but the splenium failed to develop in proportion, suggesting a defect in the neocortical commissuration of the posterior part of the hemispheres

Diffusely hypoplastic commissural plate. a Midline sagittal T2WI. The commissural plate is complete but small and poorly shaped. b Axial FLAIR. The brain appears otherwise essentially normal, without ventriculomegaly. This is in keeping with a defect of the commissuration processes

Diffusely hypoplastic commissural plate with ventriculomegaly. a Midline sagittal T1WI. The commissural plate is complete but thin with a tiny splenium. b Axial FLAIR. Diffuse ventriculomegaly without real evidence of leukomalacia: this points to a global white matter disorder that may be developmental or acquired, not a commissural disorder
Commissural agenesis associated with midline meningeal dysplasia
Interhemispheric cystic meningeal dysplasia
Classic complete or partial commissural agenesis is assumed to have causes that are intrinsic to the hemispheres: missing or defective commissural fibers, lack of cellular facilitation in the lamina reuniens and/or failure of the glial sling to form, and lack of long- or short-range attractants or repellents. Agenesis of the commissures with interhemispheric cysts is felt to have different causes, possibly related to a meningeal rather than neural disorder. There are two broad classes of interhemispheric cysts, communicating and non-communicating [48, 70, 76], the communicating cysts being expansions of the ventricular tela choroidea and the non-communicating cyst being a multiloculated meningeal cystic dysplasia. From the morphologic data in a quite large series of 25 cases, Barkovich et al. devised a classification of the callosal agenesis with cysts [76]. Type 1 refers to the cases where there is one single cystic cavity that communicates with the ventricles. Type 1 is subdivided in three subgroups: type 1a presents with macrocephaly and hydrocephalus, type 1b with macrocephaly and hydrocephalus associated with a developmental ventricular obstruction (thalamic fusion, hamartoma), and type 1c presents with microcephaly. Type 2 refers to the cases where the interhemispheric cysts are multiloculated and independent from the ventricles; it is subdivided into three subgroups also: type 2a is characterized by hydrocephalus and an essentially normal brain, type 2b affects girls and is made of multiple cysts different from CSF with frontoparietal polymicrogyria and periventricular nodular heterotopias and one or two dilated ventricles (this subgroup probably corresponds to the Aicardi syndrome) [76], and type 2c presents with multiloculated cysts, large subcortical heterotopia, and dysmorphic head and brain. It appears that these specific subcategories would concern either boys or girls and this obviously points to genetic defects [76]; however, it needs to be confirmed by studies of larger groups, as some reports may be discordant [77].
The commissural agenesis with a single ventricular diverticulation cyst is usually not associated with significant hemispheric dysplasia or malformations of cortical development. The main feature is the markedly expanded tela choroidea which involves its third ventricular component together with that of one or two lateral ventricles (so it is different from the classical high-riding third ventricular roof where the three ventricles remain separate) (Fig. 18). This implies that the tela choroidea is detached from the thalamus on one or both sides. (Given the lack of thalamic insertion of the tela choroidea with its choroidal vessels, it would be interesting to know where the vascularity of the thalami and adjacent globi pallidi comes from.) The septum pellucidum, fornices, and bundles of Probst are missing (Fig. 18c, d). It may therefore be assumed that it is this total lack of medial telencephalic medullary velum that allows the diverticulation. Such cases have been previously described as “septo-optic dysplasia with total absence of the corpus callosum” [78] or “agenesis of the corpus callosum with dehiscent fornices” [79]. When the diverticulation is bilateral, the appearance is close to that of a holoprosencephaly, except of course that the hemispheres are fully separated. The falx is commonly hypoplastic or absent. These cases may be considered as pure white matter disorders that associate the features of a complete commissural agenesis with both septal and fornical ageneses (complete lack of the ipsilateral limbic connections of the septal nuclei).

Complete commissural agenesis with septal agenesis and single communicating interhemispheric cyst (neonate). a Midline sagittal T1WI. Huge interhemispheric expansion of the roof of the third ventricle with posterior blood layering; no AC, CC, or HC. b Axial T1WI. Appearances confirm the interhemispheric cyst with blood layering, the commissural agenesis with symmetrical hemispheres, and the absence of cortical dysgenesis or heterotopia. c Anterior coronal T2WI. Complete commissural agenesis without the bundles of Probst: the cyst is a bilateral diverticulation of the tela choroidea of the third and both lateral ventricles; the falx is hypoplastic. d Posterior coronal T2WI. Similar appearance. Even though the hemispheres are well separated, the sac is an evagination of the complete prosencephalic tela choroidea. There is no medial telencephalic medullary velum (no septum, no fornix, no Probst’s bundles) and it may be speculated that the choroidal evagination results from this defect. Note that, since the tela choroidea is detached from its normal insertion, the thalami, basal ganglia, and adjacent deep white matter cannot be supplied or drained by the usual choroidal arteries and veins
In commissural agenesis with multilocular cysts, the cysts themselves result from a meningeal dysplasia and not from a lack of physical containment. The CT density and the MR signals of some of these cysts commonly are different from those of the CSF, but to the best of our knowledge there is no data in the literature regarding any histological peculiarity to explain a protein content different from that of the CSF, except in the perinatal period when intracystic bleed may occur (Fig. 19a–c). The clinical situation may be relatively simple when the brain parenchyma presents without dysplasia other than the commissural defect. Children usually are born with hydrocephalus and the size of the cysts usually increased during gestation (Fig. 19d, e); the multiplicity of the cysts makes the surgical treatment difficult, as a communication should be established to achieve a complete drainage. Most of the cases of multilocular cysts are more complex as the cysts are commonly associated with significant cerebral dysplasia. Cases with large sub-cortical heterotopias seem not to have a bundle of Probst on the affected side or an incomplete one (Fig. 20). This is not different from what can be observed in instances of unilateral dysplasia without interhemispheric cysts as well. This suggests that the hemispheric dysplasia by itself may be a causing factor, independently from the meningeal dysplasia.

Complete commissural agenesis with multiple interhemispheric cysts (neonate). a Midline sagittal T1WI. A huge interhemispheric cyst compresses the third ventricular roof. b, c Coronal and axial T2WI. Multiple interhemispheric cysts with different intensities (blood layering) expand toward the left side mostly; the right subdural collection is related to a difficult delivery (macrocephaly); no apparent cortical dysplasia or heterotopia. d, e. Same child, fetal coronal and axial HASTE about 30w GA. The interhemispheric cysts were present already, but they have significantly expanded toward term

Commissural agenesis with multiple interhemispheric cysts and cerebral dysplasia. a Coronal T2WI. The interhemispheric commissures are absent. There is a bundle of Probst on the left, but not on the right where a large subcortical heterotopia is seen instead. b Axial FLAIR. The right hemisphere is smaller, with diffuse cortical dysplasia and heterotopia adjacent to the interhemispheric cysts
The most consistent example of X-linked callosal agenesis with multiple dysplastic meningeal cysts is the Aicardi syndrome [80]. It is observed in girls only, or at least in patients with double X chromosome (i.e., boys with Klinefelter syndrome). The clinical features include infantile (or even neonatal) spasms usually without typical hypsarrhythmia and severe neurological and mental impairment. The morphological features (Fig. 21) include a partial or total callosal agenesis (with the bundles of Probst usually present), a marked asymmetry between the hemispheres (the larger hemisphere commonly corresponds to the neurological deficits and the spasms), an interhemispheric cystic meningeal dysplasia, an unlayered polymicrogyria, periventricular or subcortical nodular heterotopias, and choroid plexus cysts or papillomas. Choroidal ocular lacunae are characteristic, and ocular colobomata are common [80]. Although the syndrome may be incomplete, some features are particularly relevant to the diagnosis, such as the commissural defects, the irregular ventricular contours, the polymicrogyria, the asymmetry between the hemispheres, and the choroidal lacunae [80].

Aicardi syndrome, newborn girl. Huge right-sided choroid plexus cyst with adjacent parenchymal damage. Note the multiple subcortical heterotopias ad cortical dysplasia in the adjacent right frontal lobe and in the left parietal lobe
Interhemispheric meningeal lipomas
The second meningeal dysplasia commonly associated with a malformation of the commissures is the interhemispheric meningeal lipoma. Intracranial meningeal lipoma is believed to result from an abnormal differentiation of the meninx primitiva and as such is located in the leptomeninges [81]. It is often associated with vessels (typically dysplastic as well) and it may calcify. It is found at different specific sites (tectal/culminal cistern, tuber cinereum, cerebello-pontine angle cistern, and sylvian fissure) [81], the most common location being the depth of the interhemispheric fissure where the lipoma often extends through the choroid fissures into the choroid plexuses [81]. In this interhemispheric location, a lipoma is commonly associated with an abnormal corpus callosum. The mechanism of the malformative association is not really known. The time when the abnormal fatty differentiation of the meninges occurs in relation to the commissural agenda is not documented. It is known from previous studies on the development of the meninges [82, 83] that the subarachnoid spaces form during the embryonic period (up to stage 23, 8 weeks) [82], but also that the dorsal, interhemispheric meninge is the last to complete differentiation even beyond that time, at least until week 10 [1, 82]. As the corpus callosum forms later during week 13, one may speculate that the “bug” that leads to the meningeal mal-differentiation may also prevent the normal interhemispheric commissuration steps to occur, either because the dysplastic lipoma forms an obstacle to the crossing of the fibers or because the meningeal cells do not provide the appropriate signals to allow for the development of the midline glial sling and anterior callosum. This implies that neither the anterior nor the hippocampal commissure should be affected by the presence of the lipoma, as they cross through the lamina reuniens and not through the meninges.
It has been known for some time that, depending on the appearance (tubulonodular or curvilinear) and location (ventral or dorsal) of the lipoma, the dysplasia of the corpus callosum was different: a tubulonodular lipoma is usually located anteriorly and associated with major callosal dysplasia and a curvilinear lipoma sweeps around the posterior segment of a hardly abnormal callosal [81, 84, 85]. From what is now known of the commissural development, the hippocampal commissure develops first at 11 weeks in the lamina reuniens and is shifted dorsally by the later-forming callosum [1]. So, to explain the curvilinear lipoma, it can be assumed that the lipoma is present before the shifting, that its location was over the roof of the third ventricle (dorsal to the hippocampal commissure), that its curvilinear shape reflects the dorsal shifting of the hippocampal commissure and associated splenium, and that the lipoma did not prevent the dorsal shifting of the splenium mechanically. Inversely, it may be assumed that, to prevent the development of the callosal plate, the tubulonodular lipomas must be located more ventrally in the meninge at the level of the corticoseptal boundary, so that the development of the sling and the callosal commissuration are prevented; assumedly also, the hippocampal commissure would remain in its anterior location at the lamina reuniens. The facts correlate fairly well with the model. In the last 10 years, 33 cases of lipoma of the corpus callosum were examined with MR at our institution (Fig. 22). (The cases of cephalocele-related commissural agenesis, whose pathogenesis could be more complex, are not included.) It appears that the commissural defect does not correlate with the size or shape of the lipoma (dorsal tubulonodular lipoma can be observed with normal callosal morphology) (Fig. 22d), but it does with its location. This appears clearly if the lipoma is classified into four topographic groups: anterior, transitional (or global: covering the callosum from the front to the back), posterior, and inferior (below the hippocampal commissure) (Fig. 22a–e). The anterior lipoma (5/33, 15%) is associated with major commissural hypogenesis (Fig. 22a), the more posterior transitional lipoma (8/33, 24%) with a complete but hypoplastic commissural plate (Fig. 22b), the posterior ones (16/33, 48%) with minor shortening or tapering of the splenium (Fig. 22c, d), and the inferior ones (4/33, 12%), with minor commissural abnormalities only, if any (Fig. 22e). As a consequence, it appears that the timing of the dysplasia (likely to occur before week 13 in any case) does not matter as much as its caudo-ventral location in relation to the corticoseptal boundary and lamina reuniens.

Subgroups of interhemispheric lipoma with variable commissural dysgenesis. Midline sagittal T1WI. a Huge anterior lipoma with a commissural rudiment only; the leptomeningeal lipoma would not compromise the development of the AC and HC (imbedded into the LR), but it may prevent the development of the sling and/or callosum if located at the level of the corticoseptal boundary: the commissural rudiment therefore is likely to be a hippocampal commissure (similar to Figs. 7 and 9). b Transitional lipoma: the AC and anterior CC with the septum pellucidum are formed but short, the HC and splenium are probably present (the fornices join the CC) but hypoplastic. c Posterior lipoma: its curvinear shape reflects the dorsal displacement of the splenium and HC, which it did not prevent (the commissural plate is mildly short, however, but proportionate). d Posterior lipoma, nodular: the shape of the commissural plate is similar to that in c; the fact that the lipoma is nodular matters less than its location; in this case, it may have been more posteriorly and therefore not have been shaped by the dorsal splenial and HC shift. e Inferior lipoma: the commissural plate is essentially unaffected by the meningeal lipoma
In the first group (anterior lipoma), it would be useful to confirm with DTI that the commissural rudiment is hippocampal rather than callosal.
Malformations of the septum pellucidum
I the assessment of the midline forebrain structures, the septum pellucidum should be approached from two points of view. The first one is its relationship with the corpus callosum: the anterior callosal segment develops along the glial sling which itself forms along the corticoseptal boundary, therefore, the septum pellucidum develops together with the anterior callosum, and its appearance usually reflects the appearance of the anterior callosum. So, in cases where what seems to be a corpus callosum presents without an subjacent septum pellucidum, it is more likely to be hippocampal than callosal (Figs. 7 and 9). (On the midline sagittal cut, the pellucidum may not be apparent if the fornices are not joined on the midline, but axial and coronal cuts then would show the separated pellucidal leaves) (Fig. 11b). If the corpus callosum appears short but comprising normal segments, the hypoplastic pellucidum would point to the hypoplastic anterior callosum, while the posterior fusion of the fornices and hippocampal commissure would define the splenium (Fig. 6). The second point of view is that the septum pellucidum is made of the limbic fibers of the septocingulate perforant tract and of the longitudinal septo-hippocampal and mammillo-hippocampal fornical tracts in its lower edge; an agenesis or dysplasia therefore means an agenesis or dysplasia of these specific bundles.
The septo-optic dysplasia is not well understood. Described as such by De Morsier in 1959, it was defined by the association of dysplasia of the septum pellucidum with a hypoplasia and dysplasia of the anterior optic pathways [86]. First of all, it is often confused with the holoprosencephalies in the medical literature, although the embryology would point to two different entities: HPE corresponds to an early event affecting the development in week 5, while the fornix and septum pellucidum would not develop before weeks 9–13; HPE is characterized by the interhemispheric continuity of the cortical plate, while the fornical “fusion” in SOD concerns later-developing fibers; the gene defects implicated in HPE are different from those identified in SOD, although all may be expressed in the basal forebrain [87, 88]; finally, all HPE phenotypes have been reported in affected families, but not the septo-optic dysplasia phenotype [89]. Second, since the description by de Morsier, the frame of the disease was expanded to include the commonly associated hypothalamo-pituitary defects [90, 91] and still more to include cases of schizencephaly with optic atrophy, absent septum pellucidum, and pituitary defects [92]. This means that, like the commissural ageneses, SOD is not a malformation by itself but a malformative feature (i.e., associated defect of the anterior optic and pellucidal fiber pathways) that may belong to many different entities: the reports addressing the clinical prognosis are confusing because they address several entities under the same label [93, 94]. Nevertheless, developmental delay and hormonal deficiencies appear to be common [88]. Absence of the septum pellucidum may also be associated with bilateral polymicrogyria without clefting, or even observed without any other defect [95]. In the common septo-optic dysplasia, the anterior and hippocampal commissures, as well as the midline glia and callosal commissuration in the corticoseptal region, develop normally (weeks 9–13). The process is apparently not altered by the failure of the bilateral septum pellucidum to form, but the fornices seem to fasciculate together possibly because they are not maintained within two separate pellucidal leaves (Fig. 23). As mentioned earlier, a partial or a complete commissural agenesis rarely can be associated with an absence of the septum pellucidum and fornix, which results in a uni- or bilateral expansion of the tela choroidea (Fig. 18).

Septo-optic dysplasia. a Midline sagittal T1WI. The commissural plate is thin but essentially complete (AC not well seen); because the septum pellucidum is dehiscent, the fornix is located low toward the third ventricular roof; the optic chiasm is tiny. b Anterior coronal T2WI. Single ventricular lumen without a septum pellucidum; bulky single fornix. c Posterior coronal T2WI. Transverse HC separate from the CC
In schizencephaly, an absent septum pellucidum is fairly common and appears to be related to the topography of the cleft(s): septal defects are observed when the clefts are fronto-parietal, but not when they are temporo-occipital [96]. These pellucidal defects are independent from the clefts being uni- or bilateral, or closed versus open-lipped [96]. As the septum pellucidum is said in the literature to contain septo-limbic fibers only, it is not easy to figure out why it would be deficient in cases of developmental clefts in the lateral aspect of the cerebral mantle and why this defect would correlate with the lobar distribution of the clefts.
The most common abnormality affecting the septum pellucidum is the persistent cavum septi pellucidi. This is a normal transient feature of the late-fetal and neonatal brain, and for that reason it is often considered as a normal variant when observed later in children or adults. Yet, many reports have linked the anomaly to various disorders including developmental delays, psychiatric disorders such as schizophrenia, and even cognitive changes in boxers. In some reports, the incidence in healthy individuals was found to be 2.4% against 15.3% in cases of mental retardation and/or developmental delay [97, 98], and it may well be a non-specific marker of abnormal brain development. Rarely, the cavum (usually a common cavum septi pellucidi and Vergae, sometimes only the former and rarely only the latter) may become encysted and cause headaches and/or hydrocephalus.
The septum pellucidum may also be too small (surface area disproportionately small in relation to the anterior corpus callosum) with apparently normal fornices. It is often dysgenetic and too thick in hemimegalencephaly (see below). Such an excessive thickness may be observed also as an isolated abnormality (Fig. 24). This abnormality has no explanation given: it may be due to the presence of too many fibers, to an aberrant fascicle, or to the persistence of residual cellular material, possibly glial or neuronal, in the lumen of the cavum; it has not been correlated with any clinical condition [97].

Thick septum pellucidum. a, b Dysplastic appearance of the CC and septum pellucidum. The pellucidal thickness may be related to residual tissue in the cavum or to an aberrant fascicle (suggested by the oblique course)
Brain malformations with associated commissural disorders
Myelomeningocele–Chiari II malformations
In addition to being a disorder of the closure of the neural tube, the MMC–Chiari II malformation is a diffuse dysplasia of the neural tube and, besides the characteristic abnormalities of the posterior fossa, the abnormalities of the hemispheric white matter, especially of the commissural structures, are prominent [99]. The anterior commissure is dislocated in more than a third of the cases (38%), low on the lamina terminalis, halfway between the foramen of Monro and the optic chiasm; in an additional 7%, the upper lamina terminalis is diffusely thick, suggesting an abnormal commissuration there (an artifact related to ventricular drainage and a dysplasia of the anterior neuropore are other possibilities). The corpus callosum is grossly abnormal in 57% of the cases: hypoplasia, either posterior or diffuse, and partial agenesis, either posterior, anterior, or diffuse [99]. In 60% of the cases, a callosal ridge runs over the upper aspect of the corpus callosum, similar to what was identified as an aberrant cingular bundle in another report [100]. The corpus callosum is never completely agenetic, and the bundles of Probst are never identified. In half the cases, the septum pellucidum is hypoplastic.
Holoprosencephaly
Classically, the posterior corpus callosum noted in classic semilobar or lobar holoprosencephalies is considered as an oddity: in a model in which the callosal development would proceed from the front to the back, it is not expected to develop as the holoprosencephalic brain is not divided anteriorly. This peculiarity is, however, easier to understand if one considers that the posterior callosum forms by fasciculation along the hippocampal commissure. In partial lobar holoprosencephalies, the temporal lobes are well differentiated with well defined (if ill formed) hippocampi. These have fimbriae which follow the margin of the single ventricular opening between the cortex and the single ventricular tela choroidea (forming a pattern of single, and posterior, telencephalic medullary velum). Doing so, they travel toward the midline, cross it, and therefore connect both sides [48]. These fibers cannot proceed anteriorly toward the septal nuclei and form the fornix, but by crossing the midline in front of the tela choroidea (as they would have done in the lamina reuniens) they form a true hippocampal commissure. This allows the neocortical fibers of the divided posterior portions of the holoprosencephalic brain to cross as well and to form a true callosal splenium (Fig. 25). Because the frontal part of the brain typically remains undivided in lobar holoprosencephalies, there is no septal area, no lamina reuniens, no sulcus medianus telencephali medii, no corticoseptal boundary, and as a consequence no glial sling to form an anterior corpus callosum, even dysplastic.

Lobar holoprosencephaly. Midline sagittal FLAIR. The heavily myelinated posterior callosum (splenium and HC) is well demonstrated (ventral splenial bright signal of unknown cause); the anterior callosum is absent as the frontal lobes and anterior diencephalon are undivided
In the middle interhemispheric variant of holoprosencephaly (syntelencephaly), the presence of the posterior corpus callosum/hippocampal commissure can be explained in the same manner. However, in addition, an anterior corpus callosum may be present also because the third ventricle and lamina terminalis become differentiated with a normal lamina reuniens, anterior interhemispheric fissure, and sulcus medianus telencephali medii. A glial sling may form and the anterior callosum may develop normally (Fig. 26). Only the intermediate callosal portion lacks because the transverse interhemispheric cortical continuity prevents the completion and eventual junction of the anterior and posterior callosal segments.

Syntelencephaly (middle interhemispheric variant of holoprosencephaly). In this unusual case, the interhemispheric continuity extended posteriorly to the medial parieto-temporal cortex, preventing the formation of a HC and splenium. On the contrary, the anterior frontal lobes were well divided so that an anterior callosum could develop. In this instance, the commissure cannot be a HC as there is no septum pellucidum to carry the fornices
Craniocerebral midline defects
Along the neural tube, commissuration is primarily a basal process, and in cases of commissural agenesis other commissuration defects may be observed anywhere along the ventral cord and brainstem. In the basal forebrain, other commonly associated defects involve the anterior optic pathway [49] and the hypothalamo-pituitary axis [49]. Because the development of the corpus callosum itself is associated with the dorsalization of the hemispheres, other disorders of the dorsalization may be observed, primarily at the level of the cerebellum: a Dandy–Walker malformation (or related defect) is commonly associated with an agenesis of the corpus callosum [48, 101]. The rare rhombencephalosynapsis is often found in association with septal defects/septo-optic dysplasia [102, 103], and obviously the midline skull defects commonly include commissural agenesis or dysgenesis, especially the frontonasal dysplasia [104, 105] and the basal, notably sphenoidal cephaloceles [106].
Malformations of cortical development
All malformations of cortical development may be associated with malformations of the commissures. Migration disorders are probably the most typical. Periventricular nodular heterotopias are commonly found, sometimes extensively. Major hemispheric dysplasia with large subcortical heterotopia and cortical dysplasia are characteristic as well, associated or not with multiple interhemispheric cysts; the dysplasia often is such that one may wonder whether the failure of the commissures to cross could be due to a mechanical obstacle (the heterotopia) more than to a migrational abnormality of the cortex; quite characteristically, the Probst’s bundle typically is missing or incomplete on the dysplastic side.
Callosal agenesis is significantly associated with some subtypes of microcephaly [107]. DCX (doublecortin) related lissencephaly (X-linked on Xq22.3-q23, classic lissencephaly with predominantly anterior abnormalities) may be associated with callosal agenesis in humans and in mice [108]. Of course the agenesis is a defining feature of the ARX (Xp22.13) related lissencephaly with callosal agenesis and ambiguous genitalia, a disorder that affects the males but in which the female heterozygous relatives may present with callosal agenesis as well [109]. Other cases of lissencephaly with cerebellar hypoplasia also present with callosal agenesis [110]. In all these examples, the cortex is extensively abnormal and this may account for the failure of the commissural neurons to develop normally.
Focal cortical dysplasia is usually not associated with appreciable or specific commissural abnormality, but hemimegalencephalies may occasionally present with abnormal corpus callosum and especially with a septum pellucidum that is too thick and seems to be pulled anteriorly toward the abnormal frontal lobe [111] (Fig. 27). Again, the nature of this pellucidal hypertrophy is uncertain: embryonal residues within the cavum, aberrant fascicle related to the abnormal cellularity of the hemimegalencephaly, or lack of pruning.

Right anterior hemimegalencephaly. a Midline sagittal T1WI. Dysplastic commissural plate with thick septum pellucidum. b Coronal T2WI. Hypertrophic septum pellucidum and fornix; the appearence is heterogeneous, mostly of white matter intensity (myelinated fibers), but with islands of gray matter intensity of uncertain nature. c Axial T1WI. Markedly dysmorphic septum pellucidum obliquely oriented toward the dysplastic right frontal cortex
Occasionally, polymicrogyria may be associated with a callosal agenesis [112]. In the usual presentation of schizencephaly with lateral clefts, an abnormal thinning of the corpus callosum is often seen that correlates with the location of the clefts but without a true partial or segmental agenesis. On the contrary, in the rare cases when the clefts affect the medial aspect of the hemisphere, it appears that the corpus callosum presents with corresponding well-defined defects. It would be interesting to investigate whether this is related to the fact that the pioneer callosal fibers originate from the cingulate cortex; this would tell that segmental commissural fasciculation is not possible if the pioneer fibers did not develop along the glial sling or that the defect extended to the corticoseptal boundary and prevented the sling itself to develop there. It would also tell that a schizencephalic cleft, classified with the cortical organization disorders together with the polymicrogyria [113] and thus assumed to “occur” (be expressed) toward the end of the migration period, could be present much earlier instead, before the development of the midline glial sling and the crossing of the early pioneer cingulate fibers (weeks 11–12).
Hypertrophy of the corpus callosum is a classical marker of neurofibromatosis type 1. It has also been recently identified as a characteristic of a macrocephaly syndrome with polymicrogyria and developmental delay [114]. It has no obvious explanation in either case.
Syndromes that include commissural dysgenesis as a defining feature
When interrogated with “agenesis of the corpus callosum”, OMIM (Online Mendelian Inheritance in Man, Johns Hopkins University, March 16, 2010) lists 189 specific syndromes in which a commissural agenesis is or may be present [115]. It is beyond the scope of this review to approach this as a whole. However, in a few syndromes, brain imaging is essential in diagnosing the disorder and assessing the brain.
The CRASH syndrome (for callosal agenesis, retardation, adducted thumbs, shuffling gait, hydrocephalus) is related to a L1CAM gene defect on Xq28. L1CAM encodes for a cell adhesion molecule that is involved in the fasciculation of the axons, as well as synaptic targeting and cellular migration [116]. Multiple mutations may occur, with slightly different phenotypes all characterized by a white matter defect: callosal agenesis obviously is common, as well as impairment of the pyramidal tract (shuffling gait, spastic paraplegia), aphasia, mental retardation, ventriculomegaly (hence the term of X-linked hydrocephalus), etc. [117–119]. On imaging, the brain is characterized by a huge ventriculomegaly, a poorly developed white matter, and absent corpus callosum.
The appearance is not very different in the Walker–Warburg syndrome. This is the most severe phenotype of the group of the “cobblestone brains” that also includes the Fukuyama and the muscle-eye-brain (MEB) syndromes. All are characterized by a congenital muscular dystrophy and a neuronal migration disorder in which there is overmigration of the neurons beyond the pia limiting membrane. This results from an abnormal glycosylation of α-dystroglycans (hence the name α-dystroglycanopathies) that affects the extracellular matrix and, as a consequence, cellular proliferation, differentiation, adhesion, and migration [120]. Several gene defects have been identified: fukutin FCMD (9q31-q33), POMT1 (9q34.1), POMT2 (14q24.3), fukutin-related protein FKRP (19q13.33), and POMGNT1 (1p34.1), each with a variable phenotypic severity. The neurons overmigrate and form abnormal arrangements in the cortical and meningeal layers. This disorganization of the tissular pattern and the abnormal extracellular matrix signals in turn lead to a failure of the white matter to form properly. Of the three phenotypes, the Walker–Warburg syndrome is the most severe: irregular (cobblestone) cortical surface, disorganized cortex, thin cerebral mantle with lack of whiter matter and ventriculomegaly, absence of the commissures; the underdeveloped brainstem often has a Z shape; and the cerebellum is hypoplastic with correspondingly huge posterior fossa cisterns. The two lesser phenotypes (Fukuyama and MEB) present with less severe abnormalities and the corpus callosum is typically present.
Syndromic craniosynostoses (Apert, Crouzon, Pfeiffer mostly) present with striking cranial deformities which result in similarly striking brain deformities. Most of the brain abnormalities can be explained mechanically: for example, in Crouzon disease, the early closure on the lambdoid suture results in a small posterior fossa and in a Chiari 1 “malformation” and hydrocephalus (for review, see [121]). However, not everything can be explained mechanically and specifically; the typical occurrence of corpus callosal dysgenesis and/or septum pellucidum defects may well be intrinsically part of the syndromes (Fig. 28). Also, the patients present with abnormal, overconvoluted mesial temporal lobes with a relatively deficient white matter [121]. All syndromic craniosynostoses result from a defect of one of the FGFR genes (FGFR2 on 10q25-q26 for Apert, Crouzon and Pfeiffer; FGFR1 on 8q11.22-p12 for Pfeiffer also). It has been demonstrated that L1CAM cannot be active without a closely synergistic action of these FGFR genes [122, 123]—meaning that, in syndromic craniostenosis, the white matter disorders may well result from the same genetic abnormalities as the cranial malformations.

Apert syndrome. a Midline sagittal T1WI. Markedly dysplastic anterior callosum with septum pellucidum. The HC and splenium are absent, and the fornix and AC are small. b Coronal T1WI. The absent HC may be related to the markedly abnormal, overconvoluted mesial temporal lobes: the parahippocampal gyri are not recognized, and the white matter is scarce in these regions
Conclusion
The telencephalic commissures form one single continuous structure made of different components. Classical anatomy identifies an anterior commissure, a large corpus callosum subdivided in a rostrum, a genu, a body, an isthmus and a splenium, and a hippocampal commissure that is connected to the anterior commissure via the fornix. However, the development does not match this classical pattern. In the primitive brain, the anterior commissure connects the olfactory cortex and the hippocampal commissure connects the hippocampal cortex, both through the lamina reuniens which forms the continuity between the hemispheres. The addition of the neocortex in mammals creates a new situation. In monotremes and marsupial mammals, the neocortical fibers use the pre-existent pathways to cross: mostly the anterior commissure, possibly the hippocampal commissure as well. In placental mammals, however, in addition to the two pre-existent ones, a new path was created which is the anterior corpus callosum. This new path involves new processes of interhemispheric fusion and interhemispheric glial migration, and the bulk of the new neocortical commissure develops separately in space and time from the older ones. Not all neocortical fibers use the new path however: the lateral temporo-occipital neocortical fibers use the anterior commissure, and the neocortical fibers from the posterior part of the hemisphere use the hippocampal commissure to form the splenium. Therefore, in humans, the three commissures must be regarded as forming a developmental anatomical pattern: an anterior commissure that carries olfactory fibers as well as neocortical ones from the temporo-occipital region, a hippocampal commissure that carries fibers from the hippocampal formations together with a neocortical component (the splenium) from the postero-medial aspect of the hemispheres, and an anterior corpus callosum (anterior to the isthmus) that contain frontal associative neocortical fibers. This is clearly different from the descriptive anatomical pattern, but the malformations reflect this developmental anatomy.
The commissures form a single structure, but the processes involved in their development are many: migration of specific neurons in specific cortical layer, axons targeting specific—essentially homotopic—targets across the midline, specific cellular structures and cellular guidance factors guiding the fibers across the lamina reuniens, phylogenetically new processes of cellular migration of the midline glial sling into the primitive meninx outside of the neural tube to form a bridge to allow neocortical fibers to cross, contribution of the “channeling” glia (glial wedge, indusium griseum glia), and fasciculation factors. In such complex processes, the chances of “bugging” to occur are high and, accordingly, commissural agenesis is a common brain disorder. These commissuration factors are diverse: the variants of commissural agenesis are multiple. While the association of the commissuration factors is specific, individually each of them is not and each may participate in the formation of other structures: pure, isolated commissural agenesis is uncommon, and the abnormality is usually one feature in a constellation of cerebral or craniocerebral or syndromic defects.
Given this complexity, imaging must aim at getting as close as possible to the specific disorder, or combination of disorders in a specific patient, so that a better developmental, hopefully genetic individual understanding can be reached. This implies a careful, attentive-to-details approach to the diagnosis, with a full knowledge and a full understanding of the embryogenetic steps. The Rakic and Yakovlev 1968 paper [1] is still basic to this understanding. One must be aware that it has too often been misquoted in the subsequent literature but, more importantly, it has been largely confirmed and amazingly expanded by the modern molecular and genetic approaches in mouse and human embryos.
References
Rakic P, Yakovlev PI (1968) Development of the corpus callosum and cavum septi in man. J Comp Neurol 132:45–72
Déjerine J. Anatomie des Centres Nerveux, vol 1. Rueff, Paris, 1895. Masson, Paris, 1980, pp 119-120, 738-741 (reprint)
Silver J, Lorenz SE, Wahlsten D, Coughlin J (1982) Axonal guidance during development of the great cerebral commissures: descriptive and experimental studies, in vivo, on the role of preformed glial pathways. J Comp Neurol 210:10–29
Katz MJ, Lasek RJ, Silver J (1983) Ontophyletics of the nervous system: development of the corpus callosum and evolution of axon tracts. Proc Natl Acad Sci USA 80:5936–5940
Wahlsten D (1987) Defects of the fetal forebrain in mice with hereditary agenesis of the corpus callosum. J Comp Neurol 262:227–241
Hankin MH, Schneider BF, Silver J (1988) Death of the subcallosal glial sling is correlated with formation of the cavum septi pellucidi. J Comp Neurol 272:191–202
Koester SE, O’Leary DM (1994) Axons of early generated neurons in cingulate cortex pioneer the corpus callosum. J Neurosci 14:6608–6620
Livy DJ, Wahlsten D (1997) Retarded formation of the hippocampal commissure in embryos from mouse strains lacking a corpus callosum. Hippocampus 7:2–14
Shu T, Richards LJ (2001) Cortical axon guidance by the glial wedge during the development of the corpus callosum. J Neurosci 21:2749–2758
Richards LJ (2002) Axonal pathfinding mechanisms at the cortical midline and in the development of the corpus callosum. Braz J Med Biol Res 35:1431–1439
Shu T, Puche AC, Richards LJ (2003) Development of midline glial populations at the corticoseptal boundary. J Neurobiol 57:81–94
Shu T, Li Y, Keller A, Richards LJ (2003) The glial sling is a migratory population of developing neurons. Development 130:2929–2937
Richards LJ, Plachez C, Ren T (2004) Mechanisms regulating the development of the corpus callosum and its agenesis in mouse and human. Clin Genet 66:276–289
Lent R, Uziel D, Baudrimont M, Fallet C (2005) Cellular and molecular tunnels surrounding the forebrain commissures of human fetuses. J Comp Neurol 483:375–382
Ren T, Anderson A, Shen WB et al (2006) Imaging, anatomical and molecular analysis of callosal formation in the developing human fetal brain. Anat Record Part A 288A:191–204
Vulliemoz S, Raineteau O, Jahaudon D (2005) Reaching beyond the midline: why are human brains cross wired? Lancet Neurology 4:87–99
Abbie AA (1939) The origin of the corpus callosum and the fate of the structures related to it. J Comp Neurol 70:9–44
Ariëns Kappers CU, Huber GC, Crosby EC. The comparative anatomy of the nervous system of vertebrates including man, vol III. Hafner, New York, 1967
Sarnat HB, Netsky MG (1974) Evolution of the nervous system. Oxford University Press, New York
Romer AS, Parsons TS (1977) The vertebrate body. Saunders, Philadelphia
Aboitiz F (2003) Montiel J. One hundred million years of interhemispheric communication: the history of the corpus callosum Braz J Med Biol Res 36:409–420
Yakovlev PI (1968) Telencephalon “impar”, “semipar”, “totopar” (morphogenetic, tectogenetic, and architectonic definitions). Int J Neurol 6:245–265
Gloor P, Salanova V, Olivier A, Quesney LF (1993) The human dorsal hippocampal commissure. Brain 116:1249–1273
Amaral DG, Insausti R, Cowan WM (1984) The commissural connections of the monkey hippocampal formation. J Comp Neurol 224:307–336
Demeter S, Rosene DL, Van Hoesen GW (1985) Interhemispheric pathways of the hippocampal formation, presubiculum and entorhinal and posterior parahippocampal cortices in the rhesus monkey: the structure and organization of the hippocampal commissures. J Comp Neurol 233:30–47
Guénot M. Transfert interhémisphérique et agénésie du corps calleux. Capacités et limites de la commissure blanche antérieure. Neurochirurgie (Paris) 1998, 44(Suppl 1):113-115
Lamantia AS, Rakic P (1990) Cytological and quantitative characteristics of four cerebral commissures in the rhesus monkey. J Comp Neurol 291:520–537
Di Virgilio G, Clarke S, Pizzolato G, Schaffner T (1999) Cortical regions contributing to the anterior commissure in man. Exp Brain Res 124:1–7
Wilson CL, Isokawa M, Babb TL, Crandall PH. Functional connections in the human temporal lobe. I. Analysis of limbic system pathways using neuronal response evoked by electrical stimulation. Exp Brain Re 1990, 82:279-292
Wilson CL, Isokawa M, Babb TL et al (1991) Functional connections in the human temporal lobe. II. Evidence for a loss of functional linkage between contralateral limbic structures. Exp Brain Res 85:174–187
Spencer SS, Williamson PD, Spencer DD, Mattson RH (1987) Human hippocampal seizure spread studied by depth and subdural recording: the hippocampal commissure. Epilepsia 28:479–489
Phelps EA, Hirst W, Gazzaniga MS (1991) Deficits in recall following partial and complete commissurotomy. Cerebral cortex 1:492–498
Kier EL, Truwit CL (1997) The lamina rostralis: modification of concepts concerning the anatomy, embryology, and MR appearance of the rostrum of the corpus callosum. AJNR Am J Neuroradiol 18:715–722
Velut S, Destrieux C, Kakou M (1998) Anatomie morphologique du corps calleux. Neurochirurgie (Paris) 44(Suppl 1):17–30
Hofer S, Frahm J (2006) Topography of the human corpus callosum revisited—comprehensive fiber tractography using diffusion tensor magnetic resonance imaging. NeuroImage 32:989–994
Kier L, Truwit CL (1996) The normal and abnormal genu of the corpus callosum: an evolutionary, embryologic, anatomic and MR analysis. AJNR Am J Neuroradiol 17:1631–1641
Aboitiz F, Scheibel AB, Fisher RS, Zaidel E (1992) Fiber composition of the corpus callosum. Brain Res 598:143–153
Widjaja E, Nilsson D, Blaser S, Raybaud C (2008) White matter abnormalities in children with idiopathic developmental delay. Acta Radiol 49:589–595
Jea A, Vachhrajani S, Widjaja E et al (2008) Corpus callosotomy in children and the disconnection syndromes: a review. Childs Nerv Syst 24:685–692
De Lacoste C, Kirkpatrick JB, Ross ED (1985) Topography of the human corpus callosum. J Neuropathol Exp Neurol 44:578–591
Oh JS, Park KS, Song IC et al (2005) Fractional anisotropy-based divisions of midsagittal corpus callosum. NeuroReport 16:317–320
Déjerine J. Anatomie des Centres Nerveux, vol 2, Rueff, Paris 1901. Masson, Paris 1980, pp 263-267 (reprint)
Liss L, Mervis L (1964) The ependymal lining of the cavum septi pellucidi: a histological and histochemical study. J Neuropathol Exp Neurol 23:355–367
Lancon JA, Haines DE, Lewis AI, Parent AD (1999) Endoscopic treatment of symptomatic septum pellucidum cysts: with some preliminary observations on the ultrastructure of the cyst wall: two technical reports. Neurosurgery 45:1251–1257
Ronsin E, Grosskopf D, Perre J (1997) Morphology and immunohistochemistry of a symptomatic septum pellucidum cavum Vergae cyst in man. Acta Neurochir 139:366–372
Shu T, Shen WB, Richards LJ (2001) Development of the perforating pathway: an ipsilaterally projecting pathway between the medial septum/diagonal band of Broca and the cingulate cortex that intersects the corpus callosum. J Comp Neurol 436:411–422
Yakovlev PI, Locke S (1961) nuclei of thalamus and connections of limbic cortex. III. Cortico-cortical connections of the anterior cingulate gyrus, the cingulum, and the subcallosal bundle in the monkey. Arch Neurol 5:364–400
Johnston TB (1934) A note on the peduncle of the flocculus and the posterior medullary velum. J Anat 68:471–479
Raybaud C, Girard N. Etude anatomique par IRM des agénésies and dysplasies commissurales télencéphaliques. Corrélations cliniques et interprétation morphogénétique. Neurochirurgie (Paris) 1998, 44(Suppl 1):38-60
Larroche JC, Baudey J (1961) septi pellucidi, cavum Vergae, cavum veli interpositi: cavités de la ligne médiane. Etude anatomique et pneumoencéphalographique dans la période néonatale. Biol Neonate 3:193–236
Shaw CM, Alvord EC (1969) Cava septi pellucidi et Vergae: their normal and pathological states. Brain 92:213–224
Scoffings DJ, Kurian KM (2008) Congenital and acquired lesions of the septum pellucidum. Clin Radiol 63:210–219
Auer RN, Gilbert JJ (1982) Cavum Vergae without cavum septi pellucidi. Arch Pathol Lab Med 106:462–463
Blakemore WF, Jolly RD (1972) The subependymal plate and associated ependyma in the dog. An ultrastructural study J Neurocytol 1:69–84
Hopewell JW (1975) The subependymal plate and the genesis of gliomas. J Pathol 117:101–103
Nishio S, Fujiwara S, Tashima T et al (1990) Tumors of the lateral ventricular wall, especially the septum pellucidum: clinical presentation and variations in pathological features. Neurosurgery 27:224–230
Aldur MM, Çelik HH, Sargon MF et al (1997) Unreported anatomical variation of septum pellucidum. Clin Anat 10:245–249
Bayer SA (2006) Altman J. Atlas of central nervous system development. The human brain during the late first trimester. CRC, Boca Raton
Bayer SA (2005) Altman J. Atlas of Central Nervous System Development. The human brain during the second trimester. CRC, Boca Raton
Shen WB, Plachez C, Mongi AS, Richards LJ (2006) Identification of candidate genes at the corticoseptal boundary during development. Gene Expres Patterns 6:471–481
Silver J, Ogawa MY (1983) Postnatally induced formation of the corpus callosum in acallosal mice on glia-coated cellulose bridges. Science 220:1067–1069
Tessier-Lavigne M, Goodman CS (1996) The molecular biology of axon guidance. Science 274:1123–1133
Lanier LM, Gates MA, Witke W et al (1999) Mena is required for neurulation and commissure formation. Neuron 22:313–325
Shu T (2003) Butz KG. Plachez et al Abnormal development of forebrain midline glia and commissural projections in Nfia knock-out mice J Neurosci 23:203–212
Jovanov-Milošević N, Čuljat M, Kostović I (2009) Growth of the human corpus callosum: modular and laminar morphogenetic zones. Frontiers Neuroanat 3:1–10
Pascual M, Pozas E, Barallobre J et al (2004) Coordinated functions of netrin-1 and class 3 secreted semaphorins in the guidance of reciprocal septohippocampal connections. Mol Cell Neurosci 26:24–33
Flanagan JG, van Vactor D (1998) Through the looking glass: axon guidance at the midline choice point. Cell 92:429–432
Kaprielian Z, Imondi R, Runko E (2000) Axon guidance at the midline of the developing CNS. Anat Rec (New Anat) 261:176–197
Probst M. Über den Bau des vollständigen balkenlosen Groβhirns sowie über Microgyrie und Heterotopie den Grauen Substanz. Arch Psychiat Nervenkr 1901, 34:709-786 (quoted by [70])
Probst FP (1973) Congenital defects of the corpus callosum. Acta Radiol Suppl 331:1–152
Onufrowicz W. Das balkenlose Microcephalengehirn. Arch J Psychiat 1887, 18:305-328 (quoted by [47])
Sachs H. Das Hemisphärenmark des Menschlichen Grosshirns. I. der Hinterhauptlappen. Leipzig, 1892 (quoted by [2])
Nakata Y, Barkovich AJ, Wahl M et al (2009) Diffusion abnormalities and reduced volume of the ventral cingulum bundle in agenesis of the corpus callosum: a 3T imaging study. AJNR Am J Neuroradiol 30:1142–1148
Mufson EJ, Pandya DN (1984) Some observations on the course and composition of the cingulum bundle in the rhesus monkey. J Comp Neurol 225:31–43
Dávila-Gutiérrez G (2002) Agenesis and dysgenesis of the corpus callosum. Sem Ped Neurol 9:292–301
Barkovich AJ, Simon EM, Walsh CA (2001) Callosal agenesis with cyst. A better understanding and new classification. Neurology 56:220–227
Pavone P, Barone R, Baieli S et al (2005) Callosal anomalies with interhemispheric cysts: expanding the phenotype. Acta Paediat 94:1066–1072
Sener RN (1993) Septo-optic dysplasia associated with total absence of the corpus callosum: MR and CT features. Eur Radiol 3:551–553
De León GA, Radkowski MA, Gutierrez FA (1995) Single forebrain ventricle without prosencephaly: agenesis of the corpus callosum with dehiscent fornices. Acta Neuropathol 89:454–458
Aicardi J (1996) Aicardi syndrome. In: Guerrini R et al (eds) Dysplasias of cerebral cortex and epilepsy. Lippincott-Raven, Philadelphia, pp 211–216
Truwit CL, Barkovich AJ (1990) Pathogenesis of intracranial lipomas: an MR study in 42 patients. AJNR Am J Neuroradiol 11:665–674
Osaka K, Handa H, Matsumoto S, Yasuda M (1980) Development of the cerebrospinal fluid pathway in the normal and abnormal human embryo. Child’s Brain 6:26–38
McLone DG (1980) The subarachnoid space: a review. Child’s Brain 6:113–130
Tart RP, Quisling RG (1991) Curvilinear and tubulonodular varieties of lipoma of the corpus callosum: an MR and CT study. J Comput Assist Tomogr 15:805–810
Demaerel P, Van de Gaer P, Wilms G, Baert AL (1996) Interhemispheric lipoma with variable callosal dysgenesis: relationship between embryology, morphology and symptomatology. Eur Radiol 6:904–909
De Morsier G (1956) Etudes sur les dysraphies crânio-encéphaliques. III Agénésie du septum lucidum avec malformation du tractus optique La dysplasie septo-optique Schweiz Arch Neurol Psychiat 77:267–292
Fernandes M, Hébert JM (2008) The ups and downs of holoprosencephaly: dorsal versus ventral patterning forces. Clin Genet 73:413–23
Kelberman D, Dattani MT (2008) Septo-optic dysplasia—novel insights into the aetiology. Horm Res 69:257–265
Schachter KA, Krauss RS (2008) Murine models of holoprosencephaly. Curr Top Develop Biol 84:139–170
Hoyt WF, Kaplan SL, Grumbach MM, Glaser JS. Septo-optic dysplasia and pituitary dwarfism. Lancet 1970, 1:893-894 (letter)
Acers TE (1981) Optic nerve hypoplasia: septo-optic-pituitary dysplasia syndrome. Tr Am Ophth Soc 79:425–457
Barkovich AJ, Fram EK, Norman D (1989) Septo-optic dysplasia: MR imaging. Radiology 171:189–192
Williams J, Brodsky MC, Griebel M et al (1993) Septo-optic dysplasia: the clinical insignificance of an absent septum pellucidum. Dev Med Child Neurol 35:490–501
Belhocine O, André C, Khalifa G, Adamsbaum C (2005) Does asymptomatic septal agenesis exist? A review of 34 cases. Pediatr Radiol 35:410–418
Supprian T, Sian J, Heils A et al (1999) Isolated absence of the septum pellucidum. Neuroradiology 41:563–566
Raybaud C, Girard N, Levrier O et al (2001) Schizencephaly: correlation between the lobar topography of the cleft(s) and absence of the septum pellucidum. Childs Nerv Syst 17:217–222
Bodensteiner JB (1995) The saga of the septum pellucidum: a tale of unfunded clinical investigations. J Child Neurol 10:227–231
Bodensteiner JB, Schaefer GB, Craft JM (1998) Cavum septi pellucidi and cavum Vergae in normal and developmentally delayed populations. J Child Neurol 13:120–121
Miller E, Widjaja E, Blaser S et al (2008) The old and the new: supratentorial MR findings in Chiari II malformation. Childs Nerv Syst 24:563–575
Vachha B, Adams RC, Rollins NK (2006) Limbic tract anomalies in pediatric myelomeningocele and Chiari II malformation: anatomic correlation with memory and learning—initial investigation. Radiology 240:194–202
Raybaud C (1982) Cystic malformations of the posterior fossa—abnormalities associated with development of the roof of the fourth ventricle and adjacent meningeal structures. J Neuroradiol 9:103–133
Michaud J, Mizrahi EM, Urich H (1982) Agenesis of the vermis with fusion of the cerebellar hemispheres, septo-optic dysplasia and associated anomalies. Report of a case Acta Neuropathol (Berl) 56:161–166
Jellinger KA (2002) Rhombencephalosynapsis. Acta Neuropathol 103:305–6
Guion-Almeida ML, Richieri-Costa A, Saavedra D, Cohen MM Jr (1996) Frontonasal dysplasia: analysis of 21 cases and literature review. Int J Oral Maxillofac Surg 25:91–97
Wu E, Vargevik K, Slavotinek AM (2007) Subtypes of frontonasal dysplasia are useful in determining clinical prognosis. Am J Clin Genet Part A 143A:3069–3078
Koenig SB, Naidich TP, Lissner G (1982) The morning glory syndrome associated with sphenoidal cephalocele. Ophtalmology 89:1368–1373
Vermeulen RJ, Wilke M, Horber V, Krägeloh-Mann I (2010) Microcephaly with simplified gyral pattern. MRI classification Neurology 74:386–391
Kappeler C, Dhenain M (2007) Phan Dinh Tuy F et al. Magnetic resonance imaging and histological studies of corpus callosum and hippocampal abnormalities linked to doublecortin deficiency J Comp Neurol 500:239–254
Kitamura K, Yanazawa M, Sugiyama N et al (2002) Mutation in ARX causes abnormal development of brain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet 32:359–369
Miyata H, Chute DJ, Fink J et al (2004) Lissencephaly with agenesis of corpus callosum and rudimentary dysplastic cerebellum: a subtype of lissencephaly with cerebellar hypoplasia. Acta Neuropathol 107:69–81
Sato N, Ota M, Yagishita A et al (2008) Aberrant midsagittal fiber tracts in patients with hemimegalencephaly. AJNR Am J Neuroradiol 29:823–827
Robin NH, Taylor CJ, McDonald-McGinn et al. Polymicrogyria and deletion 22q11.2 syndrome: window to the etiology of a common cortical malformation. Am J Med Genet Part A 2006, 140A:2416-2425
Barkovich AJ, Kuzniecky RI, Jackson GD et al (2005) A developmental and genetic classification for malformations of cortical development. Neurology 65:1873–1887
Pierson TM, Zimmerman RA, Tennekoon GI, Bönnemann CG (2008) Mega-corpus callosum, polymicrogyria, and psychomotor retardation: confirmation of a syndromic entity. Neuropediatrics 39:123–127
Online Mendelian Inheritance in Man, OMIM (TM). McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), World Wide Web URL: http://www.ncbi.nlm.nih.gov/omim/
Schmid RS, Maness PF (2008) L1 and NCAM adhesion molecules as signaling co-receptors in neuronal migration and process outgrowth. Curr Opin Neurobiol 18:245–250
Fransen E, Van Camp G, Vits L, Willems PJ (1997) L1-associated diseases: clinical geneticists divide, molecular geneticists unite. Hum Mol Genet 6:1625–1632
Yamasaki M, Thompson P, Lemmon V (1997) CRASH syndrome: mutations in L1CAM correlate with severity of the disease. Neuropediatrics 28:175–178
Weller S, Gärtner J (2001) Genetic and clinical aspects of X-linked hydrocephalus (L1 disease): mutations in the L1CAM gene. Hum Mutat 18:1–12
Reed UC (2009) Congenital muscular dystrophy. Part II: a review of pathogenesis and therapeutic perspectives. Arq Neuropsiquiatr 67:343–362
Raybaud C, Di Rocco C (2007) Brain malformation in syndromic craniosynostoses, a primary disorder of white matter: a review. Childs Nerv Syst 23:1379–1388
Doherty P, Wlash F (1996) CAM-FGF receptor interaction: a model for axonal growth. Mol Cell Neurosci 8:99–111
Kamiguchi H, Lemmon V (1997) Neural cell adhesion molecule L1: signaling pathways and growth cone motility. J Neurosc Res 49:1–8
Conflict of interest statement
I declare that I have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Raybaud, C. The corpus callosum, the other great forebrain commissures, and the septum pellucidum: anatomy, development, and malformation. Neuroradiology 52, 447–477 (2010). https://doi.org/10.1007/s00234-010-0696-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00234-010-0696-3