
Abstract
Hereditary spastic paraplegia (HSP) is a genetically heterogeneous group of neurodegenerative disorders characterized by progressive lower extremity weakness and spasticity. HSP pathology involves axonal degeneration that is most pronounced in the terminal segments of the longest descending (pyramidal) and ascending (dorsal columns) tracts. In this study, we compared spinal cord magnetic resonance imaging (MRI) in 13 HSP patients with four different types of autosomal dominant hereditary spastic paraplegia (SPG3A, SPG4, SPG6, and SPG8) with age-matched control subjects. The cross-section area of HSP subjects at cervical level C2 was 59.42±12.57 mm2 and at thoracic level T9 was 28.58±5.25 mm2. Both of these values were less than in the healthy controls (p<0.001). The degree of cord atrophy was more prominent in patients with SPG6 and SPG8 who had signs of severe cord atrophy (47.60±6.58 mm2 at C2, 21.40±2.4 mm2 at T9) than in subjects with SPG3 and SPG4 (66.0±8.94 mm2 at C2, p<0.02; 31.75±2.76 mm2 at T9, p<0.001). These observations indicate that spinal cord atrophy is a common finding in the four genetic types of HSP. Spinal cord atrophy was more severe in SPG6 and SPG8 HSP subjects than in other types of HSP we studied. This may suggest a different disease mechanism with more prominent axonal degeneration in these two types of HSP when compared with HSP due to spastin and atlastin mutations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hereditary spastic paraplegia (HSP) is a group of neurodegenerative disorders characterized by progressive spasticity and weakness of the lower extremities [1]. HSP is genetically heterogeneous, and more than 20 genetic types have been identified [1, 2]. Autosomal dominant HSP (ADHSP) is the most common genetic type of HSP, accounting for more than 80% of all familial cases. Six genetic types of uncomplicated ADHSP and two genetic types of complicated ADHSP have been defined [1–3]. Mutations have been identified in five genes causing ADHSP: spastin in SPG4 linked to chromosome 2p, atlastin in SPG3A linked to chromosome 14q, Hsp60 in SPG13 linked to chromosome 2q, kinesin heavy chain (KIF5A) in SPG10 linked to 12q, and NIPA1 in SPG6 linked to 15q [4–8]. Despite this recent progress, the molecular pathogenesis of HSP is unknown.
Pathologic changes in HSP are characterized primarily by axonal degeneration that is most pronounced in the terminal segments of the longest descending (pyramidal) and ascending (dorsal columns) tracts [9–11]. Spinal cord atrophy has been observed in some patients with HSP. However, it is unknown whether this is a constant feature in all types of ADHSP or whether the degree of axonal degeneration and subsequent spinal cord atrophy varies among different genetic types of HSP.
Magnetic resonance imaging (MRI) has been used to measure spinal cord size in some HSP patients. Dürr et al. reported mild cervical spinal cord atrophy (not quantified) on MRI scans in five of 12 studied individuals [12]. Thoracic spinal cord imaging was not reported. The genetic type of HSP was not known and thus, no conclusions about the relationship between the types of ADHSP and the degree of the atrophy could be made [12]. Krabbe et al. [13] reported smaller anteroposterior diameters of the thoracic spinal cord in 16 HSP patients (later shown to be linked to chromosome 2p [14]). Qualitative analysis of spinal cord MRI studies in these patients [13, 14] were without significant abnormality, although two individuals (brothers) were considered by one radiologist to have mild atrophy of the thoracic spinal cord [14]. This indicates that quantitative measurements may be needed to appreciate spinal cord atrophy in HSP and that the presence and degree of spinal cord atrophy may be variable within a given type of HSP. Previously, we reported a clinical analysis of an SPG8 ADHSP family and noted significant spinal cord atrophy in one affected individual relatively early in the course of the disease [15]. This prompted us to compare the degree of spinal cord atrophy among patients with genetically diverse types of HSP.
Methods
Clinical analysis
We included 13 patients (six men, seven women; average age 49.3±10.8 years) who met diagnostic criteria for HSP and consented to participate in this study approved by the Institutional Review Board at the University of Michigan. Each subject had subjective complaints of gait difficulties and a positive family history of similarly affected relatives, consistent with the autosomal dominant mode of inheritance. They underwent a complete neurological examination by a neurologist (P.H. or J.K.F.), and each patient was definitely affected based on previously published criteria [3]. Information about age at symptom onset and progression of the disease was collected from each subject. Severity of the disease was graded 1–5 according to previously published criteria [15]. Briefly, grade 1 corresponds to a spastic gait without functional limitation; grade 2 represents an abnormal gait with functional limitations but not requiring consistent use of an assistive device; a grade 3 gait requires consistent use of a cane, crutches, or walker or occasional use of a wheelchair for long distances; grade 4 represents frequent use of a wheelchair (up to 50% of the time), but the individual is able to walk short distances using other assistive devices; and grade 5 marks functional impairment with inability to walk with crutches, with the individual requiring a wheelchair more than 50% of the time.
Genetic analysis
The genetic type of HSP was determined by genetic linkage analysis and/or mutation analysis of spastin and atlastin genes. We included only subjects for whom the genetic type of ADHSP was known. Linkage analysis was used to analyze HSP families with a sufficient statistical power to reach a two-point logarithm of the odds ratio (LOD score) value >3 to establish a definitive linkage. Linkage to the SPG4 locus (chromosome 2p) was tested using microsatellite markers D2S352, D2S367, and D2S2351; SPG3 locus (chromosome 14q) D14S584, D14S746, and D14S269; SPG6 (chromosome 15q) D15S118, D15S542, D15S543, and D15S128; and SPG8 (chromosome 8q) D8S266, D8S1799, D8S1138, and D8S1179. Genetic linkage analysis was performed as previously described [16]. Two-point linkage analysis was performed with the MLINK subroutine of the LINKAGE program using an autosomal dominant mode of disease inheritance and disease allele frequency of 0.001 [17]. We assigned genetic penetrance of 0.90 for LOD score calculations. Allele frequencies were not calculated from these families; instead, they were considered to be equal. All exons of spastin and atlastin genes were amplified using intronic primers as previously published [4, 5]. Polymerase chain reaction products were purified through the Sephadex G-50 columns and sequenced using the ABI PRISM dRhodamine Terminator Cycle Sequencing Ready Reaction and the ABI PRISM 3100 Genetic Analyzer (PE Applied Biosystems, Foster City, CA). DNA sequences were analyzed using SeqMan software (DNA Star, Boston, MA).
MRI analysis
Each patient underwent a noncontrast MRI of the brain and the whole spine utilizing a 1.5-T magnet. The brain was evaluated with axial and sagittal T1- and T2- weighted images; the spinal cord was evaluated with sagittal T1- and T2-weighted images and axial T1-weighted images. Axial T1 images of the spinal cord were done at the level of each vertebra and intervertebral disc space. MR images were transferred to an imaging workstation for further analysis by a neuroradiologist (O.P.E. or P.M.) who was blinded to the genetic status of each HSP subject. The brain scans were assessed for the presence of white matter signal abnormalities on T2-weighted images. The area of the corpus callosum and the whole supratentorial brain area were measured from the midsagittal view, and the corpus callosum index was calculated as previously published [18]. We measured the cross-sectional area of the spinal cord at the level of C2 and T9 from T1-weighted axial images. Three measurements for each segment were obtained, and an average value was calculated. We also selected an age- and gender-matched control group (19 women and 19 men; average age 51.7±13.9 years) who underwent MRI of the whole spine and had unremarkable neurologic examinations; the most common indication for neuroimaging in the control group was back and neck pain. The second control group (19 women and 19 men; average age 52.3±12.1 years) had normal MRI of the brain, and the most common indications for neuroimaging were headaches. The spinal cord and brain were analyzed using the same methods as in the HSP group. The cross-sectional area of the cervical and thoracic spinal cord and the corpus callosum index were compared using Student’s t-test; an association between duration of the disease and the spinal cord size was analyzed using the Spearman rank correlation test.
Results
Demographic characteristics, age at symptom onset, severity of disease, and results of genetic analyses of all patients are summarized in Table 1. We studied five SPG4 patients (one patient from each of five families), three SPG3A patients (each from different families), three SPG8 patients (from the same family), and two SPG6 patients (from the same family). Clinical descriptions of the individuals with SPG3A, SPG6, and SPG8 have been reported previously [5, 15, 19]. The SPG4 HSP patients had clinical features characteristic of uncomplicated HSP.
All patients had grossly normal MRI of the brain with the exception of subject 12, who had a remote left occipital lobe stroke; we did not detect any areas with increased signal from the deep white matter on T2-weighted images. Subjects with ADHSP did not have signs of corpus callosum atrophy, as the corpus callosum index did not differ between affected patients and healthy age- and gender-matched controls (0.0412±0.011 and 0.0417±0.013, respectively, not statistically significant).
MRI of the spinal cord revealed no signs of cord compression or abnormal cord signal in any patient with HSP. Each control subject also had a normal MRI of the spinal cord. The cross-sectional area of HSP subjects at the cervical level C2 was 59.42±12.57 mm2 and at the thoracic level was T9 28.58±5.25 mm2. Both of these values were significantly less than in healthy controls (85.66±4.25 and 41.0±7.0 mm2, respectively, both p<0.001). The degree of spinal cord atrophy was much more prominent in the subgroup of patients with SPG6 and SPG8 who had signs of severe atrophy, even in mildly affected subjects (47.60±6.58 mm2 at C2, 21.40±2.4 mm2 at T9), compared with HSP subjects with SPG3 and SPG4 (66.0±8.94 mm2 at C2, p<0.02; 31.75±2.76 mm2 at T9, p<0.001). There was no correlation between the degree of spinal cord atrophy and duration and severity of the disease in any genetic subtype of HSP (Fig. 1a–h).

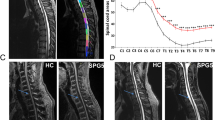
Different degrees of cord atrophy in various types of autosomal dominant hereditary spastic paraplegia. a Cervical cord of subject 3 with SPG3A. b Thoracic cord of subject 3 with SPG3A. c Cervical cord of subject 4 with SPG4. d Thoracic cord of subject 4 with SPG4. e Cervical cord of subject 9 with SPG6. f Thoracic cord of subject 9 with SPG6. g Cervical cord of subject 11 with SPG8. h Thoracic cord of subject 11 with SPG8
Discussion
Our MRI data indicate that spinal cord atrophy is a common finding in all four analyzed types of ADHSP: SPG3A, SPG4, SPG6, and SPG8. Each affected individual had a smaller cross-sectional spinal cord area than the age- and gender-matched controls. Spinal cord atrophy was more pronounced in the thoracic cord than in the cervical cord. Even though our MRI method did not allow us to separately analyze the size of various descending and ascending spinal cord tracts, the presence of a more marked atrophy in the thoracic segment suggests that the detected spinal cord atrophy is predominantly due to the degeneration of the descending (pyramidal tract) spinal cord tracts innervating the lower extremities.
Further analysis of our MRI data also indicates differences among various types of ADHSP. Early and severe spinal cord atrophy appears to be limited to SPG6 and SPG8 and was not seen in patients with SPG4 and SPG3A. Even though the number of patients with various genetic types of ADHSP is relatively small, this difference was so robust that it reached statistical significance. SPG6 and SPG8 are relatively rare, and all studied subjects were from the same families; thus, additional studies are necessary to establish whether this is a constant feature of these subtypes of ADHSP. However, our preliminary MRI data suggest that the degree of axonal loss is most prominent in these two types of ADHSP.
The degree of atrophy did not correlate with the duration or the severity of disease. Patients with ADHSP due to atlastin mutations tend to have an onset of symptoms at an early age, and indeed, all studied individuals with SPG3A had an onset of symptoms before the age of 5 years and, consequently, had the longest duration of disease within our cohort of patients; however, they were also significantly disabled and required a wheelchair for a significant amount of time [5]. The degree of spinal cord atrophy was the mildest in this type of ADHSP, suggesting that axonal dysfunction may occur also without significant axonal loss.
We also detected signs of spinal cord atrophy in patients with the most common type of ADHSP, SPG4 that is caused by mutations in spastin. Krabbe et al. studied 16 patients with ADHSP later linked to chromosome 2p [13, 14]. They did not find significant differences from the age- and gender-matched controls in the cross-sectional spinal cord area in the cervical or thoracic segments. The only difference detected was a smaller anteroposterior diameter of the spinal cord in HSP patients; however, the significance of this change is unclear, as it did not affect the cross-sectional area of the spinal cord. Moreover, the authors of this study did not provide complete demographic and clinical data other than the ranges of age and duration of symptoms (between 24 and 61 years and between 4 and 31 years, respectively); thus, it is unclear whether the discrepancies between our results and theirs may be due to the differences in patient age or disease severity. The intra- and interfamilial variability in patients with SPG4 is well known [4, 20]. This may be one potential explanation for the lack of spinal cord atrophy in Krabbe et al.’s group of patients.
Analysis of the brain did not reveal any constant abnormalities that would distinguish the patients with HSP from the control group. We did not find signs of atrophy of the corpus callosum, as the callosal index was very similar between the patients and the age- and gender-matched controls. Atrophy of the corpus callosum is a frequent finding in patients with autosomal recessive HSP linked to chromosome 15q13–q15 [21, 22]. Krabbe et al. also suggested that a mild atrophy of the corpus callosum was a feature of their SPG4 patients [13]. However, our data do not support that the corpus callosum is atrophied in ADHSP.
In summary, this study reports spinal cord atrophy among different genetic types of ADHSP and suggests that different disease mechanisms may cause various types of ADHSP. Some types, for example SPG6 and SPG8, are associated with severe and early spinal cord atrophy, whereas others, such as SPG3A and SPG4, have a marked axonal dysfunction with relatively less prominent spinal cord atrophy.
References
Fink JK (2002) Hereditary spastic paraplegia. In: Rimoin DL, Pyeritz RE, Connor JM, Korf BR (eds) Emery and Rimoin’s principles and practice of medical genetics, 4th edn. Harcourt, London, pp 3124–3145
Fink JK (2003) The hereditary spastic paraplegias: nine genes and counting. Arch Neurol 60:1045–1049
Fink JK, Heiman-Patterson T, Bird T, Cambi F, Dubé M-P, Figlewicz DA, Haines JL et al (1996) Hereditary spastic paraplegia: advances in genetic research. Neurology 46:1507–1514
Hazan J, Fonknechten N, Mavel D et al (1999) Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat Genet 23:296–303
Zhao X, Alvarado D, Rainier S et al (2001) Mutations in a novel GTPase cause autosomal dominant hereditary spastic paraplegia. Nat Genet 29:326–331
Hansen JJ, Durr A, Cournu-Rebeix I et al (2002) Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am J Hum Genet 70:1328–1332
Reid E, Kloos M, Ashley-Koch A et al (2002) A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am J Hum Genet 71:189–194
Rainier S, Chai J-H, Tokarz D, Nicholls RD, Fink JK (2003) NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6). Am J Hum Genet 273:967–971
Schwarz GA (1952) Hereditary (familial) spastic paraplegia. Arch Neurol Psychiatry 68:655–682
Sack GH, Huether CA, Garg N (1978) Familial spastic paraplegia: clinical and pathological studies in a large kindred. Johns Hopkins Med J 143:117–121
White KD, Ince PG, Lusher M et al (2000) Clinical and pathologic findings in hereditary spastic paraparesis with spastin mutation. Neurology 51:89–94
Dürr A, Brice A, Serdaru M et al (1994) The phenotype of pure autosomal dominant spastic paraplegia. Neurology 44:1274–1277
Krabbe K, Nielsen JE, Fallentin E, Fenger K, Herning M (1997) MRI of autosomal dominant pure spastic paraplegia. Neuroradiology 39:724–727
Nielsen JE, Krabbe K, Jennum P et al (1998) Autosomal dominant pure spastic paraplegia: a clinical, paraclinical and genetic study. J Neurol Neurosurg Psychiatry 64:61–66
Hedera P, DiMauro S, Bonilla E et al (1999) Phenotypic analysis of autosomal dominant hereditary spastic paraplegia linked to chromosome 8q. Neurology 53:44–50
Hedera P, Rainer S, Alvarado D et al (1999) Novel locus for autosomal dominant hereditary spastic paraplegia, on chromosome 8q. Am J Hum Genet 64:563–569
Lathrop GM, Lalouel JM, Julier C, Ott J (1985) Multipoint linkage analysis in humans: detection of linkage and estimation of recombination. Am J Hum Genet 37:482–498
Rauch RA, Jinkins JR (1994) Analysis of cross-sectional area measurements of the corpus callosum adjusted for brain size in male and female subjects from childhood to adulthood. Behav Brain Res 64:65–78
Fink JK, Sharp GB, Lange BM et al (1995) Autosomal dominant, familial spastic paraplegia, type I: clinical and genetic analysis of a large North American family. Neurology 45:325–331
Hentati A, Deng HX, Zhai H et al (2000) Novel mutations in spastin gene and absence of correlation with age at onset of symptoms. Neurology 55:1388–1390
Shibasaki Y, Tanaka H, Iwabuchi K et al (2000) Linkage of autosomal recessive hereditary spastic paraplegia with mental impairment and thin corpus callosum to chromosome 15q13–15. Ann Neurol 48:108–112
Okubo S, Ueda M, Kamiya T, Mizumura S, Terashi A, Katayama Y (2000) Neurological and neuroradiological progression in hereditary spastic paraplegia with a thin corpus callosum. Acta Neurol Scand 102:196–199
Acknowledgements
P.H. is supported by NIH K08NS42743. J.K.F. is supported by NIH R01NS33645, NIH R01NS38713, and the VA Merit Review Award. S.R. is supported by a grant from the University of Michigan Institute of Gerontology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hedera, P., Eldevik, O.P., Maly, P. et al. Spinal cord magnetic resonance imaging in autosomal dominant hereditary spastic paraplegia. Neuroradiology 47, 730–734 (2005). https://doi.org/10.1007/s00234-005-1415-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00234-005-1415-3