
Abstract
Amphipols are short amphipathic polymers that can substitute for detergents at the hydrophobic surface of membrane proteins (MPs), keeping them soluble in the absence of detergents while stabilizing them. The most widely used amphipol, known as A8-35, is comprised of a polyacrylic acid (PAA) main chain grafted with octylamine and isopropylamine. Among its many applications, A8-35 has proven particularly useful for solution-state NMR studies of MPs, for which it can be desirable to eliminate signals originating from the protons of the surfactant. In the present work, we describe the synthesis and properties of perdeuterated A8-35 (perDAPol). Perdeuterated PAA was obtained by radical polymerization of deuterated acrylic acid. It was subsequently grafted with deuterated amines, yielding perDAPol. The number-average molar mass of hydrogenated and perDAPol, ~4 and ~5 kDa, respectively, was deduced from that of their PAA precursors, determined by size exclusion chromatography in tetrahydrofuran following permethylation. Electrospray ionization–ion mobility spectrometry–mass spectrometry measurements show the molar mass and distribution of the two APols to be very similar. Upon neutron scattering, the contrast match point of perDAPol is found to be ~120 % D2O. In 1H-1H nuclear overhauser effect NMR spectra, its contribution is reduced to ~6 % of that of hydrogenated A8-35, making it suitable for extended uses in NMR spectroscopy. PerDAPol ought to also be of use for inelastic neutron scattering studies of the dynamics of APol-trapped MPs, as well as small-angle neutron scattering and analytical ultracentrifugation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
NMR studies of membrane proteins (MPs) give access to such important biological information as their secondary and tertiary structure, their dynamics, the way they recognize their ligands, etc. Traditionally, these studies are performed in aqueous detergent solutions (Kang and Li 2011). However, the destabilizing character of detergents, the high concentrations of them that is required for NMR experiments, the relatively high temperatures at which those are carried out and their long duration combine to make it difficult to prepare stable enough MP samples. This has driven the search for more stabilizing environments, such as lipid bicelles (Czerski and Sanders 2000; Sanders and Landis 1995; Sanders and Prosser 1998), lipopeptides (McGregor et al. 2003; Privé 2009), nanodiscs (Glück et al. 2009; Hagn et al. 2013; Mazhab-Jafari et al. 2013; Raschle et al. 2009, 2010; Shenkarev et al. 2010) or amphipols (Zoonens et al. 2005) (for reviews, see e.g., Catoire et al. 2014; Elter et al. 2014; Planchard et al. 2014; Popot 2010; Warschawski et al. 2011; Zoonens and Popot 2014). Whatever the environment, the size of the resulting complexes entails correlation times in excess of 10 ns. This renders structural and dynamics studies by NMR quite challenging. In addition, the presence of high concentrations of detergent and/or of lipids (in the case of bicelles or nanodiscs) generates interference signals from their 1H or natural-abundant ce 13C nuclei, which can hinder the observation of signals from the protein, in particular intramolecular 1H-1H dipolar nuclear overhauser effect (NOE) interactions. Even though some NMR pulse sequences can reduce the impact of these interference signals, the best strategy is to work with perdeuterated surfactants (see e.g. Tzitzilonis et al. 2013). This remains true when studying small organic compounds interacting with MPs, because many 13CHn chemical shifts of ligands and surfactants overlap. When the cost of labeling isotopically the ligand is prohibitive, deuteration or perdeuteration of the surfactant(s) can be the most efficient approach.
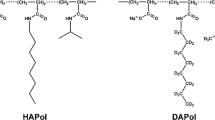
Amphipols (APols) are comprised of a polymeric backbone carrying hydrophilic and hydrophobic moieties (Popot, 2010; Popot et al. 2011; Tribet, Audebert and Popot, 1996; Zoonens and Popot, 2014). Three types of APols have been validated for NMR studies of MPs: carboxylated APols, best represented by A8-35 (Fig. 1) (Catoire et al. 2009, 2010a, b; Elter et al. 2014; Etzkorn et al. 2013; Raschle et al. 2010; Zoonens et al. 2005), sulfonated APols (Dahmane et al. 2011) and non-ionic APols (Bazzacco et al. 2012; for reviews, see Elter et al. 2014; Planchard et al. 2014; Popot 2010; Popot et al. 2011). A8-35, to date the best characterized and most extensively used APol, is comprised of a polyacrylic acid (PAA) backbone grafted with octylamine and isopropylamine side chains (Tribet et al. 1996) (Fig. 1). It has been used to-date for solution NMR studies of five MPs, namely the transmembrane β-barrel (tOmpA) of outer membrane proteins A from Escherichia coli (Zoonens et al. 2005) and from Klebsiella pneumoniae (Planchard et al. 2014; Renault 2008), OmpX from E. coli (Catoire et al. 2009, 2010b; Etzkorn et al. 2014), bacteriorhodopsin (BR) from Halobacterium salinarum (Etzkorn et al. 2013, 2014; Raschle et al. 2010) and the human BLT2 G protein-coupled receptor of leukotriene B4 (LTB4) (Catoire et al. 2010a). For some of these experiments, a partially deuterated version of A8-35 (hereafter, DAPol) has been used, in which the PAA main chain is hydrogenated, but the octylamine and isopropylamine side chains are deuterated (Gohon et al. 2004, 2006). The use of DAPol has permitted, in particular, mapping of the points of contact between the polymer and tOmpA, by comparing the width of the resonance peaks of amide protons in the presence of either DAPol or unlabeled, hydrogenated A8-35 (hereafter, HAPol) (Zoonens et al. 2005). Following DAPol-assisted folding of deuterated BLT2, measurements of transferred NOE signals led to the determination of the conformation adopted by hydrogenated leukotriene B4 (LTB4) when bound to its specific recognition site at the surface of the receptor (Catoire et al. 2010a), an approach that may be widely applicable (Banères, Popot and Mouillac, 2011; Catoire et al. 2011). Part of the magnetization transfer signals between LTB4 protons could not be observed, however, because they were masked by signals originating from the hydrogenated backbone of DAPol (Catoire et al. 2010a) (reviewed in Planchard et al. 2014).

In the present article, we describe the synthesis and properties of a perdeuterated form of A8-35 (perDAPol). Synthesis of an exact mimic of HAPol required re-examination of the average degree of polymerization of the commercial hydrogenated PAA used for HAPol and DAPol synthesis. This was followed by the synthesis of an equivalent deuterated PAA, which was obtained by radical telomerization of its perdeuterated acrylic acid precursor. The deuterated PAA was then successively grafted with deuterated octylamine and deuterated isopropylamine. Upon dissolution into aqueous buffers, the resulting perDAPol self-organizes into particles with the same behavior in aqueous SEC as those formed by HAPol and DAPol. The mass distribution of the three forms of A8-35 was studied by electrospray ionization–ion mobility spectrometry–mass spectrometry (ES-IMS-MS) (Leney et al. 2012) and found to be comparable. The contrast match point (CMP) of perDAPol was established by small-angle neutron scattering (SANS). Homonuclear and heteronuclear NMR spectroscopy was performed on the three forms of A8-35. Partial deuteration reduces proton signals of A8-35 by ~68 %, perdeuteration by ~94 %. The applications of perDAPol to NMR, inelastic neutron scattering (INS), SANS and analytical ultracentrifugation (AUC) studies of MPs are discussed.
Materials and Methods
Materials
Tetrahydrofuran (THF), purchased from VWR (FontenaysousBois, France), was distilled over calcium hydride before use. Poly(acrylic acid) (PAA) and other chemicals were from Sigma-Aldrich (Saint Quentin Fallavier, France). A single and identical weight-average molar mass (\(\bar{M}_{w}\) ≈ 5,000 g mol−1) was given by the purchaser for all PAA batches used and/or cited in this paper. Acrylic acid-d4 was from Cil (Courbevoie, France) and used as received. A8-35 (batch FGH29), A8-75 and DAPol-4 were synthesized and characterized as described in refs. (Gohon et al. 2004, 2006).
Buffer
Tris buffer: Tris/HCl 20 mM, NaCl 100 mM, pH 8.0.
Methods
Synthesis of a Perdeuterated Poly(acrylic acid) Precursor
The overall scheme of the synthesis of perdeuterated PAA and perDAPol is shown in Fig. 2. In a 25-mL two-neck round-bottom flask, acrylic acid-d4 (1.06 g, 14 mmol) was dissolved into 9 mL of a 8:1 water/THF mixture and the solution freed from O2 with argon by multiple cycles of freeze–pump–thaw degassing under vacuum. Prior to polymerization, the solution was flushed with a stream of argon. Thioglycolic acid (TGA) (0.074 g, 0.8 mmol) and 1 mL (26.2 mg, 0.16 mmol) of a previously degassed 0.16 M 2,2′-azoisobutyronitrile (AIBN) (recrystallized twice in ethanol) solution in THF were injected into the aqueous mixture. The flask was sealed and kept under vigorous stirring in an oil bath at 65 °C for 4 h. It was then quickly brought to 0 °C (ice bath) and the polymerization reaction quenched by flushing the mixture with air and cooling at −30 °C (dry ice in ethanol). The flask was brought to room temperature, and the solution added dropwise to 100 mL of 0.2 N NaOH in ethanol under vigorous stirring. The precipitate was filtered, rinsed with 100 mL of absolute ethanol and dissolved in 10 mL of water purified on an A10 Advantage Millipore system (mQ water). After acidification with 1.5 mL of concentrated HCl, the water was evaporated under reduced pressure and the dry PAA triturated in 20 mL of THF. The precipitated sodium chloride was removed by filtration, and the filtrate concentrated to a 10-mL final volume. The THF solution was added dropwise to 100 mL of petroleum ether (or n-heptane) under vigorous stirring, and the resulting precipitate recovered after filtration. The polymer was dried under vacuum and dissolved in 20 mL of mQ water. Freeze-drying of the solution yielded 1.0 g (94 % yield) of the expected perdeuterated PAA (PAA-d-3, batch 3).

Synthesis of perDAPol, starting from acrylic acid-d4, and permethylation of polymers for SEC analysis. a TGA, AIBN in 80:20 water/THF at 65 °C, 4 h; b n-octylamine-d17, DCI, NMP, 60 °C, 1 h, then RT 12 h, then isopropylamine-d7, DCI, NMP, 50 °C, 1 h, then RT 12 h, then NaOH; c H3O+; d TMSCHN2, THF:MeOH 9:1
Synthesis of perDAPol
PAA-d-3 (0.86 g) (11.46 mmol carboxylic acid) was dissolved into 17 mL of N-methylpyrrolidone (NMP) and stirred at 60 °C in an oil bath for 4 h. Octylamine-d17 (0.418 g, 2.86 mmol) and dicyclohexylcarbodiimide (DCI) (0.59 g, 2.86 mmol) in solution in NMP (2 mL and 4 mL, respectively) were successively added dropwise to the reaction medium (Fig. 2). After complete addition of the reagents, the mixture was kept at 60 °C for 1 h, then brought to room temperature and stirred overnight. The precipitate of dicyclohexylurea (DCU) was removed by filtration and 1-N-hydroxybenzotriazole (HOBt) (0.62 g, 4.73 mmol) added to the filtrate, which was heated at 50 °C. Isopropylamine-d7 (0.3 g, 4.54 mmol) and DCI (0.994 g, 4.82 mmol) were added to the reaction medium, which was kept at 50 °C for a further hour after complete addition of the reactants (Gohon et al. 2004, 2006). After overnight stirring at room temperature, followed by filtration, the filtrate was treated with sodium methoxide (1.5 g, 27.8 mmol) and stirred for 15 min before being poured into 300 mL of mQ water. The mixture was stirred for 30 min, filtered, and the filtrate added dropwise to 20 mL of 2 N HCl under vigorous stirring. The precipitate was recovered by filtration and suspended into 100 mL of water under vigorous stirring; 1 mL of a 30 % NaOH solution in mQ water was added. The resulting solution was centrifuged (5,000×g for 30 min), and the supernatant filtered on Millipore filters. After three further cycles of precipitation in acidic solution/dissolution in basic medium, the solution was dialyzed against mQ water for 4 h and freeze-dried to yield 1.43 g (90% yield) of the expected perdeuterated A8-35 (perDAPol, batch 1) as a white solid.
Analysis of the Chemical Composition of the Polymers
The chemical composition of the polymers was analyzed by 1H and 13C NMR spectroscopy as previously described (Gohon et al. 2004, 2006). Results are reported in Table 1.
Characterization of the Molar Mass of the Polymers
Estimation of the Average Molar Mass of the Polymers by SEC Analysis
-
SEC analysis of PAA in aqueous buffer
SEC analyses of PAAs in aqueous buffer were carried out as described in refs. (Gohon et al. 2006; Tribet et al. 1996). In both cases, PAAs were injected under their sodium salt form on a Shodex OH pack column (Waters) calibrated with poly(ethylene oxide) (PEO) standards (“universal” calibration) and eluted with a 0.5 M LiNO3 aqueous solution at 40 °C. Injections were monitored by double detection [refractive index (RI) and differential viscometry (dV) detection]. SEC experiments were employed to determine the weight-average molar mass \(\bar{M}_{w}\) and the number-average molar mass \(\bar{M}_{n}\) of the PAAs but, as described below (see “Results”), masses were overestimated.
-
SEC analyses of PAA in organic solvent.
Prior to SEC analysis, the AA units of all PAAs were modified by methylation of the carboxylic acid groups using trimethylsilyldiazomethane according to the method described by Couvreur et al. 2003. Briefly, 50 mg of the PAA was dissolved in 10 mL of a 9:1 THF/methanol mixture and treated with 0.5 mL of trimethylsilyldiazomethane 2 M in diethyl ether (Presser and Hüfner 2004). The solvent was evaporated under reduced pressure, and the permethylated polymer, poly(methyl acrylate), was dried under vacuum for 12 h.
Measurements were performed with a Viscotek TDAmax system from Malvern Instruments that consists of an integrated solvent and sample delivery module (GPCmax) and a Tetra Detector Array (TDA) including a differential detector RI, a right (90 °) and a low (7 °) angle light scattering (LS) detector (RALS/LALS), a four-capillary differential viscometer and a diode array UV detector. THF was used as the mobile phase at a flow rate of 1 mL min−1 and toluene as a flow rate marker. All polymers were injected (100 μL of solution) at a concentration of 10 g L−1 after filtration through a 0.45-μm pore-size membrane. The separation was carried out on three Polymer Laboratories columns (3 × PLgel 5-μm Mixed C; 300 × 7.5 mm) and a guard column (PL gel 5 μm). Columns and detectors were maintained at 40 °C. The OmniSEC 4.6.2 software was used for data acquisition and data analysis. \(\bar{M}_{n}\), \(\bar{M}_{w}\), and the molar mass dispersity, Đ = \(\bar{M}_{w}\)/\(\bar{M}_{n}\), were calculated with a calibration curve based on narrow PS standards (from Polymer Standard Services), using only the RI detector (simple detection). Indeed, regarding the Mark–Houwink–Sakurada parameters in THF, the polystyrene calibration is appropriate for poly(methyl acrylate) (Couvreur et al. 2003; Velasquez et al. 2013). In addition, \(\bar{M}_{n}\) was also calculated using the combined signals from RALS/LALS, RI and the viscometer (triple detection). All molar mass were recalculated and expressed for the sodium salt of each polymer (see Tables).
Estimation of the Average Molar Mass and Dispersity of the Polymers by Mass Spectrometry
ES-IMS-MS experiments were performed on the sodium salt of the polymers on a Synapt HDMS mass spectrometer (Micromass UK Ltd./Waters, Manchester, UK) as described in ref. (Leney et al. 2012). Samples were introduced into the instrument by direct infusion using gold-coated borosilicate capillaries. A capillary voltage of 1.8 kV was applied, and a source temperature of 80 °C was used. A cone voltage of 70 V was used, and trap and transfer collision voltages were both set to 5 V. Ion mobility separation was performed by ramping the wave height from 4.5–28.5 V at a speed of 400 m s−1. An aqueous solution of CsI was used for m/z calibration. All data were acquired over the m/z range 500–5,000 and the raw data processed by use of MassLynx v.4.1 and Driftscope v.3.0 software (Micromass UK Ltd/Waters Corp., Manchester, UK).
Average masses are expressed as the number-average molar mass, \(\bar{M}_{n}\), and the weight-average molar mass, \(\bar{M}_{w}\), with
and
where N i is the number of moles, M i is the molar mass and m i the mass of species i.
Characterization of the association behavior of APols in aqueous buffer
APols were dissolved at 100 g L−1 in mQ water. The stock solution was diluted at 10 g L−1 in Tris buffer and kept under stirring for 48 h at room temperature; 80 µL of the resulting sample were injected at room temperature on an Äkta purifier 10 FPLC system (GE Healthcare) equipped with a calibrated Superose 12 10/300 GL column (GE Healthcare) (see Gohon et al. 2004, 2006, for detailed procedure). The flow rate was set at 0.5 mL min−1. Injection buffer was 0.1 M NaCl, 20 mM Tris/HCl, pH 8. The elution profile was monitored at 220 nm.
Small-Angle Neutron Scattering (SANS)
Sample Preparation
PerDAPol was dissolved at 7.2 g L−1 either in Tris/HCl 20 mM, pH 8.0, 100 mM NaCl, 2 mM EDTA, H2O or in Tris/HCl 20 mM, pD 7.6 100 mM NaCl, 2 mM EDTA, D2O. UV spectra of the two solutions confirmed that their concentrations were identical. The H2O and D2O samples were mixed in the appropriate proportions to yield the desirable ratios of H2O/D2O.
SANS Experiments
SANS experiments were performed on the Bio-SANS instrument of the High Flux Isotope Reactor of the Oak Ridge National Laboratory (Lynn et al. 2006). SANS data were taken at sample-to-detector distances of 1.1 and 6.8 m. The neutron wavelength, λ, was set to 6 Å, and the wavelength spread, Δλ/λ, was set to 0.15. The two instrument settings provided a q-range from 0.006–0.70 Å−1. Scattered neutrons were collected by a 1 m × 1 m two-dimensional (2D) linear position-sensitive detector having 192 × 256 pixels (Berry et al. 2012). The solutions and associated solvent backgrounds were measured at 20 °C in 1-mm path-length quartz cells (Hellma U.S.A., Plainview, NY, USA). The 2D data were corrected for detector pixel sensitivity, dark current and scattering by the buffer. The 2D reduced data were azimuthally averaged to yield the 1D scattering intensity I(q) versus q, where q = 4π sin(θ)/λ and 2θ is the scattering angle from the incident beam. The 1D profiles from the two detector distances used on Bio-SANS were merged to produce the full scattering intensity profile using software developed at the National Institute of Standards and Technology (Kline 2006). A constant background representing the difference in incoherent scattering resulting from the differences in hydrogen content of the samples and backgrounds was subtracted from the 1D profile (Fig. 5a).
The radius of gyration, R g, was determined with the Guinier approximation shown in Eq. 1 (Guinier and Fournet 1955):
where I(0) is the forward scattering intensity. A linear fit of ln(I(q)) versus q 2 (“Guinier plot”) (Fig. 5b) provides I(0) and R g from the intercept and slope, respectively. The molar mass of the particle is provided by the Guinier fitting through the relation in Eq. 2:
where N A is Avogadro’s number, C is the concentration of the samples in solution, the neutron scattering length densities of the sample and buffer are ρ and ρ buffer, respectively, and V s is the specific volume of the scattering particle.
The contrast match point of perDAPol is determined by plotting the square root of the forward scattering intensities as a function of the D2O ratio (v/v) (Fig. 5c). The intercept of the linear fitting of the plot with the y axis (zero intensity) determines the contrast match point.
Results
Re-examination of the Degree of Polymerization of the Hydrogenated PAA used for Synthesizing A8-35
The average degree of polymerization of the hydrogenated PAA (PAA-1) used for synthesizing A8-35 was determined by SEC analysis in organic solvent (Table 1). This study made it possible to clarify the difference between the degree of polymerization estimated in ref. (Tribet et al. 1996) (\(\overline{DP}_{w}\) ≈70, i.e. \(\overline{DP}_{n}\) ≈35 assuming a molar mass dispersity of 2; PAACT-1 in Table 1), using SEC in aqueous solution with a RI calibration based on PEO standards and a differential viscometry (dV) detection and that found later, using a similar procedure, (\(\overline{DP}_{w}\) ≈130, \(\overline{DP}_{n}\) ≈80) (Gohon et al. 2004) (PAACT2 in Table 1). The present data were obtained by SEC of permethylated PAA in an apolar organic solvent (THF) using either a simple detection mode (differential RI detector) or a triple detection (possessing as a key element a static light scattering detector). The results tally with the estimate of ref. (Tribet et al. 1996), suggesting that the \(\overline{DP}_{n}\) of the PAA was overestimated in ref. (Gohon et al. 2004) (see Table 1). Several factors that may explain the inconsistency between the estimates of refs. (Tribet et al. 1996) and (Gohon et al. 2004) are indicated in the “Discussion” section. On the basis of the new measurements, the \(\bar{M}_{n}\)of the sodium salt of hydrogenated A8-35 is expected to be ~4 kDa, rather than 8–10 kDa as assumed in refs. (Gohon et al. 2004, 2006; Table 2). The lower estimate was confirmed qualitatively by MS analysis of the final products (see below). As discussed below, this revision has no practical consequence under most circumstances, where it is the mass of the APol particle or MP-associated APol that matters, not the number of molecules it is comprised of (see “Discussion”).
Synthesizing a Deuterated PAA with a Matching Length
In order to produce a perdeuterated version of A8-35 (perDAPol) that be chemically as similar as possible to the unlabeled form (HAPol), it has to be synthesized starting from a perdeuterated PAA of similar chain length (\(\overline{DP}_{n}\) ≈ 35) and molar mass dispersity (Đ ≈ 2) as the unlabeled PAA and according the same protocol (hydrophobic modification). Perdeuterated PAA was synthesized by radical polymerization of the perdeuterated acrylic acid (AAd4) in the presence of a telogen. The protocol used was adapted from that described by Loubat, Javidan and Boutevin, 2000. Polymerization was initiated by heat decomposition of AIBN, TGA being used as the telogen or chain transfer reagent. The use of TGA allows to limit the growth of the macroradicals formed during the propagation step of the polymerization and yields polymers of restricted length, namely telomers (see refs. Loubat et al. 2000, and Pavia et al. 1992, for further details). With this approach, the only parameters involved in the control of the number-average degree of polymerization (\(\overline{DP}_{n}\)) are the initial concentration of TGA, [TA]0, and the transfer constant C t of the telogen. These three terms are related by Eq. 3 (Loubat et al. 2000), which is accurate only at the beginning of the reaction (degree of advancement of the reaction <10 %):
In the ideal case, C t = 1 and the final length of the telomer \(\overline{DP}_{n}\) ≈ [AA]0/[TA]0, this relation assuming that TA and AA are consumed in the same proportion during the reaction. It has been reported that this ideal condition is obtained with TGA when the polymerization of AA is performed in a 80:20 water/THF mixture (Loubat et al. 2000). Our first attempts at telomerizing AAd4 using these conditions gave unsatisfactory results (unexpected \(\overline{DP}_{n}\) and/or poor yields), but sample PAA-d-3 (Table 3) yielded finally the desired \(\overline{DP}_{n}\) (~35). Subsequent experiments gave similar results as those reported by (Loubat et al. 2000) (Table 3), illustrating that the method could be mastered only after several attempts.
Synthesis, Purification and Chemical Analysis of perDAPol
The synthesis and purification of perDAPol were carried out according to the same procedure as used to synthesize HAPol (Gohon et al. 2004, 2006). Perdeuterated PAA was hydrophobically modified in two steps in NMP, first with perdeuterated octylamine and then with perdeuterated isopropylamine, in the presence of DCI as a coupling reagent. Purification of the polymer was achieved by four cycles of precipitation/dissolution in aqueous solution. Dialysis of the final basic solution followed by freeze-drying yielded perDAPol quantitatively as its sodium salt. The overall degree of modification was estimated by 13C NMR spectroscopy to be close to 65 % (leaving ~35 % of free carboxylates), as expected for 100 % grafting efficiency. The relative amount of grafted octylamine and isopropylamine could not be accurately determined by the same technique as for HAPol because of 13C-D heteronuclear coupling. Indeed, the relaxation time (τ R) of each carbon depends on its degree of deuteration (the higher this degree, the lower τ R). This makes it difficult to derive the degree of grafting of each type of chain from the intensity of the signals, as is done for hydrogenated A8-35 (Gohon et al. 2004, 2006). Decoupling deuterium and 13C could permit to circumvent this problem. This has not been implemented here. In our experience, however, the amount of grafted alkylamine has always been found to be close to that aimed for. The extent of grafting in perDAPol can therefore be safely assumed to be close to 25 % octylamine and 40 % isopropylamine, consistent with the solution behavior of the polymer (see below).
ESI-IMS-MS Comparison of the Mass Distribution of A8-35, DAPol and perDAPol
The heterogeneity of APols and other polymers makes their study challenging. However, the advent of electrospray ionization–ion mobility spectrometry–mass spectrometry (ESI-IMS-MS) has significantly improved the capability of MS to study complex polymeric mixtures (Weidner and Trimpin 2010). IMS separates ions based on their drift time though a neutral buffer gas under the influence of a weak electric field, the drift time depending on the ions’ shape (collision cross-sectional area) and charge state (Kanu et al. 2008). Coupled with MS, this provides an extra dimension of separation which is particularly useful in the study of polymers.
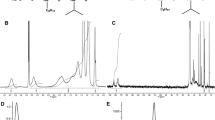
The three-dimensional (3D) ESI-IMS-MS spectra of A8-35 and perDAPol are shown in Fig. 3. Ions with +1, +2, +3 and +4 charge states that overlap in MS can be separated by IMS, observed and identified in the spectra, as indicated. There are also low-intensity ions with higher charge states that cannot be assigned reliably in the mass spectra. The ESI-IMS-MS data indicate that, as expected, the two APols have broad mass distributions. Note that, as species with higher mass-to-charge ratios (m/z) are less readily desolvated and detected, the distribution observed may be skewed toward low masses (Hernandez and Robinson 2007). Nevertheless, the approach is extremely useful on a comparative basis: The similar mass distributions observed in the two spectra indicate that A8-35 and perDAPol are highly comparable in terms of their heterogeneity and composition.

ESI-IMS-MS spectra of a A8-35 and b perDAPol (1 g L−1 in 100 mM ammonium hydrogen carbonate, pH 8.0). The contour plots indicate the drift time of ions in the IMS cell (ms) as a function of m/z
The weight-average (\(\bar{M}_{w}\)) and number-average (\(\bar{M}_{n}\)) molar masses of each APol were calculated from the MS data by first converting from m/z to mass and then proceeding as described in ref. (Gohon et al. 2006). For A8-35, \(\bar{M}_{w}\) = 4.8 kDa and \(\bar{M}_{n}\) = 3.8 kDa, corresponding to a polydispersity index Ð = \(\bar{M}_{n}\)/\(\bar{M}_{w}\) ≈ 1.26. For perDApol, \(\bar{M}_{w}\)= 5.2 kDa, \(\bar{M}_{n}\) = 4.2 kDa, Ð ≈ 1.24. The most abundant ions in the spectra correspond to species with molecular masses of 3.4 kDa (A8-35) and 3.6 kDa (perDAPol). These values compare well with the revised mass estimates of A8-35 (Table 2), even though they may be skewed, as outlined above, due to high mass polymer species potentially being ionized and detected less efficiently, and to the inability to assign conclusively all the low intensity high m/z species. The difference between the two values of Ð, 1.24 and 1.26, is much smaller than the experimental error, indicating a comparable molar mass dispersity. They are, however, lower than the value expected, Ð ≈ 2, possibly due to the skewing effect mentioned above. Furthermore, the similarity of A8-35 and perDAPol in terms of chain length is confirmed by their mass ratios: 1.088 for \(\bar{M}_{w}\), 1.101 for \(\bar{M}_{n}\), 1.059 for the most abundant ions, to be compared with the value of 1.081 expected from the extent of deuteration if the degrees of polymerization are identical. MS, therefore, confirms that HAPol and perDAPol are essentially identical but for isotopic labeling.
Self-assembly of perDAPol in Aqueous Buffer
The behavior of perDAPol in aqueous buffer was characterized by SEC on a Superose 12 column (Gohon et al. 2004, 2006). The elution profile of perDAPol is identical to that obtained for A8-35 (Fig. 4). In both cases, one main sharp peak is followed by a smaller, broader one. The main peak at V e ≈ 12 mL comprises the particles formed by self-association of the polymer in aqueous buffer, whose Stokes radius R S is ~3 nm (Gohon et al. 2006). Its sharpness—width at half height (WHH) ≈ 1 mL—reflects the narrow size distribution of the particles. The smaller but broader peak (WHH ≈ 2.1 mL) observed at V e ≈ 14.2 mL is characteristic of A8-35 samples and indicates the presence of smaller (R S ≈ 2 nm), heterogeneous species, which have not been formally identified. They may be short, probably poorly hydrophobized polymers that subsist in solution under their monomeric state, or loosely associated with the particles.

Overlay of the chromatograms obtained after the injection of HAPol (unlabeled A8-35) (circles) or perDAPol (batch 1) (squares) in Tris buffer on a Superose 12 10–300 GL column. The elution profile was monitored at 220 nm. The two profiles have been normalized to the same maximum. a.u. arbitrary units. V 0, V T excluded and total volumes, respectively
Neutron Scattering contrast match point
SANS is widely used to determine the mass, shape and arrangement of components in soluble particles (Zaccai and Jacrot, 1983). For MPs, being able to contrast match the surfactant by adjusting the H2O/D2O ratio in the buffer is an important asset. The contrast match point (CMP) of perDAPol was determined by measuring the intensity scattered at zero angle [I(0)] by perDAPol particles as a function of the percentage of D2O in the solution (Fig. 5). The CMP obtained by extrapolation of I(0) to zero intensity, ~120.5 % D2O, is in perfect agreement with that expected theoretically (~120 %) on the basis of the composition of perDAPol (this work) and the specific volume of A8-35 (Gohon et al. 2004) (G. Zaccai, personal communication).

SANS analysis of perDAPol particles. a SANS by perDAPol at different D2O/H2O ratios. b Guinier plots of the data in A. Dashed vertical lines define the region chosen (~0.6 ≤ (q × R g) ≤ ~1.7) to determine the molar mass and R g of the particles. All samples were at 7.2 g L−1. c Determination of the contrast match point
Less expected are the smaller R g (1.9 ± 0.2 nm) and mass (~18.8 kDa) obtained for perDAPol particles from the Guinier plots in Fig. 5b, which do not tally with those determined previously for HAPol and DAPol (2.4 ± 0.2 nm and 39–40 ± 5 kDa, respectively) (Gohon et al. 2006). The two measurements are consistent one with another in indicating ~2 × smaller particles, but it is highly unlikely that such an effect originates from isotopic labeling. Further investigations will be needed to establish its origin (possibly, but somewhat surprisingly, the effect of one component of the buffer, EDTA, which was not present in earlier SANS studies; about the effect of EDTA on the dispersity of A8-35 particles and MP/A8-35 complexes, see Picard et al. 2006; Catoire et al. 2010b).
Membrane Protein Trapping by perDAPol
In order to check on the ability of perDAPol to trap MPs, BLT2, a G protein-coupled receptor of leukotriene B4 (Yokomizo et al. 2000), was folded and trapped using either HAPol or perDAPol, according to the procedure described in refs. (Catoire et al. 2010a; Dahmane et al. 2009). Comparative SEC analysis of the BLT2/HAPol and BLT2/perDAPol complexes revealed almost identical particle sizes (Stokes radii of ~5.4 and ~5.5 nm, respectively) and size distributions (peak width at mid-height ~1.7 mL) (Fig. 6). These experiments suggest that folding and trapping procedures that were formerly defined for various PMs using HAPol, e.g. those used for GPCRs (Dahmane et al. 2009), can be reproduced with perDAPol without the need of a new optimization.

Comparative size exclusion chromatography analysis of human G protein-coupled receptor BLT2 folded and trapped in either fully protonated A8-35 (HAPol, dashed line) or perDAPol (solid line) at 1:5 w/w BLT2/A8-35 ratio. Injections of 100-μL samples onto a Superose 12 10–300GL column. Elution profiles were recorded at 220 nm. Protein concentration was ~1 g L−1. The equilibration and running buffer for both experiments was Tris/HCl (pH 8.0) supplemented with 100 mM NaCl. V 0, excluded volume; V T, total volume. Asterisk indicates the elution peak of residual salts. Elution volumes correspond to apparent Stokes radii of 5.4 and 5.5 nm for BLT2/HAPol and BLT2/perDAPol complexes, respectively
Comparison of HAPol, DAPol and perDAPol 2D 1H,1H NOESY and 1H,13C HSQC NMR Spectra
2D 1H,1H NOE spectroscopy (NOESY) and 1H,13C heteronuclear single quantum correlation (HSQC) spectra of HAPol, DAPol and perDAPol solutions, recorded at identical APol concentrations (20 g L−1 in pure D2O), are shown in Figs. 7 and 8. In 1D and 2D 1H homonuclear experiments (Fig. 7), 1H signals are reduced, as compared to HAPol, by 68 % for DAPol and by 94 % for perDAPol. In particular, between 0 and 2.5 ppm, the intensity of 1H signals is considerably reduced in perDAPol, as shown by the 1D rows extracted from the 2D NOESY spectra (Fig. 7d–f).

1H 1D a and 2D NOESY b homonuclear NMR spectra of HAPol, DAPol and perDAPol. In (a), the inset describes the chemical structure of APol A8-35. Circled numbers refer to chemically equivalent 1H whose resonances are shown in the 1D spectra displayed in a. One-dimensional spectra corresponding to the rows labeled C–F in (b) are shown to the right (panels c-f, respectively). All data were acquired at 400 MHz 1H Larmor frequency, 25 °C, at the same concentration of polymers (30 g L−1) solubilized in pure D2O

The 2D 1H,13C HSQC spectra show that substituting HAPol or DAPol with perDAPol entails a substantial reduction in the 1H,13C correlation peaks that could mask amino acid resonances, particularly above 24 ppm in the 13C dimension (Fig. 8). Note, however, that in the case of methyl-TROSY-based experiments (Plevin and Boisbouvier 2012; Sprangers, Velyvis and Kay 2007), the interference by HAPol would be severe, but that by DAPol limited (areas indicated by colored ellipses in Fig. 8).
Discussion
Revisitation of the Mass Distribution of A8-35 Molecules
The aim of the present work was the synthesis of a perdeuterated version of A8-35 that would be chemically as similar as possible to hydrogenated A8-35. This implied the prior synthesis of perdeuterated PAA with the same average size as the commercial PAA used as starting material to synthesize the hydrogenated version. Previous estimates of the size of commercial PAA, performed by SEC in aqueous solution using double detection, RI and differential viscometry (dV) detection and a “universal” PEO calibration, yielded scattered results (Gohon et al. 2004, 2006; Tribet et al. 1996). Here, we have reinvestigated the average molar mass and mass distribution of PAA, deuterated or not, by a more appropriate method, namely SEC in organic solvent following permethylation, and examined the molar mass distribution of the resulting APols by ESI-IMS-MS. SEC profiles monitored by two detection modes were used to calculate the average molar masses and mass distribution of the samples: A simple detection mode (differential RI detector) or a triple-detection one (possessing as a key element a static light scattering detector). It should be noted here that the accurate and reliable mass determination of low molar mass chains species is very difficult by light scattering (weak signals with strong noise), which may lead to overestimated average values. As regards the simple RI detection, it has been shown that in THF, a polystyrene calibration is valid for poly(methyl acrylate) samples (Couvreur et al. 2003), which simplifies analyses of methylated PAA samples. SEC analyses consistently indicate that the \(\overline{DP}_{n}\) of commercial PAA is close to ~35, which, assuming Ð ≈ 2 (cf. Table 1, PAA-1), is consistent with the \(\overline{DP}_{w}\) of ~70 estimated in ref. (Tribet et al. 1996), but lower than the \(\overline{DP}_{n}\) of ~80 (deduced from \(\bar{M}_{n}\) ≈ 7.5 kDa) found, also for commercial PAA, in subsequent measurements (Gohon et al. 2004, 2006). The difference may stem from (1) the difficulty of determining accurately the molar mass of PAA by SEC using a “universal” calibration based on a PEO standards, due to differences in interaction parameters (Gohon et al. 2004; Tribet et al. 1996) (the procedure used in these works was initially developed to characterize neutral polymers and not polyelectrolytes), (2) a slight drift of the semi-logarithmic “universal” calibration curve, (3) the reproducibility of the preparation of the different batches of PAA at the industrial scale and in the laboratory and/or possibly other unidentified causes. The revised value of \(\overline{{M_{n} }}\) is ~2 × smaller than that (8–10 kDa, depending on the source of PAA) assumed previously. Note that a similar revision also applies to sulfonated amphipols (SAPols), which are synthesized starting from the same PAA (Dahmane et al. 2011).
Revising the average molar mass of A8-35 does not affect any of the conclusions previously published regarding the mass of A8-35 particles or the mass ratio of APol to MP in complexes. Simply, an A8-35 particle formed in buffer solution, whose mass was determined to be ~40 kDa (Gohon et al. 2006), is now concluded to comprise ~9 rather than ~4 molecules, as had been previously estimated. The mass of A8-35 bound by BR, which is ~54 kDa (Gohon et al. 2008), corresponds to 12–13 molecules, not ~6. For functionalized derivatives of A8-35 such as fluorescent (Fernandez et al. 2014; Giusti et al. 2012; Opačić et al. 2014; Zoonens et al. 2007) or tagged (Charvolin et al. 2009; Giusti et al. 2014; Le Bon et al. 2014a) APols [reviewed in ref. (Le Bon, Popot and Giusti, 2014b)], the number of fluorophores or tags per particle or complex is not affected either. There are very few circumstances where knowledge of the actual number of APol molecules in a particle or complex is needed. This would be the case, e.g., should one want to calculate the entropy of dissociation of a particle or a MP-bound A8-35 layer into individual molecules (Giusti et al. 2012; Tribet et al. 2009), as well as for some other thermodynamic calculations (Pocanschi et al. 2013). Knowledge of the actual molar mass is also necessary should one want to make sure that most of the individual molecules are labeled or tagged, which is seldom relevant but can be of use under certain circumstances.
Synthesis and Chemical Characterization of perDAPol
The first and limiting step of the synthesis of perDAPol consisted in producing a perdeuterated PAA precursor whose molar mass be close to that of the perhydrogenated PAA. Perdeuterated PAA was synthesized by free radical telomerization of commercial perdeuterated acrylic acid according to a procedure adapted from that described in ref. (Loubat et al. 2000). Although this method is well established and described in details, a considerable period of development was needed before obtaining the desired perdeuterated PAA, which was achieved on a multi-gram scale. Perdeuterated PAA was then easily converted to perDAPol according to the classic procedure of PAA hydrophobization (Gohon et al. 2004, 2006). The molar mass of the polymer could be deduced from that of the perdeuterated PAA precursor, as determined by SEC. Because of the perdeuteration, however, NMR spectra did not permit to assess whether the extent of octylamine and isopropylamine grafting was exactly that aimed for. The grafting procedure, however, is very reliable and large deviations from the nominal composition highly unlikely.
ESI-IMS-MS spectra of A8-35 and perDAPol cannot ascertain absolute masses, because ionization and detection yields depend on the mass of the polymers, but, used as a comparative method, they do show that the two polymers have the same average mass, which is close to that expected from the mass of the PAA, and very similar mass distributions.
Solution Properties of perDAPol
Above a concentration of ~0.002 g L−1 (Giusti et al. 2012), A8-35 self-assembles in aqueous solutions into small, well-defined globular particles, with an average mass of ~40 kDa, which have been extensively characterized by light scattering, SEC, AUC and small-angle neutron scattering (Gohon et al. 2004, 2006), as well as modeled by molecular dynamics (Perlmutter et al. 2011). As expected from its chemical composition, perDAPol was found to behave, according to SEC, exactly as hydrogenated A8-35. Upon SANS, however, perDAPol particles appeared smaller than previously observed for A8-35 and DAPol (Gohon et al. 2006), for reasons that have not been identified yet but are unlikely to be related to isotopic labeling and may stem from differences in buffer composition. Whatever the case may be, MP trapping with perDAPol proceeded with the same efficacy as with HAPol or DAPol and, according to SEC, yielded MP/APol complexes of the same apparent size and dispersity.
Foreseeable Applications
The development of perDAPol was primarily undertaken in view of NMR applications. In previous work, determination of the structure of BLT2-bound LTB4 was possible using DAPol, because a sufficient number of protons in the ligand have chemical shifts that differ from those of the backbone protons of A8-35 (Catoire et al. 2010a). Nevertheless, the conformation of some regions of the molecule remained less precisely defined, because NOE signals between aliphatic protons of the ligand were masked by those from the APol. This problem would have been crippling for other ligands, such as the heptadecanoid 12-HHT (12S-hydroxyheptadeca-5Z, 8E, 10E-trienoic acid) (Catoire et al. 2011; Hamberg, Svensson and Samuelsson, 1974). For such studies, perDAPol appears as a promising new tool, given the very low intensity of the signals originating from the few remaining 1H nuclei. Indeed, the protonated backbone of DAPol gives rise to intense signals between 0.8 and 2.6 ppm (Fig. 7b), creating important distortions of the baseline of the NOESY spectrum. Baseline corrections carried out after the Fourier transform can affect the volumes of weak ligand cross-peaks that are located close to DAPol ones, which affects the accuracy of peak volume measurements. With perDAPol, no such baseline treatment will be needed.
As regards MP studies, APols signals can interfere with those from α-carbon-linked protons and CHn moieties of amino acid side chains, as illustrated in Fig. 8. The use of perDAPol can be very advantageous, especially when protein 13C signals need to be observed above ~25 ppm (Fig. 7). Among various approaches that emerged during the past 10 years to study large proteins or protein complexes, one of the best strategies is the use of 13CH3 methyl groups immersed in a perdeuterated environment (Plevin and Boisbouvier 2012; Sprangers et al. 2007). Unfortunately, most of the 13CH3 methyl groups in amino acids resonate between 0 and 2.5 ppm and 10 and 30 ppm in the 1H and 13C dimensions, respectively (Fig. 8), i.e., in a region where many signals from the CHn groups of the alkyl chains of surfactants are localized. In the case of methyl-TROSY-based experiments, the use of perDAPol rather than DAPol would not make much of a difference, because in this region, i.e., between 0.6 and 1.4 ppm in the 1H dimension, the remaining 1H signals are more or less the same.
Previous work has shown that the resolution of 2D 1H, 15N TROSY spectra collected on APol-trapped MPs is sufficient for to obtain insights into membrane protein structure and dynamics at least up to the size of BR (27 kDa) (Bazzacco et al. 2012; Catoire et al. 2009, 2010b; Dahmane et al. 2011; Elter et al. 2014; Etzkorn et al. 2013; Planchard et al. 2014; Zoonens et al. 2005). A8-35 permits the identification of most amide protons, but not, however, of those whose exchange with the solvent is too rapid at the slightly alkaline pH (typically, pH 8) that is optimal for use of this APol. Hence, sequence-specific assignment of backbone 1HN, 15N and 13Cα chemical shifts using TROSY-based HNCA (Salzmann et al. 1999) and HNCOCA (Kay et al. 1990) experiments should be realized with MPs trapped in a pH-insensitive APol, such as SAPol (Dahmane et al. 2011) or non-ionic APol (NAPol) (Bazzacco et al. 2012), at least for amide protons located in solvent-exposed loops. Concerning the identification of intramolecular 1H dipolar interactions through 3D 15N-edited and 13C-edited NOESY experiments, the choice of the APol to be used would again depend on how exchangeable the protons investigated are. DAPol and perDAPol would be highly recommended to observe methyl–methyl interactions, for instance, whereas NAPol or SAPol would be needed to observe NOEs involving loop 1HN backbone protons, provided that the NOE cross-peaks are not hidden by NAPol or SAPol signals. This begs the question of SAPol and NAPol perdeuteration. Because their solubility is ensured by sugar residues, NAPol would be extremely costly to perdeuterate. SAPol, on the other hand, could be perdeuterated following the route described here for obtaining perDAPol. A remaining concern is that SAPol, probably because of its high charge density (~75 charges per 100 U), appears less stabilizing than A8-35 or NAPol (Bazzacco et al. 2012; Dahmane et al. 2011; Picard et al. 2006; see however Huynh et al. 2014). It will probably be adequate for many MPs, but a higher stabilizing power could probably be achieved by lowering its charge density to the level of that of A8-35 (i.e., ~35 charges per 100 U). This, however, will require the development of a different synthesis protocol. Phosphocholine-based APols (Diab et al. 2007a, b) could in principle be perdeuterated, but their use for NMR studies remains to be established. For a more detailed discussion of the applications of APols to NMR studies of MPs, see ref. (Planchard et al. 2014).
Deuterated APols have other uses than in NMR studies. DAPol has been used, for instance, to modulate the contrast of APol with the solvent in both AUC and SANS experiments (Gohon et al. 2004, 2006, 2008; Sverzhinsky et al. 2014). The benefit of perdeuteration for AUC experiments would be limited, because the extra contrast afforded over DAPol is small, the expected hydrostatic density of perDAPol being ~1.336 g L−1 versus 1.307 g L−1 for DAPol and 1.236 g L−1 for HAPol (Gohon et al. 2004). On the contrary, the CMP of perDAPol in SANS experiments is ~120.5 % D2O (present work), versus 85.1 and 23.1 % for DAPol and HAPol, respectively (Gohon et al. 2004, 2006). This is potentially highly interesting, because it makes it possible, by mixing perDAPol and HAPol, to achieve a CMP of 100 % D2O, providing ideal conditions of high contrast and low inelastic scattering noise for MP SANS studies. Equally interesting would be the use of perDAPol for INS studies of the dynamics of APol-trapped MPs. This is an important issue, because damping of MP dynamics by APols seems to be involved both in MP stabilization and in some functional effects [for discussions, see refs. (Perlmutter et al. 2014; Popot et al. 2011; Zoonens and Popot 2014)]. Current INS data have been obtained on DAPol particles and MP/DAPol complexes, in which case the hydrogenated backbone of DAPol contributes to the signal (Tehei et al. 2014). The use of perDAPol would make it possible to totally eliminate the contribution of APol protons.
Abbreviations
- 1D, 2D, 3D:
-
One-, two- and three-dimensional, respectively
- A8-35:
-
Sodium poly(acrylate)-based amphipol with a weight-average molar mass close to 8 kDa and containing 35 % of free carboxylate
- A8-75:
-
Sodium poly(acrylate)-based amphipol with a weight-average molar mass close to 8 kDa and containing 75 % of free carboxylate
- AA:
-
Acrylic acid
- AAd4 :
-
Acrylic acid-d4
- AIBN:
-
2,2′-Azoisobutyronitrile
- APol:
-
Amphipol
- AUC:
-
Analytical ultracentrifugation
- CMP:
-
Neutron scattering contrast match point
- C t :
-
Transfer constant
- DAPol:
-
A8-35 with perdeuterated side chains
- DCI:
-
Dicyclohexylcarbodiimide
- DCU:
-
N,N-Dicyclohexylurea
- \(\overline{{DP_{n} }}\) :
-
Average degree of polymerization in number
- dV:
-
Differential viscometry
- Đ:
-
Molar mass dispersity
- ESI:
-
Electrospray ionization
- HAPol:
-
Hydrogenated A8-35
- HSQC:
-
Heteronuclear single quantum correlation
- HOBt:
-
1-N-Hydroxybenzotriazole
- IMS:
-
Ion mobility spectrometry
- INS:
-
Inelastic neutron scattering
- LTB4 :
-
Leukotriene B4
- \(\overline{{M_{n} }}\) :
-
Number-average molar mass
- mQ water:
-
Water purified on a A10 advantage millipore system
- MS:
-
Mass spectrometry
- \(\overline{{M_{w} }}\) :
-
Weight-average molar mass
- NAPol:
-
Non-ionic amphipol
- NMP:
-
N-Methylpyrrolidone
- NOE:
-
Nuclear Overhauser effect
- NOESY:
-
NOE spectroscopy
- PAA:
-
Poly(acrylic acid)
- perDAPol:
-
Perdeuterated A8-35
- PEO:
-
Poly(ethylene oxide)
- PS:
-
Polystyrene
- RI:
-
Refractive index
- R S :
-
Stokes radius
- SANS:
-
Small-angle neutron scattering
- SAPol:
-
Sulfonated amphipol derived from A8-75
- SEC:
-
Size-exclusion chromatography
- TA:
-
Transfer agent
- TGA:
-
Thioglycolic acid
- THF:
-
Tetrahydrofuran
- TMSCHN2 :
-
Trimethylsilyldiazomethane
- WHH:
-
Width at half-height
References
Banères J-L, Popot J-L, Mouillac B (2011) New advances in production and functional folding of G protein-coupled receptors. Trends Biotechnol 29:314–322
Bazzacco P, Billon-Denis E, Sharma KS, Catoire LJ, Mary S, Le Bon C, Point E, Banères J-L, Durand G, Zito F, Pucci B, Popot J-L (2012) Non-ionic homopolymeric amphipols: application to membrane protein folding, cell-free synthesis, and solution NMR. Biochemistry 51:1416–1430
Berry KD, Bailey KM, Beal J, Diawara Y, Funk L, Steve Hicks J, Jones AB, Littrell KC, Pingali SV, Summers PR, Urban VS, Vandergriff DH, Johnson NH, Bradley BJ (2012) Characterization of the neutron detector upgrade to the GP-SANS and Bio-SANS instruments at HFIR. Nucl Instr Meth Phys Res A 693:179–185
Catoire LJ, Zoonens M, van Heijenoort C, Giusti F, Popot J-L, Guittet E (2009) Inter- and intramolecular contacts in a membrane protein/surfactant complex observed by heteronuclear dipole-to-dipole cross-relaxation. J Magn Res 197:91–95
Catoire LJ, Damian M, Giusti F, Martin A, van Heijenoort C, Popot J-L, Guittet E, Banères J-L (2010a) Structure of a GPCR ligand in its receptor-bound state: leukotriene B4 adopts a highly constrained conformation when associated to human BLT2. J Am Chem Soc 132:9049–9057
Catoire LJ, Zoonens M, van Heijenoort C, Giusti F, Guittet E, Popot J-L (2010b) Solution NMR mapping of water-accessible residues in the transmembrane β-barrel of OmpX. Eur Biophys J 39:623–630
Catoire LJ, Damian M, Baaden M, Guittet E, Banères J-L (2011) Electrostatically-driven fast association and perdeuteration allow detection of transferred cross-relaxation for G protein-coupled receptor ligands with equilibrium dissociation constants in the high-to-low nanomolar range. J Biomol NMR 50:191–195
Catoire LJ, Warnet XL, Warschawski DE (2014) Micelles, bicelles, amphipols, nanodiscs, liposomes or intact cells: The hitch-hiker guide to membrane protein study by NMR. In: Mus-Veteau I (ed) Membrane protein production for structural analysis. Springer, Berlin (in press)
Charvolin D, Perez J-B, Rouvière F, Giusti F, Bazzacco P, Abdine A, Rappaport F, Martinez KL, Popot J-L (2009) The use of amphipols as universal molecular adapters to immobilize membrane proteins onto solid supports. Proc Natl Acad Sci USA 106:405–410
Couvreur L, Lefay C, Belleney J, Charleux B, Guerret O, Magnet S (2003) First nitroxide-mediated controlled free-radical polymerization of acrylic acid. Macromolecules 36:8260–8267
Czerski L, Sanders CR (2000) Functionality of a membrane protein in bicelles. Anal Biochem 284:327–333
Dahmane T, Damian M, Mary S, Popot J-L, Banères J-L (2009) Amphipol-assisted in vitro folding of G protein-coupled receptors. Biochemistry 48:6516–6521
Dahmane T, Giusti F, Catoire LJ, Popot J-L (2011) Sulfonated amphipols: synthesis, properties and applications. Biopolymers 95:811–823
Diab C, Tribet C, Gohon Y, Popot J-L, Winnik FM (2007a) Complexation of integral membrane proteins by phosphorylcholine-based amphipols. Biochim Biophys Acta 1768:2737–2747
Diab C, Winnik FM, Tribet C (2007b) Enthalpy of interaction and binding isotherms of non-ionic surfactants onto micellar amphiphilic polymers (amphipols). Langmuir 23:3025–3035
Elter, S, Raschle, T, Arens, S, Gelev, V, Etzkorn, M, Wagner, G (2014). The use of amphipols for NMR structural characterization of 7-TM proteins (submitted for publication)
Etzkorn M, Raschle T, Hagn F, Gelev V, Rice AJ, Walz T, Wagner G (2013) Cell-free expressed bacteriorhodopsin in different soluble membrane mimetics: biophysical properties and NMR accessibility. Structure 21:394–401
Etzkorn, M, Zoonens, M, Catoire, LJ, Popot, J-L, Hiller, S (2014). How amphipols embed membrane proteins: Global solvent accessibility and interaction with a flexible protein terminus. J Membr Biol (in press)
Fernandez, A, Le Bon, C, Baumlin, N, Giusti, F, Crémel, G, Popot, J-L, Bagnard, D (2014) In vivo characterization of the biodistribution profile of amphipols (submitted for publication)
Giusti F, Popot J-L, Tribet C (2012) Well-defined critical association concentration and rapid adsorption at the air/water interface of a short amphiphilic polymer, amphipol A8-35: a study by Förster resonance energy transfer and dynamic surface tension measurements. Langmuir 28:10372–10380
Giusti, F, Kessler, P, Westh Hansen, R, Della Pia, EA, Le Bon, C, Mourier, G, Popot, J-L, Martinez, KL, Zoonens, M (2014). Synthesis of polyhistidine- or imidazole-bearing amphipols and their use for immobilized metal affinity chromatography and surface plasmon resonance studies of membrane proteins (in preparation)
Glück JM, Wittlich M, Feuerstein S, Hoffmann S, Willbold D, Koenig BW (2009) Integral membrane proteins in nanodiscs can be studied by solution NMR spectroscopy. J Am Chem Soc 131:12060–12061
Gohon Y, Pavlov G, Timmins P, Tribet C, Popot J-L, Ebel C (2004) Partial specific volume and solvent interactions of amphipol A8-35. Anal Biochem 334:318–334
Gohon Y, Giusti F, Prata C, Charvolin D, Timmins P, Ebel C, Tribet C, Popot J-L (2006) Well-defined nanoparticles formed by hydrophobic assembly of a short and polydisperse random terpolymer, amphipol A8-35. Langmuir 22:1281–1290
Gohon Y, Dahmane T, Ruigrok R, Schuck P, Charvolin D, Rappaport F, Timmins P, Engelman DM, Tribet C, Popot J-L, Ebel C (2008) Bacteriorhodopsin/amphipol complexes: structural and functional properties. Biophys J 94:3523–3537
Guinier A, Fournet G (1955) Small-angle scattering of X-rays. John Wiley and sons, New York
Hagn F, Etzkorn M, Raschle T, Wagner G (2013) Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J Am Chem Soc 135:1919–1925
Hamberg M, Svensson J, Samuelsson B (1974) Prostaglandin endoperoxides. A new concept concerning the mode of action and release of prostaglandins. Proc Natl Acad Sci USA 71:3824–3828
Hernandez H, Robinson CV (2007) Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat Protocols 2:715–726
Huynh, KW, Cohen, MR, Moiseenkova-Bell, VY (2014). Application of amphipols for structure-functional analysis of TRP channels (submitted for publication)
Kang CB, Li Q (2011) Solution NMR study of integral membrane proteins. Curr Opin Struct Biol 15:560–569
Kanu AB, Dwivedi P, Tam M, Matz L, Hill HH Jr (2008) Ion mobility: mass spectrometry. J Mass Spectrom 43:1–22
Kay LE, Ikura M, Tschudin R, Bax A (1990) Three-dimensional triple resonance NMR spectroscopy of isotopically enriched proteins. J Magn Reson 89:496–514
Kline SR (2006) Reduction and analysis of SANS and USANS data using IGOR Pro. J Appl Crystall 39:895–900
Le Bon, C, Della Pia, EA, Giusti, F, Lloret, N, Zoonens, M, Martinez, KL, Popot, J-L (2014a). Synthesis of an oligonucleotide-derivatized amphipol and its use to trap and immobilize membrane proteins. Nucleic Acids Res (in press)
Le Bon, C, Popot, J-L, Giusti, F (2014b). Labeling and functionalizing amphipols for biological applications. J Membr Biol (in press)
Leney AC, McMorran LM, Radford SE, Ashcroft AE (2012) Amphipathic polymers enable the study of functional membrane proteins in the gas phase. Anal Chem 84:9841–9847
Loubat C, Javidan A, Boutevin B (2000) Etude de la télomérisation de l’acide acrylique par les mercaptans. 1. Télomérisation de l’acide acrylique par l’acide thioglycolique. Influence de la nature du solvant sur la valeur de la constante de transfert et de k p/√k te. Macromol Chem Phys 201:2845–2852
Lynn GW, Heller W, Urban V, Wignall GD, Weiss K, Myles DAA (2006) A dedicated facility for neutron structural biology at Oak Ridge National Laboratory. Physica B 385–386:880–882
Mazhab-Jafari MT, Marshall CB, Stathopulos PB, Kobashigawa Y, Stambolic V, Kay LE, Inagaki F, Ikura M (2013) Membrane-dependent modulation of the mTOR activator Rheb: NMR observations of a GTPase tethered to a lipid-bilayer nanodisc. J Am Chem Soc 135:3367–3370
McGregor C-L, Chen L, Pomroy NC, Hwang P, Go S, Chakrabartty A, Privé GG (2003) Lipopeptide detergents designed for the structural study of membrane proteins. Nat Biotechnol 21:171–176
Opačić, M, Giusti, F, Broos, J, Popot, J-L (2014). Isolation of Escherichia coli mannitol permease, EIImtl, trapped in amphipol A8-35 (submitted for publication)
Pavia AA, Pucci B, Riess JG, Zarrif L (1992) New perfluoroalkyl telomeric non-ionic surfactants: synthesis, physicochemical and biological properties. Makromol Chem 193:2505–2517
Perlmutter JD, Drasler WJ, Xie W, Gao J, Popot J-L, Sachs JN (2011) All-atom and coarse-grained molecular dynamics simulations of a membrane protein stabilizing polymer. Langmuir 27:10523–10537
Perlmutter, JD, Popot, J-L, Sachs, JN (2014). Molecular dynamics simulations of a membrane protein/amphipol complex (submitted for publication)
Picard M, Dahmane T, Garrigos M, Gauron C, Giusti F, le Maire M, Popot J-L, Champeil P (2006) Protective and inhibitory effects of various types of amphipols on the Ca2+-ATPase from sarcoplasmic reticulum: a comparative study. Biochemistry 45:1861–1869
Planchard, N, Point, E, Dahmane, T, Giusti, F, Renault, M, Le Bon, C, Durand, G, Milon, A, Guittet, E, Zoonens, M, Popot, J-L, Catoire, LJ (2014). The use of amphipols for solution NMR studies of membrane proteins: advantages and limitations as compared to other solubilizing media. J Membr Biol (in press)
Plevin, MJ, Boisbouvier J (2012). Isotope-labelling of methyl groups for NMR studies of large proteins. In M Clore and J Potts (eds) Recent developments in biomolecular NMR, Royal Society of Chemistry pp 1–24
Pocanschi C, Popot J-L, Kleinschmidt JH (2013) Folding and stability of outer membrane protein A (OmpA) from Escherichia coli in an amphipathic polymer, amphipol A8-35. Eur Biophys J 42:103–118
Popot J-L (2010) Amphipols, nanodiscs, and fluorinated surfactants: three non-conventional approaches to studying membrane proteins in aqueous solutions. Annu Rev Biochem 79:737–775
Popot J-L, Althoff T, Bagnard D, Banères J-L, Bazzacco P, Billon-Denis E, Catoire LJ, Champeil P, Charvolin D, Cocco MJ, Crémel G, Dahmane T, de la Maza LM, Ebel C, Gabel F, Giusti F, Gohon Y, Goormaghtigh E, Guittet E, Kleinschmidt JH, Kühlbrandt W, Le Bon C, Martinez KL, Picard M, Pucci B, Rappaport F, Sachs JN, Tribet C, van Heijenoort C, Wien F, Zito F, Zoonens M (2011) Amphipols from A to Z. Annu Rev Biophys 40:379–408
Presser A, Hüfner A (2004) Trimethylsilyldiazomethane. A mild and efficient reagent for the methylation of carboxylic acids and alcohols in natural products. Monatsch Chem 135:1015–1022
Privé G (2009) Lipopeptide detergents for membrane protein studies. Curr Opin Struct Biol 19:1–7
Raschle T, Hiller S, Yu TY, Rice AJ, Walz T, Wagner G (2009) Structural and functional characterization of the integral membrane protein VDAC-1 in lipid bilayer nanodiscs. J Am Chem Soc 131:17777–17779
Raschle T, Hiller S, Etzkorn M, Wagner G (2010) Nonmicellar systems for solution NMR spectroscopy of membrane proteins. Curr Opin Struct Biol 20:471–479
Renault M (2008) Etudes structurales et dynamiques de la protéine membranaire KpOmpA par RMN en phase liquide et solide, Ph. D. Thesis, Université Paul Sabatier, Toulouse
Salzmann M, Wider G, Pervushin K, Wüthrich K (1999) Improved sensitivity and coherence selection for [15N,1H]-TROSY elements in triple resonance experiments. J Biomol NMR 15:181–184
Sanders CR II, Landis GC (1995) Reconstitution of membrane proteins into lipid-rich bilayered mixed micelles for NMR studies. Biochemistry 34:4030–4040
Sanders CR, Prosser RS (1998) Bicelles: a model membrane system for all seasons? Structure 6:1227–1234
Shenkarev ZO, Lyukmanova EN, Paramonov AS, Shingarova LN, Chupin VV, Kirpich-ni-kov MP, Blommers MJ, Arseniev AS (2010) Lipid-protein nanodiscs as reference medium in detergent screening for high-resolution NMR studies of integral membrane proteins. J Am Chem Soc 132:5628–5629
Sprangers R, Velyvis A, Kay LE (2007) Solution NMR of supramolecular complexes: providing new insights into function. Nat Meth 4:697–703
Sverzhinsky, A, Qian, S, Yang, L, Allaire, M, Moraes, I, Ma, D, Chung, JW, Zoonens, M, Popot, J-L, Coulton, JW (2014). Amphipol-trapped ExbB—ExbD membrane protein complex from Escherichia coli: A biochemical and structural case study (submitted for publication)
Tehei, M, Giusti, F, Popot, J-L, Zaccai, G (2014). Thermal fluctuations in amphipol A8-35 measured by neutron scattering (submitted for publication)
Tribet C, Audebert R, Popot J-L (1996) Amphipols: polymers that keep membrane proteins soluble in aqueous solutions. Proc Natl Acad Sci USA 93:15047–15050
Tribet C, Diab C, Dahmane T, Zoonens M, Popot J-L, Winnik FM (2009) Thermodynamic characterization of the exchange of detergents and amphipols at the surfaces of integral membrane proteins. Langmuir 25:12623–12634
Tzitzilonis C, Eichmann C, Maslennikov I, Choe S, Riek R (2013) Detergent/nanodisc screening for high-resolution NMR studies of an integral membrane protein containing a cytoplasmic domain. PLoS ONE 8:e54378
Velasquez E, Pembouong G, Rieger J, Stoffelbach F, Boyron O, Charleux B, D’Agosto F, Lansalot M, Dufils P-E, Vinas J (2013) Poly(vinylidene chloride)-based amphiphilic block copolymers. Macromolecules 46:664–673
Warschawski DE, Arnold AA, Beaugrand M, Gravel A, Chartrand E, Marcotte I (2011) Choosing membrane mimetics for NMR structural studies of transmembrane proteins. Biochim Biophys Acta 1808:1957–1974
Weidner SM, Trimpin S (2010) Mass spectrometry of synthetic polymers. Anal Chem 82:4811–4829
Yokomizo T, Kato K, Terawaki K, Izumi T, Shimizu T (2000) A second leukotriene B(4) receptor, BLT2. A new therapeutic target in inflammation and immunological disorders. J Exp Med 192:421–432
Zaccai G, Jacrot B (1983) Small-angle neutron scattering. Annu Rev Biophys Bioeng 12:139–157
Zoonens, M, Popot, J-L (2014). Amphipols for each season (submitted for publication)
Zoonens M, Catoire LJ, Giusti F, Popot J-L (2005) NMR study of a membrane protein in detergent-free aqueous solution. Proc Natl Acad Sci USA 102:8893–8898
Zoonens M, Giusti F, Zito F, Popot J-L (2007) Dynamics of membrane protein/amphipol association studied by Förster resonance energy transfer. Implications for in vitro studies of amphipol-stabilized membrane proteins. Biochemistry 46:10392–10404
Acknowledgments
Particular thanks are due to Alain Fradet (UPMC - CNRS, IPCM) for his support and his comments on the manuscript, to Gaëlle Pembouong and Marion Chenal (same laboratory) for assistance with SEC analyses, to Christophe Tribet (Ecole Normale Supérieure, Paris) for his kind help at interpreting the results of the SEC experiments and to the Biotechnology and Biological Sciences Research Council of the UK for funding for the Synapt HDMS mass spectrometer (BB/E012558/1), ANC (BB/K000659/1) and TGW (BB/K501827/1). This work was supported by the French Centre National de la Recherche Scientifique (CNRS), by Université Paris-7 Denis Diderot, by grant “DYNAMO,” ANR-11-LABX-0011-01 from the French “Initiative d’Excellence” program, by the Office of Biological and Environmental Research, US Department of Energy (Bio-SANS, operated by ORNL’s Center for Structural Molecular Biology) and the Scientific User Facilities Division, Office of Basic Energy Sciences, US Department of Energy (ORNL’s High Flux Isotope Reactor).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Giusti, F., Rieger, J., Catoire, L.J. et al. Synthesis, Characterization and Applications of a Perdeuterated Amphipol. J Membrane Biol 247, 909–924 (2014). https://doi.org/10.1007/s00232-014-9656-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00232-014-9656-x