
Abstract
Current cancer management strategies fail to adequately treat malignancies with multivariable dose-restricting factors such as systemic toxicity and multi-drug resistance limiting therapeutic benefit, quality of life and complete long-term remission rates. The targeted delivery of a therapeutic compound aims to enhance its circulation and cellular uptake, decrease systemic toxicity and improve therapeutic benefit with disease specificity. The transferrin peptide, its receptor and their biological significance, has been widely characterised and vastly relevant when applied to targeting strategies. Utilising knowledge about the physiological function of the transferrin–transferrin receptor complex and the efficiency of its receptor-mediated endocytosis provides rationale to continue the development of transferrin-targeted anticancer modalities. Furthermore, multiple studies report an upregulation in expression of the transferrin receptor on metastatic and drug resistant tumours, highlighting its selectivity to cancer. Due to the increased expression of the transferrin receptor in brain glioma, the successful delivery of anticancer compounds to the tumour site and the ability to cross the blood brain barrier has shown to be an important discovery. Its significance in the development of cancer-specific therapies is shown to be important by direct conjugation and immunotoxin studies which use transferrin and anti-transferrin receptor antibodies as the targeting moiety. Such conjugates have demonstrated enhanced cellular uptake via transferrin-mediated mechanisms and increased selective cytotoxicity in a number of cancer cell lines and tumour xenograft animal models. In addition, incubation of chemotherapy-insensitive cancer cells with transferrin-targeted conjugates in vitro has resulted in a reversal of their drug resistance. Transferrin immunotoxins have also shown similar promise, with a diphtheria toxin mutant covalently bound to transferrin (Tf-CRM107) currently involved in human clinical trials for the treatment of glioblastoma. Despite this, the inability to translate preliminary research into a clinical setting has compelled research into novel targeting strategies including the use of nanoparticulate theory in the design of drug delivery systems. The main objective of this review is to evaluate the importance of the transferrin–transferrin receptor complex as a target for cancer therapy through extensive knowledge of both the physiological and pathological interactions between the complex and different cell types. In addition, this review serves as a summary to date of direct conjugation and immunotoxin studies, with an emphasis on transferrin as an important targeting moiety in the directed delivery of anticancer therapeutic compounds.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transferrin Protein and its Receptor
A 78 kDa-monomeric glycoprotein, transferrin is an important chelator with a primary function of serum iron transportation (Huebers and Finch 1987). It has the capacity to reversibly bind two atoms of ferric iron (Fe3+) with high affinity (1022 M−1 at pH 7.4) (Aisen et al. 1978). The free transferrin peptide (apotransferrin) exhibits a conformational change following iron binding (diferric transferrin or holotransferrin) which has been demonstrated to be significant in its selective recognition by the transferrin receptor (Richardson and Ponka 1997). Richardson and co-workers have found that differic transferrin has a higher affinity (an approximate 10- to 100-fold increase) for the receptor compared to that of apotransferrin in physiological conditions. Transferrin is the main protein in the regulation and distribution of circulating iron, with iron required for various biological processes including DNA synthesis, cellular metabolism and proliferation (Brandsma et al. 2011). Additionally, its importance in the maintenance of systemic and cellular iron homeostasis is highlighted by its ability to be internalised upon binding to a cell surface transferrin receptor (Singh 1999).
The transferrin receptors have been identified in monoclonal antibody studies, and extensively characterised using numerous biochemical techniques (Schneider et al. 1982, 1984; Huebers and Finch 1987). The first transferrin receptor (denoted, TfR1 in most literature) is involved in iron uptake and cell growth regulation (Neckers and Trepel 1986). Its primary structure, elucidated through nucleotide sequencing of complimentary DNA clones, consists of two identical glycosylated subunits with an approximate mass of 95 kDa each linked by two disulphide bonds to form a dimer (McClelland et al. 1984). Each polypeptide subunit (of a length of 760 amino acids) is made up of a short N-terminal cytoplasmic domain, a hydrophobic transmembrane domain and a large, globular extracellular C-terminal domain that contains the binding site for transferrin (Zerial et al. 1986; Daniels et al. 2006a). Since each subunit may bind a transferrin peptide, the receptor has the capacity to internalise up to four ferric ions during one cycle of transferrin-mediated endocytosis. A second transferrin receptor denoted TfR2 has been of recent discovery (Calzolari et al. 2010). It is a homologue of TfR1 displaying 45–66 % similarity (range dependent on literature) in the extracellular domain, with differentiation between the receptors found predominately in tissue distribution (Trinder and Baker 2003; Kawabata et al. 2000). While TfR2 is expressed mainly within the tissues responsible for iron metabolism regulation such as the liver and small intestine, TfR1 is ubiquitously expressed on most active proliferating cell types (Deaglio et al. 2002; Gatter et al. 1983; Jefferies et al. 1984). Interestingly, TfR2 has been shown to have a significantly lower affinity for transferrin (25-fold decrease) in comparison to TfR1, with its expression not correlated to iron levels in cells (Kawabata et al. 2000).
Intracellular Uptake Pathway of Transferrin
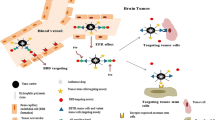
The main mechanism involved in the cellular uptake of iron mediated by transferrin is clathrin-mediated endocytosis. The fate of the transferrin–transferrin receptor complex following endocytosis has been subject to extensive investigation (El Hage Chahine et al. 2012; Luck and Mason 2012; Mayle et al. 2012; Steere et al. 2012). These studies have resulted in the elucidation of two distinct pathways, the first involving the recycling of the complex back to the cell surface, and the second leading to its lysosomal degradation (Mayle et al. 2012) (Fig. 1).

Transferrin receptor-mediated endocytosis. Following the binding of iron-loaded transferrin to its cell-surface receptor, initiation of endocytosis begins with the formation of clathrin-coated pits, and the migration of the complex into the cytoplasm. Subsequently, transferrin indiscriminately enters into two populations of early endosomes that only differ in maturation kinetics: dynamic and static early endosomes. It is within the early endosomes where the dissociation of ferric ions from transferrin occurs due to a high pH within the interior of the endosomal compartment. Furthermore, the transferrin complex is sorted to its corresponding degradation or recycling pathways in the early endosome. Approximately, 85–95 % of the complex follows the recycling pathway, which involves the recycling of the transferrin–transferrin receptor back to the cell surface via either a fast route (direct transportation from the early endosome) or slow route (trafficked to the recycling endosome before transportation to surface). The degradation pathway which accounts for the route taken by approximately 5–15 % of the transferrin receptors has been shown to be a physiological process. Lysosomal degradation occurs through two mechanisms with the first involving the maturation of the early endosome into a late endosome, and the second relating to the recycling endosome. All transferrin receptors eventually follow the degradation pathway for receptor turnover
Following the binding of iron-loaded transferrin to its corresponding receptor, the ligand-receptor complex activates a cascade, believed to be important in the mediation of its specific internalisation via clathrin-mediated endocytosis (Yashunsky et al. 2009). Initiation of endocytosis begins with vesicle budding and formation of clathrin-coated pits on the cell surface (Mayle et al. 2012). Dynamin, an important component of this process is required to complete endocytosis, mediating vesicle separation from the cell membrane and migration to the cytoplasm (Conner and Schmid 2003). Within the cytoplasm, the clathrin coat is rapidly removed by adenosine triphosphate (ATP)-dependent uncoating enzymes for reuse by the cell (Ciechanover et al. 1983; Dautry Varsat et al. 1983).
Subsequently, transferrin indiscriminately enters into two populations of early endosomes that differ only in maturation kinetics. Studies by Lakadamyali et al. 2006, established the presence of these two distinct populations—dynamic and static. Identification of static and dynamic endosomes using real-time live-cell fluorescent imaging and Rab GTPase markers suggest that these endosomes exhibit either slow or fast maturation kinetics, respectively. Rab5, a Rab GTPase specifically associated with early endosomes, was fluorescently labelled in order to visualise the trafficking of numerous ligands including transferrin. Results indicate that a small fraction of early endosomes (Rab5-positive; dynamic endosomes) mature rapidly into late endosomes (observed through the acquisition of late endosome-specific marker, Rab7) within ~30 s of formation. Conversely, the majority of the early endosome population (or static population) fails to acquire Rab7 within 100 s of formation. The sorting process for directing the transferrin-bound receptor to either of these populations is believed to originate at the cell surface, and be indiscriminate, with no preference for static or dynamic endosomes. Despite this, transferrin naturally becomes enriched in the static early endosome population due to the greater number of such endosomes as compared to their dynamic counterparts. The mechanism by which ligand sorting occurs at the cell surface, and within the intracellular compartments, remains largely unclear. Dissociation of ferric ions from transferrin occurs within the interior of the endosomal compartment, which is maintained at a pH of ~5–5.5 by V-type ATPases that pump protons into the lumen from the cytoplasm (El Hage Chahine et al. 2012). These ions are then transferred to the cytoplasm by a process involving reduction to the ferrous state. At the low pH of the endosome, the transferrin receptor retains a high affinity for the iron-free transferrin.
Within the early endosome, the transferrin complex is further sorted to its corresponding degradation or recycling pathways. Approximately 85–95 % of the complex follows the recycling monensin-resistant pathway (Stein and Sussman 1986; Jin and Snider 1993),which involves the recycling of the internalised transferrin–transferrin receptor back to the cell surface through either a fast route (direct transportation back to the plasma membrane), or slow route (trafficked to the recycling endosome before returning to the surface). The alternative degradation monensin-sensitive pathway, involving the transport of ~5–15 % of the complex, is not yet completely understood (Jin and Snider 1993).
The recycling pathway from early endosomes has been found to be mediated by various GTPases and their regulators, which in turn determines the route in which the transferrin receptor complex is recycled back to the cell surface. Specifically, Rab4 is important in the regulation of the transferrin receptor cycle by mediating the trafficking pathway (McCaffrey et al. 2001). Overexpression of Rab4 was shown to cause an increase in the delivery of the transferrin receptor complex to the recycling endosome (slow recycling pathway). The fast recycling pathway from the early endosome, which involves the bypassing of the endocytic recycling compartment and the direct transport of the complex to the cell surface, has been shown to be mediated by Rab8 and Rab35. Conversely, Rab11 has been found to be responsible for directing transferrin through the slow recycling pathway, delivering the complex to the endocytic recycling compartment from early endosomes. Following transport (either by the fast or slow recycling pathway) to the neutral environment of the cell surface (pH 7.4), the receptor affinity for unbound transferrin is significantly decreased (~50-fold) (Dautry Varsat et al. 1983). Dissociation of iron-free transferrin from the receptor occurs rapidly, with the unbound receptor subsequently available to repeat another cycle of endocytosis (Klausner et al. 1983a, b). Clathrin-mediated endocytosis and the recycling pathway of the transferrin–transferrin receptor complex is a rapid and efficient process, with reports estimating the mean transit time to be in the order of 10–20 min (Bleil and Bretscher 1982; Hopkins and Trowbridge 1983).
The small proportion of transferrin receptors that are segregated from the recycling pathway and directed to the lysosomal pathway is likely due to its relatively long half-life (19 ± 6 h) (Rutledge et al. 1991). This pathway is thought to be physiological however, with all receptors eventually transported via this pathway for receptor turnover (Mayle et al. 2012). To demonstrate, treatment of mouse embryonic fibroblast cells with cycloheximide, an inhibitor of protein synthesis causes a significant decrease in receptor signalling (Matsui and Fukuda 2011; Matsui et al. 2011). Conversely, treatment of these cells with an inhibitor of lysosomal degradation (bafilomycin A1) results in a significant increase in transferrin signalling. It is proposed that the degradation of the transferrin receptor, not yet fully elucidated, consists of two distinct pathways that the cell may use dependent on its condition. The first is thought to be induced by an increase in the cellular uptake of iron under selective conditions (mainly, pathological) and based around the maturation of the early endosome into a late endosome. It is from here that the receptor is transported to the lysosome for degradation in an attempt to reduce the amount of iron within the cell. The second, thought to occur in physiological conditions has been shown to be regulated by the GTPase, Rab12. Overexpression of a constitutively active mutant of Rab12 in mouse embryonic fibroblast cells causes a reduction in the amount of transferrin receptor protein. In contrast, cells with a complete knockdown of functional Rab12 causes an increase in the amount of protein. It has been also found that Rab12 is colocalised in transferrin-positive recycling endosomes and partially in lysosomes, but is absent within early and late endosomes. Collectively, these results indicate that Rab12 regulates the degradation of the transferrin receptor in a distinct pathway to that of conventional mechanisms, based on the trafficking of the receptor complex from the recycling endosome to the lysosome.
Cancer Specificity of the Transferrin Receptor and Transferrin-Mediated Endocytosis
Transferrin Receptor Expression Under Physiological Conditions
The transferrin receptor is ubiquitously expressed on a variety of normal tissue reflecting the cellular metabolic requirements for iron (Gatter et al. 1983). Gatter and co-workers observed low expression levels on nonproliferating cells, including vascular endothelial cells of the brain capillaries, hepatocytes, Kupffer cells of the liver, and cells of the endocrine pancreas, seminiferous tubules of the testes, cells of the pituitary gland, luminal membranes of the breast and tubules of the kidney. Correlation between receptor expression and the requirement for increased cellular proliferation has also been demonstrated with the transferrin receptor expressed at greater levels on cells with a high proliferation rate including those of the basal layer of the skin, the endothelium of brain capillaries and the crypts of the intestinal villi (Gatter et al. 1983; Jefferies et al. 1984; Trowbridge and Omary 1981; Sutherland et al. 1981). Activated peripheral blood mononuclear cells also express high levels of the receptor (Woith et al. 1993). Additionally, although results found no transferrin receptors on normal circulating lymphocytes, stimulation of these cells to proliferate by mitogenic plant lectins led to the expression of transferrin receptors in order to accommodate for the increased iron requirements (Hammarstrom et al. 1982). During foetal development where proliferation is essential, transferrin receptors are highly expressed on cells that require large amounts of iron, including those of the placental trophoblasts which are responsible for the delivery of iron to the foetus, and maturing erythroid cells which require iron for heme synthesis (Galbraith et al. 1980; Sieff et al. 1982). Interestingly, normal mature erythroid cells have been found to not express transferrin receptor (Calzolari et al. 2004).
The expression of transferrin receptors has been shown to be regulated by the availability of iron. Studies in cell culture reveal that a negative correlation between iron availability and receptor expression exists (Mattia et al. 1984; Bridges and Cudkowicz 1984; Ward et al. 1984). In detail, in the presence of a permeable iron chelator, desferrioxamine, transferrin receptor expression is increased 2- to 5-fold (Mattia et al. 1984; Bridges and Cudkowicz 1984). Conversely, in the presence of exogenous iron, such as hemin or ferric ammonium citrate, a significant decrease in the number of transferrin receptors ensues (Ward et al. 1984; Rouault et al. 1985). It has been suggested that this regulation may be achieved by the ability of increased iron within the cellular stores to downregulate the expression of transferrin receptors by destabilisation of the receptor mRNA (Ward et al. 1984; Rouault et al. 1985).
The Upregulation of Transferrin Receptor in Cancer
Multiple studies have shown an upregulation in expression of the transferrin receptor on metastatic and drug resistant tumours when compared to their normal counterparts, including those of the pancreas, colon, lung and bladder (Ryschich et al. 2004; Calzolari et al. 2007; Prutki et al. 2006; Kondo et al. 1990; Seymour et al. 1987). For example, Singh et al. 2011 evaluated the expression of the transferrin receptor in a spectrum of normal to malignant breast tissue samples to observe the association between overexpression and malignant transformation. It was shown that normal and benign lesions had significantly lower expression compared with premalignant lesions and invasive carcinoma. Interestingly, the highest expression of the receptor was found in the more aggressive phentoypes of breast cancer, including high-grade ductal carcinoma in situ and low oestrogen receptor positive-tumours). It is hypothesised that transferrin overexpression in cancer may be attributed to the increased iron requirement, with iron an important cofactor of the ribonucleotide reductase enzyme involved in the synthesis of DNA in rapidly dividing cells (Daniels et al. 2006a). In addition, the transferrin receptor displays a high turnover on tumour cells due to their increased iron consumption (Hopkins and Trowbridge 1983).
Correlation between increased transferrin receptor expression and the level of malignancy has also been determined in a range of cancer cases, including those patients with bladder transitional cell carcinoma, breast cancer, glioma, lung adenocarcinoma, chronic lymphocytic leukaemia and non-Hodgkin’s lymphoma (Habeshaw et al. 1983; Kondo et al. 1990; Singh et al. 2011; Seymour et al. 1987; Prior et al. 1990; Das Gupta and Shah 1990). Specifically, high expression of transferrin receptor in a number of tumours demonstrate a higher rate of recurrence than those with low receptor expression (Seymour et al. 1987), observe a higher degree of histopathologic differentiation (Kondo et al. 1990), and/or results in a poorer prognosis (Wrba et al. 1986). Increased transferrin receptor expression has also been detected on peripheral blood mononuclear cells from patients with lymphoma, myeloma or leukaemia in comparison to those taken from healthy subjects (Yeh et al. 1984). Significance of the transferrin receptor and its associated pathways in targeting strategies.
As cancer therapy becomes progressively more specific with the discovery of biological targets that are either uniquely expressed or exhibit an upregulated expression pattern on tumour cells, current research strategies aim to exploit the differences between malignant and normal cells. To date, the transferrin receptor has been the most extensively studied cellular target in anticancer research. Its significance may be explained by its physiological nature and role in cancer.
Described previously, the expression of the transferrin receptor is significantly upregulated in cancer, increasing several hundred-fold in various tumour cell lines and malignant tissue (Hamilton et al. 1979; Galbraith et al. 1980; Trowbridge and Omary 1981; Faulk et al. 1980; Schulman et al. 1981; Panaccio et al. 1987). Although the receptor has an established physiological function within the tissue, its aberrant stimulation and overexpression in cancer allows for its use as a recognition structure for the active targeting of tumour cells (Daniels et al. 2006a). Furthermore, transferrin-mediated endocytosis and the subsequent recycling pathway are highly efficient, with the entire cycle complete in 10 min on average (Dautry Varsat et al. 1983; Klausner et al. 1983a). Interestingly, it has been observed that this cycle may only take 4–5 min to complete in the K562 cancer cell line (Klausner et al. 1983a). This rapid recycling leads to high turnover rates, with reports suggesting that due to the number of receptors expressed on cells (over 150,000 on K562 cells) in conjunction with rapid endocytosis, approximately 2x104 transferrin molecules may be internalised per minute per cell (Ciechanover et al. 1983). It is thus hypothesised that targeting the transferrin receptor through its endocytic mechanisms for the delivery of anticancer compounds will enable efficient and selective uptake, enhanced therapeutic cellular concentrations and ultimately, increased drug efficacy in malignant cells.
Its function in iron transportation and importance in cellular growth and proliferation also allows for the capacity to directly inhibit the action of the receptor through the antagonistic properties of monoclonal antibodies (Taetle et al. 1986). A number of studies have shown the capacity of anti-transferrin receptor antibodies to limit cell growth in cultured, normal and malignant cells (Mendelsohn et al. 1983; Taetle et al. 1983; Trowbridge and Lopez 1982). The use of anti-transferrin antibodies in targeted drug delivery strategies, mainly for the delivery of therapeutics across the blood brain barrier, has also been reported (Friden et al. 1991; Ulbrich et al. 2009; Paris-Robidas et al. 2011). For example, OX26 is a mouse monoclonal antibody against rat TfR1 which has been explored as a targeting moiety for the delivery of therapeutics into the brain (Friden et al. 1991). Friden and co-workers, observed the potential of OX26 conjugation to the hydrophilic anticancer drug methotrexate by a hydrozone bond (six molecules of methotrexate were attached per antibody). Following intravenous administration to rats, the conjugate was shown to cross the blood brain barrier via transferrin receptor-mediated transcytosis, and had the capacity to deliver the drug to the brain parenchyma. By targeting the receptor with its antibody as the delivery method for anticancer therapies, the capacity to selectively treat malignancies and decrease cellular growth due to their own inhibitory effects, is a significant concept which requires further investigation.
Due to the high expression of transferrin receptors on the luminal membrane of brain endothelial cells, the delivery of therapeutics into the brain via transferrin-mediated transcytosis has been widely investigated (Dufes et al. 2013). It has been shown that transferrin is the major peptide involved in the transportation of iron from the blood into the brain (Burdo et al. 2003). The mechanisms involved in the subsequent release of iron from the transferrin-transferrin receptor complex and its subsequent delivery to brain cells remains to be elucidated, although it has been shown that there is an increase in transferrin receptor expression on malignant brain tissue (Recht et al. 1990). Multiple studies have shown the capacity of transferrin and anti-transferrin receptor antibodies to cross the blood brain barrier (Descamps et al. 1996; Ulbrich et al. 2009; Friden et al. 1991). The inability of many free drugs to cross the blood brain barrier is a major limitation in the treatment of many brain disorders, including glioma (Dufes et al. 2013). Coupled with the knowledge that the transferrin complex may cross the blood brain barrier, numerous delivery strategies including direct linkage of therapeutic compound to transferrin/anti-transferrin receptor antibody, and/or encapsulation in transferrin-modified carriers have been designed to overcome such an inadequacy.
The development of multi-drug resistance (MDR) following chronic administration of chemotherapy is a common phenomenon in cancer. There are two mechanisms of resistance: (1) the impaired delivery of anticancer drug to tumour cells, and (2) the genetic and epigenetic alterations in malignant cells that affect their drug sensitivity (Gottesman et al. 2002). The active targeting of cancer with transferrin has been shown to have the capacity to overcome these two resistance mechanisms. Impaired drug delivery may result from poor biodistribution, increased metabolism and elimination, which results in low drug concentrations in the blood and therefore, a reduced amount at the tumour site (Jain 2001). Through the efficiency of transferrin-mediated endocytosis, enhanced delivery of compounds into tumour cells has been demonstrated in multiple studies (Chang et al. 2012; Yoon et al. 2009; Nam et al. 2013; Wu et al. 2007; Chiu et al. 2006; Liu et al. 2013). For example, transferrin-targeted PLGA nanoparticles have been observed to internalise into F98 glioma cells via both transferrin-mediated caveolae- and clathrin-dependent endocytosis (Chang et al. 2012). Most interesting was the finding that transferrin conjugation significantly increased nanoparticle stability and accumulation within brain tissue in vivo as compared to their nontargeted (BSA conjugated) counterparts. In another study, Liu et al. 2013 engineered a transferrin-modified PEG-PLA nanoparticle for the encapsulation of doxorubicin. Results indicated a 2.07-fold increase in intracellular drug concentrations when C6 glioma cells were incubated with the targeted nanoparticles compared to a nontargeted formulation. In addition, biodistribution and inhibition of tumour growth was demonstrated to be significantly higher following administration of transferrin-modified nanoparticles as compared to free doxorubicin and the nontargeted carrier in vivo. An innate insensitivity to chemotherapeutic compounds in tumour cells through genetic mutations and epigenetic alterations is a major limitation in current cancer management strategies. The capacity to evade immune response and cell signalling, through the conjugation or encapsulation of drug for effective delivery within tumour cells is of high importance. By exploiting knowledge of the transferrin receptor cycle, active targeting with transferrin has shown to be highly effective in reversing MDR (Chiu et al. 2006; Wu et al. 2007). A transferrin-conjugated liposomal formulation encapsulating doxorubicin and verapamil has been studied to determine its capacity to sensitise doxorubicin-resistant K562 cells to treatment (Wu et al. 2007). Incubation with the targeted liposomes demonstrated its high specificity to K562 cells as compared to free drugs and nontargeted formulation. The most important finding, however, was the capability of the transferrin-modified liposomal system to overcome drug resistance due to an increase of time in circulation, and internalisation via the transferrin receptor.
Transferrin-Conjugated and -Targeted Approaches for Cancer Therapy: Current Insight
Direct Conjugation of Transferrin and its Associated Receptor Antibodies to Anticancer Drugs and the Development of Immunotoxins
Targeting allows for enhanced drug circulation time and cellular uptake, decreased systemic toxicity, and improved cellular targeting in order to effectively deliver therapeutic compounds to disease site (Kim et al. 2013). Although challenging, conjugation with a variety of ligands, such as peptides, lipids, polymers and other small molecules through covalent or noncovalent binding may impart these desired properties (Mout et al. 2012). The two main mechanisms used for the formulation of targeted therapeutic strategies are based on either passive or active processes. While passive targeting relies on the size and physical properties of the delivery system to target the tumour microenvironment, active targeting requires covalent linking with targeting moieties that bind to a specific antigen and/or receptor on the tumour cell surface. Given the significant role of the transferrin receptor cycle in cell growth and the overexpression of the receptor on tumour cells, the ability to successfully deliver chemotherapeutic compounds via transferrin-mediated endocytosis would theoretically lead to malignant cell death (Mendelsohn and Baselga 2000; Krenning et al. 1993; Kersten et al. 2000; Ferrara et al. 2005; Hicklin and Ellis 2005).
The direct conjugation of transferrin to chemotherapeutic compounds has been widely investigated for their selective and enhanced delivery into malignant cells (Table 1). Perhaps the greatest example, and the most extensively studied is the doxorubicin–transferrin conjugate (Faulk et al. 1990; Berczi et al. 1993; Faulk et al. 1991; Munns et al. 1998; Singh et al. 1998). Doxorubicin, an anthracyclin antibiotic is used in the treatment of a variety of cancers including, breast, ovarian, sarcomas, lymphomas and acute leukaemias (Speth et al. 1988). Despite the capacity to treat such a range of malignancies, the administration of doxorubicin is dose-limiting with the drug found to accumulate in the heart causing cardiotoxicity (Carvalho et al. 2013). Thus, the ability to target its delivery to the tumour site becomes increasingly important in order to enhance its therapeutic index. The covalent linkage of transferrin to doxorubicin yields a conjugate that exhibits cytotoxic effects in multiple cancer cell lines including K562 (human myelogenous leukaemia), HL-60 (human promyelocytic), MCF-7 (breast cancer), HeLa (human cervical cancer) and Hep2 (human liver carcinoma) cell lines (Faulk et al. 1991; Berczi et al. 1993; Lemieux and Page 1994; Singh et al. 1998; Sun et al. 1992). Dependent on the cell line, the doxorubicin–transferrin conjugate has been observed to enhance cytotoxicity by 3- to 10-fold compared to free doxorubicin (Daniels et al. 2006b). Kratz and co-workers have also indicated that conjugation of transferrin to doxorubicin also displays cytotoxic effects in MDA-MB-468 (breast cancer), LXFL-529 (lung carcinoma) and U937 (leukaemia) cell lines (Kratz et al. 1998). Most interesting, however, was the finding that doxorubicin–transferrin conjugates were significantly less cytotoxic than free doxorubicin in human umbilical vein endothelial cells (HUVEC). Selectivity to tumour cells (to those overexpressing the transferrin receptor) has thus been demonstrated, with the inability of the conjugate to accumulate within normal cells also demonstrated in normal human fibroblasts (SBL3F cell line) (Lubgan et al. 2009).
Multiple studies have indicated the capacity to overcome drug resistance with the administration of doxorubicin using a direct transferrin conjugation for enhanced cellular uptake (Berczi et al. 1993; Lemieux and Page 1994; Singh et al. 1998; Wang et al. 2000; Lubgan et al. 2009; Szwed et al. 2013). Specifically, drug resistance has been demonstrated to be overcome in various cancer cell lines in vitro, including HL-60, K562 and MCF-7 cells (Berczi et al. 1993; Lemieux and Page 1994). Modification of the transferrin–doxorubicin conjugate through its saturation with antineoplastic drug gallium nitrate has also shown important properties for the reversal of drug resistance (Wang et al. 2000). Administration of the transferrin–gallium–doxorubicin conjugate to doxorubicin-sensitive MCF-7 cells displayed similar inhibitory effects to free drug. Resistance was shown to be overcome in the MDR MCF-7 cell line, with a decrease in the IC50 (concentration required to inhibit 50 % of cell growth) by approximately 100-fold and a decrease in the expression of multidrug resistance protein. This reversal of resistance may be due to the ability of the conjugate to enter cells through transferrin-mediated mechanisms, with the cell unable to recognise doxorubicin in its conjugated state. Wang and co-workers showed that free doxorubicin accumulated in the cytoplasm of drug-resistant cells for its eventual efflux. Conjugation with transferrin and gallium allowed for the accumulation of drug into the nucleus of resistant cells, eliciting therapeutic effects such as cell growth inhibition, and ultimately cell death.
The systemic administration of doxorubicin–transferrin conjugates into in vivo models of cancer has also produced promising results (Kratz et al. 2000; Singh et al. 1998). The conjugate was tested against nude mice bearing mesothelioma tumours in their peritoneal cavity, with results indicating its therapeutic benefit (Singh et al. 1998). Survival was prolonged in mice given the conjugate compared to those mice injected with the free drug. Additionally, an acid-sensitive transferrin and albumin doxorubicin conjugate was evaluated in a xenograft mamma carcinoma model MDA-MB-435 in comparison to free doxorubicin (Kratz et al. 2000). The conjugate showed significantly reduced systemic toxicity (reduced lethality and body weight loss) with similar or slightly improved antitumour activity compared to free drug. At a dose of 12 mg/kg, mortality in the free doxorubicin-treatment group was ~80 %, with no mortality observed within the conjugate-treated group. At a concentration of 3 × 12 mg/kg, the conjugate was shown to significantly improve antitumour activity compared to free doxorubicin (given at its optimal dose of 2 × 8 mg/kg). This finding showed that the conjugate may be administered at a larger dose than free doxorubicin, through its transferrin-targeting capacity and avoidance of normal cell toxicity.
Delivery of other therapeutic compounds via transferrin-mediated endocytosis using their direct conjugation with transferrin has been attempted in order to enhance their cellular uptake and increase their therapeutic benefit (Hoshino et al. 1995; Karagiannis et al. 2006b; Jiang et al. 2007; Kim et al. 2012). Cisplatin, a platinum-based alkylating agent, which inhibits DNA synthesis and is used in the clinical treatment of metastatic testicular and ovarian cancer, as well as advanced bladder carcinoma (Daniels et al. 2006b). The chemical conjugation of cisplatin to transferrin has been shown to enhance its anticancer activity in A431 (human epidermoid cancer), MCF-7 and HeLa cell lines (Head et al. 1997; Hoshino et al. 1995). An in vivo study using mice inoculated with B16 melanoma cells demonstrated prolonged circulation, and enhanced cell growth inhibition in comparison to free drug (Hoshino et al. 1995). The cisplatin–transferrin conjugate is currently undergoing human clinical trials (MPTC-63) (Head et al. 1997). A phase I clinical trial resulted in a 36 % (4 out of 11 patients) response rate in breast cancer patients with advanced disease, including one complete response to MPTC-63 and minor adverse effects observed. This same clinical trial also showed that 87.5 % (7 out of 8 patients) of patients had a partial response to MPTC-63 in combination with the iron chelator deferoxamine mesylate. Most recently, a case study using a cisplatin-transferrin complex (targeted chemotherapy) in combination with vaccine immunotherapy in a female with stage IV breast cancer showed that treatment resulted in complete remission of malignant ascites (Elliott and Head 2006).
The specific delivery using transferrin as the targeting moiety, of a iodinated DNA minor groove-binding bibenzimidazole, previously shown to sensitise cells to UVA irradiation (Martin et al. 1990; Karagiannis et al. 2006a), demonstrates the ability to exploit transferrin-mediated mechanisms (Karagiannis et al. 2006b). Acid-labile hydrazone-linked conjugate proved to be the most efficient in mediating UVA-induced phototoxicity in the K562 cell line. These results imply binding of the conjugate to cell-surface receptors, internalisation and degradation of the conjugate-receptor complex, with release and translocation of the ligand to nuclear DNA. Coincubation with excess native transferrin led to inhibition of ligand-mediated phototoxicity, indicating that cellular uptake of the conjugate is mediated by the transferrin receptor with specificity.
In addition, successful conjugation of cytokines including tumour necrosis factor-α (TNF-α) and TNF-related apoptosis-inducing ligand (TRAIL) to transferrin have allowed for their selective delivery to the site of the tumour (Jiang et al. 2007; Kim et al. 2012). Jiang et al. 2007 showed that conjugation of TNF-α to PEGylated, thiolated transferrin possessed a similar affinity to the transferrin receptor to that of native transferrin. It remained in circulation significantly longer than free TNF-α, with higher intratumoral TNF-α levels following administration to the S180 sarcoma mouse model. In addition, tumour growth was significantly inhibited. Similarly, the PEGylated transferrin-TRAIL conjugate demonstrated enhanced biodistribution patterns and increased antitumour effects in mice bearing B16F10 murine melanomas or HCT116 colon cancer (Kim et al. 2012). Tumour accumulation of conjugate was increased 5.2-fold than free TRAIL, with the suppression of tumour growth 3.6-fold higher.
The development of immunotoxins, a chimeric protein consisting of a targeting moiety covalently linked to a toxin, has aimed to eliminate cancer cells with specificity (Madhumathi and Verma 2012). A number of toxic compounds have been used in such research, with the induction of cell death caused by either their interference with components involved in cellular processes, their capacity to modify the cell membrane, or through the induction of apoptotic protein expression (Shapira and Benhar 2010). Conjugation with transferrin or an anti-transferrin receptor antibody has allowed for tumour-specific targeting. There are two main groups of toxins used in immunotoxin development strategies: plant and bacterial toxins (Choudhary et al. 2011).
Plant toxins, including ricin and saporin, are known as ribosome inactivating proteins and have been shown to inactivate ribosomal activity by cleaving the N-glycosidic bond of the adenine residue in 28S ribosomal RNA (rRNA) causing protein synthesis inhibition (Choudhary et al. 2011). Conjugation of plant toxins with transferrin or anti-transferrin receptor antibody allows for the cellular uptake of the toxin via transferrin receptor-mediated pathways (Raso and Basala 1984; Martell et al. 1993; Bergamaschi et al. 1988; Ippoliti et al. 1995) (Table 2). Martell and co-workers demonstrated a positive correlation between transferrin receptor expression and sensitivity to an anti-transferrin receptor antibody-ricin immunotoxin (Martell et al. 1993). Successful uptake was also shown with the ricin immunotoxin observed to accumulate within the cytoplasm, and a transferrin–saporin conjugate delivered to the endosome (Martell et al. 1993; Ippoliti et al. 1995). Increased protein synthesis inhibition and enhanced cytotoxicity has also been reported in numerous studies with both ricin and saporin transferrin-targeted immunotoxins in multiple cancer cell lines (Trowbridge and Domingo 1981; Raso and Basala 1984; Martell et al. 1993; Daniels et al. 2007). For example, ricin was conjugated to human transferrin via a disulphide bond and subsequently tested in a human leukaemia (CEM) cell line (Raso and Basala 1984). The immunotoxin displayed enhanced cytotoxicity, with a 10,000-fold increase when compared to free ricin. Furthermore, following coincubation with native transferrin, this cytotoxic activity was lost indicating selectivity and transferrin-mediated cellular uptake. Incubation of saporin-sensitive and -resistant malignant lymphoblast cells with a chimeric human anti-transferrin receptor antibody–saporin conjugate resulted in enhanced cytotoxic effects in sensitive cells, and a reversal of resistance in saporin-insensitive cells in comparison to free toxin (Daniels et al. 2007). In vivo studies of ricin immunotoxin formulations have shown promise, with inhibition of cell growth in murine models of both human melanoma (M21) and glioblastoma (U251) (Trowbridge and Domingo 1981; Laske et al. 1994). A phase I human clinical trial of a ricin immunotoxin (conjugated to the monoclonal antibody, 454A12) in patients with leptomeningeal spread of systemic neoplasia found that following administration four out of the eight patients responded to treatment, with a 50 % or greater reduction of tumour cell counts in their lumbar cerebrospinal fluid (Laske et al. 1997b).
Pseudomonas exotoxin and diphtheria toxin are the most commonly used bacterial toxins in anticancer therapeutic research though their capacity to inhibit protein synthesis by inactivating elongation factor-2 (EF-2) through ADP ribosylation (Madhumathi and Verma 2012). The selective targeting of transferrin-expressing cancer cells through the direct conjugation of bacterial toxins with transferrin and its associated receptor antibodies compared to free toxin, has been demonstrated in various cancer cell lines including human ovarian cancer (A1847), breast cancer (MCF-7) and cervical carcinoma (HeLa) (Pirker et al. 1985, 1988; Trowbridge and Domingo 1981). Interestingly, metastatic and aggressive malignant tumour cells are extremely sensitive to bacterial immunotoxins due to an increase in transferrin receptor expression (Shinohara et al. 2000; Martell et al. 1993). O’Keefe and Draper 1985 report the potential of a diphtheria toxin–transferrin conjugate when administered to thymidine kinase-deficient mouse L cells. A reduction in protein synthesis by 50 % was observed 24 h post administration, with selectivity indicated through an inhibition of cytotoxic activity following transferrin or anti-transferrin receptor antibody treatment. Furthermore, enhanced cytotoxicity and increased survival of a murine xenograft model of human mesothelioma (H-MESO-1) following treatment with a transferrin–diphtheria toxin mutant (CRM107) conjugate was shown in conjunction with doxorubicin administration (Griffin et al. 1992).
The most important immunotoxin to date is the chemical conjugate transferrin-CRM107 (Tf-CRM107) which has entered clinical trials following initial success in preliminary studies. CRM107 is a mutant form of diphtheria toxin that contains two amino acid mutations in the B chain which results in its inability to bind to the cell surface (Johnson et al. 1989). Phase I clinical trials with Tf-CRM107 involving its delivery by intratumoral infusion to patients with malignant brain tumours were conducted with promising results (Laske et al. 1997a, b). Nine out of fifteen (60 %) patients who could be evaluated responded to treatment with at least a 50 % reduction in tumour volume measured by magnetic resonance imaging (MRI). Two patients displayed a complete response, with one having no tumour for 23 months post treatment. Administration of the drug (<1.0 μg/mL) did not cause any local toxicity in patients while anti-tumour activity was present. At high doses, however, Tf-CRM107 was shown to cause local toxicity in all patients treated. Following these promising results, phase II clinical trials were conducted for the treatment of refractory and recurrent glioblastoma multiforme or anaplastic astrocytoma (Weaver and Laske 2003). Following two infusions, five patients completely responded to treatment, seven partially responded and nine displayed stabilised disease in a cohort of 34 evaluable patients. Symptomatic progressive cerebral oedema resulted in eight out of the 44 patients enrolled, and seizures were observed in three patients who responded to anticonvulsant therapy. Phase III clinical trials were initiated, however, one was withdrawn prior to recruitment of patients due to probability analysis determining that it was unlikely that Tf-CRM107 would meet FDA criteria for efficacy (NCT00083447; clinicaltrials.gov). Another phase III study comparing Tf-CRM107 with standard treatment of glioblastoma was completed in 2008, however, results have not been released (NCT00088400; clinicaltrials.gov). A phase I trial involving Tf-CRM107 in children with progressive or recurrent glioblastoma multiforme or anaplastic astrocytoma was also initiated, however, the status in unknown due to unverified information (NCT00052624; clinicaltrials.gov).
Despite such promise, limitations have been observed in direct conjugation and immunotoxin studies. A limitation of using naturally occurring peptides, including transferrin is its innate sensitivity to pH and enzymatic degradation, which has prompted studies investigating the design of more stable analogues, and synthesis of evasive nanoparticulate delivery systems to prolong their circulating half-life in order to optimise formulations for clinically-relevant applications. Furthermore, the inability to conjugate a significant amount of therapeutic compound to the transferrin peptide itself also serves as a significant limitation (Karagiannis et al. 2006b). Enhanced delivery, and ultimately drug efficacy, is therefore hindered by the inability to deliver high concentrations of drug into the target cell in one cycle of transferrin-mediated endocytosis. Due to these limitations, a novel way in which to deliver therapeutic compounds to the disease site was required. Through the ability to modify the surface of nanoparticulate systems with targeted moieties including transferrin, and the capacity to encapsulate high concentrations of therapeutic compound within such a carrier, nano-based drug delivery has shown the most promise to date.
Transferrin-Targeted Nano-Based Systems for the Delivery of Therapeutic Compounds to Tumour Cells
The rapidly evolving and expanding discipline of nanotechnology is a science of engineering material and systems on a molecular scale. Due to their unique size-dependent physical and chemical properties (Whitesides 2003), and the exploitable nature of each property, development of functional nanoparticles may be designed for a range of applications. In cancer biology, nanoparticulate theory is expected to be useful for various therapeutic and diagnostic strategies, with nanoparticles in the size range of 1–200 nm demonstrating important and often unique interactions with biological systems (Jiang et al. 2008). Limitations in current cancer treatment strategies including systemic toxicity and MDR, coupled with varied results in studies involving direct conjugation studies and bare nanoparticles, provide a basis for the development of surface-modified, disease-targeted nanocarriers.
Surface functionalisation with the transferrin peptide itself or anti-transferrin receptor antibody has proved to be promising in the development of functionally active and targeted nanoparticulate systems. The ability to covalently conjugate transferrin-targeting moieties to the surface of nanoparticles with relative ease has enabled its potential use in cancer-specific drug delivery and diagnostic applications due to the overexpression of its receptor on numerous malignant tissues (Dufes et al. 2013; Brigger et al. 2002). In vitro and in vivo studies have demonstrated the significance of the transferrin or associated receptor antibody modification in targeted-nanoparticle formulations, as high drug encapsulation in conjunction with enhanced cellular uptake (via transferrin-mediated endocytosis) improves selective cytotoxicity (Sahoo et al. 2004; Koppu et al. 2010; Liu et al. 2013; Huang et al. 2013; Ulbrich et al. 2009). Specifically, nanoparticle formulations such as one consisting of poly (lactic-co-glycolic acid) (PLGA) loaded with paclitaxel were conjugated to transferrin using an epoxy compound emphasise the potential advantages in developing targeted drug delivery strategies (Sahoo et al. 2004). Sahoo and co-workers evaluated the activity of their nanoparticulate system in human prostate cancer PC3 cells in vitro which resulted in a 70 % inhibition of proliferation in comparison to both free paclitaxel (35 % inhibition) and its nontargeted counterpart (25 % inhibition). Administration of the paclitaxel-loaded targeted nanoparticles (at a concentration of 24 mg/kg) to nude mice following their inoculation with PC3 cells demonstrated the full capability of the formulation, with complete tumour regression and increased survival. Continuing these studies, the same formulation was tested in human breast cancer cell lines MCF-7 and MCF-7/ADR drug resistant cells showed significant anti-proliferative activity in vitro compared to unconjugated nanoparticles and free drug (Sahoo and Labhasetwar 2005). Positive correlations between this activity and increased cellular uptake compared to its nontargeted counterpart display the importance of actively targeted malignant cells using the transferrin-transferrin receptor mechanism.
To date, four transferrin-conjugated nano-based drug delivery systems for the encapsulation of anticancer therapeutic compounds have progressed into human clinical trials (van der Meel et al. 2013). All are currently in early stage (Phase I and/or II) trials for the treatment of solid malignancies. Despite this apparent lack of development, the difficulty in designing clinically relevant nanomedicines with optimal properties, coupled with the novelty of the research field highlights the complexity of targeting cancer with specificity.
Conclusion
Through the extensive knowledge gained by studies involved in understanding the interactions between the transferrin peptide, its receptor and subsequent cellular fate, the ability to design clinically relevant targeting strategies using the complex has proven to be difficult. The increased expression of the transferrin receptor on a number of malignant tissue including that of the brain, theoretically allows for the tumour-specific and -directed delivery of therapeutic compounds following direct conjugation or encapsulation within a carrier. Direct conjugation and immunotoxin studies, using both the transferrin peptide and anti-transferrin receptor antibodies as the targeting moiety, display the potential of using such a pathway for enhanced and selective drug delivery. Due to the highly efficient endocytic and recycling mechanisms observed during cellular uptake of native transferrin, an increase in cellular uptake has been demonstrated both in vitro and in vivo. Furthermore, enhanced and selective cytotoxicity in both anticancer drug-sensitive and -resistant tumour cell lines highlights the capacity to overcome limitations of current cancer management strategies. Collectively, extensive knowledge in cancer biology (and the role of transferrin) and promising results obtained in direct conjugation, immunotoxin and nanoparticulate studies provide rationale to continue the development of transferrin-targeted therapies.
References
Aisen P, Leibman A, Zweier J (1978) Stoichiometric and site characteristics of the binding of iron to human transferrin. J Biol Chem 253(6):1930–1937
Batra JK, Jinno Y, Chaudhary VK, Kondo T, Willingham MC, FitzGerald DJ, Pastan I (1989) Antitumor activity in mice of an immunotoxin made with anti-transferrin receptor and a recombinant form of Pseudomonas exotoxin. Proc Natl Acad Sci USA 86(21):8545–8549
Batra JK, Fitzgerald DJ, Chaudhary VK, Pastan I (1991) Single-chain immunotoxins directed at the human transferrin receptor containing Pseudomonas exotoxin A or diphtheria toxin: anti-TFR(Fv)-PE40 and DT388-anti-TFR(Fv). Mol Cell Biol 11(4):2200–2205
Bejaoui N, Page M, Noel C (1991) Cytotoxicity of transferrin–daunorubicin conjugates on small cell carcinoma of the lung (SCCL) cell line NCI-H69. Anticancer Res 11(6):2211–2213
Berczi A, Barabas K, Sizensky JA, Faulk WP (1993) Adriamycin conjugates of human transferrin bind transferrin receptors and kill K562 and HL60 cells. Arch Biochem Biophys 300(1):356–363
Bergamaschi G, Cazzola M, Dezza L, Savino E, Consonni L, Lappi D (1988) Killing of K562 cells with conjugates between human transferrin and a ribosome-inactivating protein (SO-6). Br J Haematol 68(3):379–384
Bleil JD, Bretscher MS (1982) Transferrin receptor and its recycling in HeLa cells. EMBO J 1(3):351–355
Brandsma M, Jevnikar A, Ma S (2011) Recombinant human transferrin: beyond iron binding and transport. Biotechnol Adv 29(2):230–238
Bridges KR, Cudkowicz A (1984) Effect of iron chelators on the transferrin receptor in K562 cells. J Biol Chem 259(21):12970–12977
Brigger I, Dubernet C, Couvreur P (2002) Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev 54(5):631–651
Burdo JR, Antonetti DA, Wolpert EB, Connor JR (2003) Mechanisms and regulation of transferrin and iron transport in a model blood–brain barrier system. Neuroscience 121(4):883–890
Calzolari A, Deaglio S, Sposi N, Petrucci E, Morsilli O, Gabbianelli M, Malavasi F, Peschle C, Testa U (2004) Transferrin receptor 2 protein is not expressed in normal erythroid cells. Biochem J 381(3):629–634
Calzolari A, Oliviero I, Deaglio S, Mariani G, Biffoni M, Sposi N, Malavasi F, Peschle C, Testa U (2007) Transferrin receptor 2 is frequently expressed in human cancer cell lines. Blood Cells Mol Dis 39(1):82–91
Calzolari A, Larocca L, Deaglio S, Finisguerra V, Boe A, Raggi C, Ricci Vitani L, Pierconti F, Malavasi F, De Maria R, Testa U, Pallini R (2010) Transferrin receptor 2 is frequently and highly expressed in glioblastomas. Transl Oncol 3(2):123–134
Carvalho FS, Burgeiro A, Garcia R, Moreno AJ, Carvalho RA, Oliveira PJ (2013) Doxorubicin-induced cardiotoxicity: from bioenergetic failure and cell death to cardiomyopathy. Med Res Rev. doi:10.1002/med.21280
Chang J, Paillard A, Passirani C, Morille M, Benoit J-P, Betbeder D, Garcion E (2012) Transferrin adsorption onto PLGA nanoparticles governs their interaction with biological systems from blood circulation to brain cancer cells. Pharm Res 29(6):1495–1505
Chiu S-J, Liu S, Perrotti D, Marcucci G, Lee R (2006) Efficient delivery of a Bcl-2-specific antisense oligodeoxyribonucleotide (G3139) via transferrin receptor-targeted liposomes. J Control Release 112(2):199–207
Choudhary S, Mathew M, Verma RS (2011) Therapeutic potential of anticancer immunotoxins. Drug Discov Today 16(11–12):495–503
Ciechanover A, Schwartz AL, Dautry-Varsat A, Lodish HF (1983) Kinetics of internalization and recycling of transferrin and the transferrin receptor in a human hepatoma cell line. Effect of lysosomotropic agents. J Biol Chem 258(16):9681–9689
Cimini A, Mei S, Benedetti E, Laurenti G, Koutris I, Cinque B, Cifone MG, Galzio R, Pitari G, Di Leandro L, Giansanti F, Lombardi A, Fabbrini MS, Ippoliti R (2012) Distinct cellular responses induced by saporin and a transferrin–saporin conjugate in two different human glioblastoma cell lines. J Cell Physiol 227(3):939–951
Conner S, Schmid S (2003) Regulated portals of entry into the cell. Nature 422(6927):37–44
Daniels T, Delgado T, Rodriguez J, Helguera G, Penichet M (2006a) The transferrin receptor part I: biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol 121(2):144–158
Daniels TR, Delgado T, Helguera G, Penichet ML (2006b) The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clinical Immunology 121(2):159–176. doi:10.1016/j.clim.2006.06.006
Daniels TR, Ng PP, Delgado T, Lynch MR, Schiller G, Helguera G, Penichet ML (2007) Conjugation of an anti transferrin receptor IgG3-avidin fusion protein with biotinylated saporin results in significant enhancement of its cytotoxicity against malignant hematopoietic cells. Mol Cancer Ther 6(11):2995–3008
Das Gupta A, Shah VI (1990) Correlation of transferrin receptor expression with histologic grade and immunophenotype in chronic lymphocytic leukemia and Non-Hodgkin’s lymphoma. Hematol Pathol 4(1):37–41
Dautry Varsat A, Ciechanover A, Lodish HF (1983) pH and the recycling of transferrin during receptor-mediated endocytosis. Proc Natl Acad Sci USA 80(8):2258–2262
Deaglio S, Capobianco A, Calı A, Bellora F, Alberti F, Righi L, Sapino A, Camaschella C, Malavasi F (2002) Structural, functional, and tissue distribution analysis of human transferrin receptor-2 by murine monoclonal antibodies and a polyclonal antiserum. Blood 100(10):3782–3789. doi:10.1182/blood-2002-01-0076
Descamps L, Dehouck MP, Torpier G, Cecchelli R (1996) Receptor-mediated transcytosis of transferrin through blood–brain barrier endothelial cells. Am J Physiol 270(4):H1149–H1158
Dufes C, Al Robaian M, Somani S (2013) Transferrin and the transferrin receptor for the targeted delivery of therapeutic agents to the brain and cancer cells. Therapeutic delivery 4(5):629–640
El Hage Chahine J-M, Hémadi M, Ha-Duong N-T (1820) Hémadi M, Ha-Duong NT (2012) Uptake and release of metal ions by transferrin and interaction with receptor 1. Biochimica et Biophysica Acta (BBA) 3:334–347. doi:10.1016/j.bbagen.2011.07.008 General Subjects
Elliott R, Head J (2006) Complete resolution of malignant ascites in stage IV breast cancer by peritoneal drainage and innovative chemoimmunotherapy: a case report. Cancer Biother Radiopharm 21(2):138–145
Faulk WP, Hsi BL, Stevens PJ (1980) Transferrin and transferrin receptors in carcinoma of the breast. Lancet (Lond) 2(8191):390–392
Faulk WP, Taylor CG, Yeh CJ, McIntyre JA (1990) Preliminary clinical study of transferrin–adriamycin conjugate for drug delivery to acute leukemia patients. Mol Biother 2(1):57–60
Faulk WP, Barabas K, Sun IL, Crane FL (1991) Transferrin–adriamycin conjugates which inhibit tumor cell proliferation without interaction with DNA inhibit plasma membrane oxidoreductase and proton release in K562 cells. Biochem Int 25(5):815–822
Ferrara N, Hillan KJ, Novotny W (2005) Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun 333(2):328–335. doi:10.1016/j.bbrc.2005.05.132
Friden PM, Walus LR, Musso GF, Taylor MA, Malfroy B, Starzyk RM (1991) Anti-transferrin receptor antibody and antibody-drug conjugates cross the blood–brain barrier. Proc Natl Acad Sci USA 88(11):4771–4775
Galbraith GM, Galbraith RM, Temple A, Faulk WP (1980) Demonstration of transferrin receptors on human placental trophoblast. Blood 55(2):240–242
Gatter KC, Brown G, Trowbridge IS, Woolston RE, Mason DY (1983) Transferrin receptors in human tissues: their distribution and possible clinical relevance. J Clin Pathol 36(5):539–545
Gottesman MM, Fojo T, Bates SE (2002) Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2(1):48–58
Griffin TW, Stocl M, Collins J, Fernandes J, Maher VE (1992) Combined antitumor therapy with the chemotherapeutic drug doxorubicin and an anti-transferrin receptor immunotoxin: in vitro and in vivo studies. J Immunother 11(1):12–18
Habeshaw JA, Lister TA, Stansfeld AG, Greaves MF (1983) Correlation of transferrin receptor expression with histological class and outcome in non-Hodgkin lymphoma. Lancet (Lond) 1(8323):498–501
Hamilton TA, Wada HG, Sussman HH (1979) Identification of transferrin receptors on the surface of human cultured cells. Proceedings of the National Academy of Sciences of the United States of America. Proc Natl Acad Sci USA 76(12):6406–6410
Hammarström ML, Axelsson B, Ivansen M, Hammarström S, Perlmann P (1982) Transferrin receptors on mitogen-stimulated human thymus-derived lymphocytes. Scand J Immunol 16(4):355–360
Head JF, Wang F, Elliott RL (1997) Antineoplastic drugs that interfere with iron metabolism in cancer cells. Adv Enzyme Regul 37:147–169
Hicklin DJ, Ellis LM (2005) Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol 23(5):1011–1027. doi:10.1200/jco.2005.06.081
Hopkins CR, Trowbridge IS (1983) Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J Cell Biol 97(2):508–521
Hoshino T, Misaki M, Yamamoto M, Shimizu H, Ogawa Y, Toguchi H (1995) In vitro cytotoxicities and in vivo distribution of transferrin–platinum(II) complex. J Pharm Sci 84(2):216–221
Huang Y, He L, Liu W, Fan C, Zheng W, Wong Y-S, Chen T (2013) Selective cellular uptake and induction of apoptosis of cancer-targeted selenium nanoparticles. Biomaterials 34(29):7106–7116. doi:10.1016/j.biomaterials.2013.04.067
Huebers HA, Finch CA (1987) The physiology of transferrin and transferrin receptors. Physiol Rev 67(2):520–582
Ippoliti R, Lendaro E, D’Agostino I, Fiani ML, Guidarini D, Vestri S, Benedetti PA, Brunori M (1995) A chimeric saporin–transferrin conjugate compared to ricin toxin: role of the carrier in intracellular transport and toxicity. FASEB J 9(12):1220–1225
Jain RK (2001) Delivery of molecular and cellular medicine to solid tumors. Adv Drug Deliv Rev 46(1–3):149–168
Jefferies WA, Brandon MR, Hunt SV, Williams AF, Gatter KC, Mason DY (1984) Transferrin receptor on endothelium of brain capillaries. Nature 312(5990):162–163
Jiang YY, Liu C, Hong MH, Zhu SJ, Pei YY (2007) Tumor cell targeting of transferrin–PEG–TNF-alpha conjugate via a receptor-mediated delivery system: design, synthesis, and biological evaluation. Bioconjug Chem 18(1):41–49
Jiang W, KimBetty YS, Rutka JT, ChanWarren CW (2008) Nanoparticle-mediated cellular response is size-dependent. Nat Nano 3(3):145–150. http://www.nature.com/nnano/journal/v3/n3/suppinfo/nnano.2008.30_S1.html
Jin M, Snider MD (1993) Role of microtubules in transferrin receptor transport from the cell surface to endosomes and the Golgi complex. J Biol Chem 268(24):18390–18397
Karagiannis T, Lobachevsky P, Martin R (2006a) DNA targeted UVA photosensitization: characterization of an extremely photopotent iodinated minor groove binding DNA ligand. J Photochem Photobiol B 83(3):195–204
Karagiannis TC, Lobachevsky PN, Leung BK, White JM, Martin RF (2006b) Receptor-mediated DNA-targeted photoimmunotherapy. Cancer Res 66(21):10548–10552
Kawabata H, Germain RS, Vuong PT, Nakamaki T, Said JW, Koeffler HP (2000) Transferrin receptor 2-alpha supports cell growth both in iron-chelated cultured cells and in vivo. J Biol Chem 275(22):16618–16625
Kersten S, Desvergne B, Wahli W (2000) Roles of PPARs in health and disease. Nature 405(6785):421–424
Kim TH, Jo YG, Jiang HH, Lim SM, Youn YS, Lee S, Chen X, Byun Y, Lee KC (2012) PEG–transferrin conjugated TRAIL (TNF-related apoptosis-inducing ligand) for therapeutic tumor targeting. J Control Release 162(2):422–428. doi:10.1016/j.jconrel.2012.07.021
Kim S, Saha K, Kim C, Rotello V (2013) The role of surface functionality in determining nanoparticle cytotoxicity. Acc Chem Res 46(3):681–691
Klausner RD, Ashwell G, van Renswoude J, Harford JB, Bridges KR (1983a) Binding of apotransferrin to K562 cells: explanation of the transferrin cycle. Proc Natl Acad Sci USA 80(8):2263–2266
Klausner RD, Van Renswoude J, Ashwell G, Kempf C, Schechter AN, Dean A, Bridges KR (1983b) Receptor-mediated endocytosis of transferrin in K562 cells. J Biol Chem 258(8):4715–4724
Kondo K, Noguchi M, Mukai K, Matsuno Y, Sato Y, Shimosato Y, Monden Y (1990) Transferrin receptor expression in adenocarcinoma of the lung as a histopathologic indicator of prognosis. Chest 97(6):1367–1371
Koppu S, Oh YJ, Edrada-Ebel R, Blatchford DR, Tetley L, Tate RJ, Dufès C (2010) Tumor regression after systemic administration of a novel tumor-targeted gene delivery system carrying a therapeutic plasmid DNA. J Control Release 143(2):215–221. doi:10.1016/j.jconrel.2009.11.015
Kratz F, Beyer U, Roth T, Tarasova N, Collery P, Lechenault F, Cazabat A, Schumacher P, Unger C, Falken U (1998) Transferrin conjugates of doxorubicin: synthesis, characterization, cellular uptake, and in vitro efficacy. J Pharm Sci 87(3):338–346
Kratz F, Roth T, Fichiner I, Schumacher P, Fiebig HH, Unger C (2000) In vitro and in vivo efficacy of acid-sensitive transferrin and albumin doxorubicin conjugates in a human xenograft panel and in the MDA-MB-435 mamma carcinoma model. J Drug Target 8(5):305–318
Krenning EP, Kwekkeboom DJ, Bakker WH, Breeman WA, Kooij PP, Oei HY, van Hagen M, Postema PT, de Jong M, Reubi JC (1993) Somatostatin receptor scintigraphy with [111In-DTPA-D-Phe1]- and [123I-Tyr3]-octreotide: the Rotterdam experience with more than 1000 patients. Eur J Nucl Med 20(8):716–731
Lakadamyali M, Rust M, Zhuang X (2006) Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell 124(5):997–1009
Laske DW, Ilercil O, Akbasak A, Youle RJ, Oldfield EH (1994) Efficacy of direct intratumoral therapy with targeted protein toxins for solid human gliomas in nude mice. J Neurosurg 80(3):520–526
Laske DW, Muraszko KM, Oldfield EH, DeVroom HL, Sung C, Dedrick RL, Simon TR, Colandrea J, Copeland C, Katz D, Greenfield L, Groves ES, Houston LL, Youle RJ (1997a) Intraventricular immunotoxin therapy for leptomeningeal neoplasia. Neurosurgery 41(5):1039–1049
Laske DW, Youle RJ, Oldfield EH (1997b) Tumor regression with regional distribution of the targeted toxin TF-CRM107 in patients with malignant brain tumors. Nat Med 3(12):1362–1368
Lemieux P, Page M (1994) Sensitivity of multidrug-resistant MCF-7 cells to a transferrin-doxorubicin conjugate. Anticancer Res 14(2A):397–403
Liu G, Mao J, Jiang Z, Sun T, Hu Y, Zhang C, Dong J, Huang Q, Lan Q (2013) Transferrin-modified doxorubicin-loaded biodegradable nanoparticles exhibit enhanced efficacy in treating brain glioma-bearing rats. Cancer Biother Radiopharma 28(9):691–696
Lubgan D, Jozwiak Z, Grabenbauer GG, Distel LV (2009) Doxorubicin–transferrin conjugate selectively overcomes multidrug resistance in leukaemia cells. Cell Mol Biol Lett 14(1):113–127
Luck A, Mason A (2012) Transferrin-mediated cellular iron delivery. Curr Top Membr 69:3–35
Madhumathi J, Verma RS (2012) Therapeutic targets and recent advances in protein immunotoxins. Curr Opin Microbiol 15(3):300–309. doi:10.1016/j.mib.2012.05.006
Martell LA, Agrawal A, Ross DA, Muraszko KM (1993) Efficacy of transferrin receptor-targeted immunotoxins in brain tumor cell lines and pediatric brain tumors. Cancer Res 53(6):1348–1353
Martin RF, Murray V, D’Cunha G, Pardee M, Kampouris E, Haigh A, Kelly DP, Hodgson GS (1990) Radiation sensitization by an iodine-labelled DNA ligand. Int J Radiat Biol 57(5):939–946
Matsui T, Fukuda M (2011) Small GTPase Rab12 regulates transferrin receptor degradation: implications for a novel membrane trafficking pathway from recycling endosomes to lysosomes. Cell Logist 1(4):155–158
Matsui T, Itoh T, Fukuda M (2011) Small GTPase Rab12 regulates constitutive degradation of transferrin receptor. Traffic 12(10):1432–1443
Mattia E, Rao K, Shapiro DS, Sussman HH, Klausner RD (1984) Biosynthetic regulation of the human transferrin receptor by desferrioxamine in K562 cells. J Biol Chem 259(5):2689–2692
Mayle K, Le A, Kamei D (2012) The intracellular trafficking pathway of transferrin. Biochimica et biophysica acta 1820(3):264–281
McCaffrey MW, Bielli A, Cantalupo G, Mora S, Roberti V, Santillo M, Drummond F, Bucci C (2001) Rab4 affects both recycling and degradative endosomal trafficking. FEBS Lett 495(1–2):21–30. doi:10.1016/S0014-5793(01)02359-6
McClelland A, Kühn LC, Ruddle FH (1984) The human transferrin receptor gene: genomic organization, and the complete primary structure of the receptor deduced from a cDNA sequence. Cell 39(2):267–274
Mendelsohn J, Baselga J (2000) The EGF receptor family as targets for cancer therapy. Oncogene 19(56):6550–6565
Mendelsohn J, Trowbridge I, Castagnola J (1983) Inhibition of human lymphocyte proliferation by monoclonal antibody to transferrin receptor. Blood 62(4):821–826
Mout R, Moyano D, Rana S, Rotello V (2012) Surface functionalization of nanoparticles for nanomedicine. Chem Soc Rev 41(7):2539–2544
Munns J, Yaxley J, Coomer J, Lavin MF, Gardiner RA, Watters D (1998) Evaluation of the potential of transferrin–adriamycin conjugates in the treatment of bladder cancer. Br J Urol 82(2):284–289
Nam J-P, Park S-C, Kim T-H, Jang J-Y, Choi C, Jang M-K, Nah J-W (2013) Encapsulation of paclitaxel into lauric acid-O-carboxymethyl chitosan-transferrin micelles for hydrophobic drug delivery and site-specific targeted delivery. Int J Pharm 457(1):124–135
Neckers LM, Trepel JB (1986) Transferrin receptor expression and the control of cell growth. Cancer Invest 4(5):461–470
O’Keefe DO, Draper RK (1985) Characterization of a transferrin–diphtheria toxin conjugate. J Biol Chem 260(2):932–937
Panaccio M, Zalcberg JR, Thompson CH, Leyden MJ, Sullivan JR, Lichtenstein M, McKenzie IF (1987) Heterogeneity of the human transferrin receptor and use of anti-transferrin receptor antibodies to detect tumours in vivo. Immunol Cell Biol 65(6):461–472
Paris-Robidas S, Emond V, Tremblay C, Soulet D, Calon F (2011) In vivo labeling of brain capillary endothelial cells after intravenous injection of monoclonal antibodies targeting the transferrin receptor. Mol Pharmacol 80(1):32–39. doi:10.1124/mol.111.071027
Pirker R, FitzGerald DJP, Hamilton TC, Ozols RF, Willingham MC, Pastan I (1985) Anti-transferrin receptor antibody linked to Pseudomonas exotoxin as a model immunotoxin in human ovarian carcinoma cell lines. Cancer Res 45(2):751–757
Pirker R, FitzGerald DJP, Willingham MC, Pastan I (1988) Enhancement of the activity of immunotoxins made with either ricin a chain or pseudomonas exotoxin in human ovarian and epidermoid carcinoma cell lines. Cancer Res 48(14):3919–3923
Prior R, Reifenberger G, Wechsler W (1990) Transferrin receptor expression in tumours of the human nervous system: relation to tumour type, grading and tumour growth fraction. Virchows Archiv 416(6):491–496
Prutki M, Poljak Blazi M, Jakopovic M, Tomas D, Stipancic I, Zarkovic N (2006) Altered iron metabolism, transferrin receptor 1 and ferritin in patients with colon cancer. Cancer Lett 238(2):188–196
Raso V, Basala M (1984) A highly cytotoxic human transferrin–ricin A chain conjugate used to select receptor-modified cells. J Biol Chem 259(2):1143–1149
Recht L, Torres CO, Smith TW, Raso V, Griffin TW (1990) Transferrin receptor in normal and neoplastic brain tissue: implications for brain-tumor immunotherapy. J Neurosurg 72(6):941–945. doi:10.3171/jns.1990.72.6.0941
Richardson DR, Ponka P (1997) The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim Biophys Acta 1331(1):1–40
Rouault T, Rao K, Harford J, Mattia E, Klausner RD (1985) Hemin, chelatable iron, and the regulation of transferrin receptor biosynthesis. J Biol Chem 260(27):14862–14866
Rutledge EA, Mikoryak CA, Draper RK (1991) Turnover of the transferrin receptor is not influenced by removing most of the extracellular domain. J Biol Chem 266(31):21125–21130
Ryschich E, Huszty G, Knaebel HP, Hartel M, Büchler MW, Schmidt J (2004) Transferrin receptor is a marker of malignant phenotype in human pancreatic cancer and in neuroendocrine carcinoma of the pancreas. Eur J Cancer 40(9):1418–1422
Sahoo SK, Labhasetwar V (2005) Enhanced antiproliferative activity of transferrin-conjugated paclitaxel-loaded nanoparticles is mediated via sustained intracellular drug retention. Mol Pharm 2(5):373–383. doi:10.1021/mp050032z
Sahoo SK, Ma W, Labhasetwar V (2004) Efficacy of transferrin-conjugated paclitaxel-loaded nanoparticles in a murine model of prostate cancer. Int J Cancer 112(2):335–340. doi:10.1002/ijc.20405
Schneider C, Sutherland R, Newman R, Greaves M (1982) Structural features of the cell surface receptor for transferrin that is recognized by the monoclonal antibody OKT9. J Biol Chem 257(14):8516–8522
Schneider C, Owen MJ, Banville D, Williams JG (1984) Primary structure of human transferrin receptor deduced from the mRNA sequence. Nature 311(5987):675–678
Schulman HM, Wilczynska A, Ponka P (1981) Transferrin and iron uptake by human lymphoblastoid and K-562 cells. Biochem Biophys Res Commun 100(4):1523–1530
Scott CF Jr, Goldmacher VS, Lambert JM, Jackson JV, McIntyre GD (1987) An immunotoxin composed of a monoclonal antitransferrin receptor antibody linked by a disulfide bond to the ribosome-inactivating protein gelonin: potent in vitro and in vivo effects against human tumors. J Natl Cancer Inst 79(5):1163–1172
Seymour GJ, Walsh MD, Lavin MF, Strutton G, Gardiner RA (1987) Transferrin receptor expression by human bladder transitional cell carcinomas. Urol Res 15(6):341–344
Shapira A, Benhar I (2010) Toxin-based therapeutic approaches. Toxins 2(11):2519–2583
Shinohara H, Fan D, Ozawa S, Yano S, Van Arsdell M, Viner JL, Beers R, Pastan I, Fidler IJ (2000) Site-specific expression of transferrin receptor by human colon cancer cells directly correlates with eradication by antitransferrin recombinant immunotoxin. Int J Oncol 17(4):643–651
Sieff C, Bicknell D, Caine G, Robinson J, Lam G, Greaves MF (1982) Changes in cell surface antigen expression during hemopoietic differentiation. Blood 60(3):703–713
Singh M (1999) Transferrin As A targeting ligand for liposomes and anticancer drugs. Curr Pharm Des 5(6):443–451
Singh M, Atwal H, Micetich R (1998) Transferrin directed delivery of adriamycin to human cells. Anticancer Res 18(3A):1423–1427
Singh M, Mugler K, Hailoo D, Burke S, Nemesure B, Torkko K, Shroyer K (2011) Differential expression of transferrin receptor (TfR) in a spectrum of normal to malignant breast tissues: implications for in situ and invasive carcinoma. Appl Immunohistochem Mol Morphol 19(5):417–423
Speth PAJ, Hoesel QGCM, Haanen C (1988) Clinical pharmacokinetics of doxorubicin. Clin-Pharmacokinet 15(1):15–31. doi:10.2165/00003088-198815010-00002
Steere A, Byrne S, Chasteen ND (1820) Mason A (2012) Kinetics of iron release from transferrin bound to the transferrin receptor at endosomal pH. Biochim Biophys Acta 3:326–333
Stein BS, Sussman HH (1986) Demonstration of two distinct transferrin receptor recycling pathways and transferrin-independent receptor internalization in K562 cells. J Biol Chem 261(22):10319–10331
Sun IL, Sun EE, Crane FL, Morre DJ, Faulk WP (1992) Inhibition of transplasma membrane electron transport by transferrin–adriamycin conjugates. Biochim Biophys Acta 23(1):84–88
Sutherland R, Delia D, Schneider C, Newman R, Kemshead J, Greaves M (1981) Ubiquitous cell-surface glycoprotein on tumor cells is proliferation-associated receptor for transferrin. Proc Natl Acad Sci 78(7):4515–4519
Szwed M, Matusiak A, Laroche-Clary A, Robert J, Marszalek I, Jozwiak Z (2013) Transferrin as a drug carrier: cytotoxicity, cellular uptake and transport kinetics of doxorubicin transferrin conjugate in the human leukemia cells. Toxicol Vitro. doi:10.1016/j.tiv.2013.09.013
Taetle R, Honeysett JM, Trowbridge I (1983) Effects of anti-transferrin receptor antibodies on growth of normal and malignant myeloid cells. Int J Cancer 32(3):343–349
Taetle R, Castagnola J, Mendelsohn J (1986) Mechanisms of growth inhibition by anti-transferrin receptor monoclonal antibodies. Cancer Res 46(4):1759–1763
Trinder D, Baker E (2003) Transferrin receptor 2: a new molecule in iron metabolism. Int J Biochem Cell Biol 35(3):292–296
Trowbridge IS, Domingo DL (1981) Anti-transferrin receptor monoclonal antibody and toxin-antibody conjugates affect growth of human tumour cells. Nature 294(5837):171–173
Trowbridge IS, Lopez F (1982) Monoclonal antibody to transferrin receptor blocks transferrin binding and inhibits human tumor cell growth in vitro. Proc Natl Acad Sci USA 79(4):1175–1179
Trowbridge IS, Omary MB (1981) Human cell surface glycoprotein related to cell proliferation is the receptor for transferrin. Proc Natl Acad Sci 78(5):3039–3043
Ulbrich K, Hekmatara T, Herbert E, Kreuter J (2009) Transferrin- and transferrin-receptor-antibody-modified nanoparticles enable drug delivery across the blood–brain barrier (BBB). Eur J Pharma Biopharma 71(2):251–256. doi:10.1016/j.ejpb.2008.08.021
van der Meel R, Vehmeijer LJC, Kok RJ, Storm G, van Gaal EVB (2013) Ligand-targeted particulate nanomedicines undergoing clinical evaluation: current status. Adv Drug Deliv Rev 65(10):1284–1298. doi:10.1016/j.addr.2013.08.012
Wang F, Jiang X, Yang DC, Elliott RL, Head JF (2000) Doxorubicin–gallium–transferrin conjugate overcomes multidrug resistance: evidence for drug accumulation in the nucleus of drug resistant MCF-7/ADR cells. Anticancer Res 20(2A):799–808
Ward JH, Jordan I, Kushner JP, Kaplan J (1984) Heme regulation of HeLa cell transferrin receptor number. J Biol Chem 259(21):13235–13240
Weaver M, Laske DW (2003) Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) therapy of malignant gliomas. J Neurooncol 65(1):3–13
Whitesides GM (2003) The ‘right’ size in nanobiotechnology. Nat Biotechnol 21(10):1161–1165
Woith W, Nüsslein I, Antoni C, Dejica DI, Winkler TH, Herrmann M, Pirner K, Kalden JR, Manger B (1993) A soluble form of the human transferrin receptor is released by activated lymphocytes in vitro. Clin Exp Immunol 92(3):537–542
Wrba F, Ritzinger E, Reiner A, Holzner JH (1986) Transferrin receptor (TrfR) expression in breast carcinoma and its possible relationship to prognosis. An immunohistochemical study. Virchows Archiv A 410(1):69–73
Wu J, Lu Y, Lee A, Pan X, Yang X, Zhao X, Lee R (2007) Reversal of multidrug resistance by transferrin-conjugated liposomes co-encapsulating doxorubicin and verapamil. J Pharm Pharma Sci 10(3):350–357
Wu L, Wu J, Zhou Y, Tang X, Du Y, Hu Y (2012) Enhanced antitumor efficacy of cisplatin by tirapazamine–transferrin conjugate. Int J Pharm 431(1–2):190–196. doi:10.1016/j.ijpharm.2012.04.032
Yashunsky V, Shimron S, Lirtsman V, Weiss A, Melamed Book N, Golosovsky M, Davidov D, Aroeti B (2009) Real-time monitoring of transferrin-induced endocytic vesicle formation by mid-infrared surface plasmon resonance. Biophys J 97(4):1003–1012
Yeh CJ, Taylor CG, Faulk WP (1984) Transferrin binding by peripheral blood mononuclear cells in human lymphomas, myelomas and leukemias. Vox Sang 46(4):217–223
Yoon D, Chu DSH, Ng C, Pham E, Mason A, Hudson D, Smith V, MacGillivray RTA, Kamei D (2009) Genetically engineering transferrin to improve its in vitro ability to deliver cytotoxins. J Control Release 133(3):178–184
Zerial M, Melancon P, Schneider C, Garoff H (1986) The transmembrane segment of the human transferrin receptor functions as a signal peptide. EMBO J 5(7):1543–1550
Acknowledgments
The support of the Australian Institute of Nuclear Science and Engineering (AINSE) is acknowledged. TCK was the recipient of AINSE awards. TCK is supported by an Australian Research Council Future Fellowship and the Epigenomic Medicine Laboratory is supported by McCord Research. Supported in part by the Victorian Government’s Operational Infrastructure Support Program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tortorella, S., Karagiannis, T.C. Transferrin Receptor-Mediated Endocytosis: A Useful Target for Cancer Therapy. J Membrane Biol 247, 291–307 (2014). https://doi.org/10.1007/s00232-014-9637-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00232-014-9637-0