
Abstract
Studies have reported that Na,K-ATPase interacts with E-cadherin to stabilize (AJs) and regulate the expression of claudins, the main proteins present in the tight junction (TJ) in epithelial cells containing caveolae. However, the role of this ATPase in the regulation of the AJ and TJ proteins in colorectal cancer cells as well as the molecular events underlying this event in a caveolae-independent system remain undefined. In the present study, we used ouabain, a classic drug known to inhibit Na,K-ATPase, and Caco-2 cells, which are a well-established human colorectal cancer model that does not exhibit caveolae. We demonstrated that ouabain treatment resulted in a reduction of the β1 Na,K-ATPase protein and cell redistribution of the AJ proteins E-cadherin and β-catenin, as well as the α1 Na,K-ATPase subunit. Furthermore, ouabain increased claudin-3 protein levels, impaired the TJ barrier function and increased cell viability and proliferation during the early stages of treatment. Additionally, the observed ouabain-induced events were dependent on the activation of ERK1/2 signaling; but in contrast to previous studies, this signaling cascade was caveolae-independent. In conclusion, our findings strongly suggest that α1 and β1 Na,K-ATPase downregulation and ERK1/2 activation induced by ouabain are interlinked events that play an important role during cell–cell adhesion loss, which is an important step during the tumor progression of colorectal carcinomas.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In epithelial cells, the apical junctional complex (AJC), which consists of tight junctions (TJs) and adherens junctions (AJs), is the structure responsible for the establishment and maintenance of functional adhesive interactions between neighboring cells (Miyoshi and Takai 2008). E-Cadherin, the most important AJ protein, binds to Armadillo family proteins that include β-catenin and plakoglobin (γ-catenin), which modulate the interaction with α-catenin in a dynamic fashion. The downregulation or mislocalization of E-cadherin causes a loss of cell–cell adhesion, which occurs during epithelial tumorigenesis (Yap et al. 2007). Integral TJ membrane proteins include claudin family proteins, occludin and the junctional adhesion molecule. These proteins interact with the ZO-1, ZO-2 and ZO-3 adaptor proteins of the zonula occludens family to link TJs with the actin cytoskeleton. The claudin protein family is currently composed of 27 members and regulates the functions of TJs (Mineta et al. 2011). A myriad of diseases, including cancer, have been associated with changes in the expression, stability and subcellular localization of claudin family members (Ikari et al. 2011; Takizawa et al. 2012).
Na,K-ATPase is an enzyme that uses the energy from ATP hydrolysis to catalyze the active transport of Na+ and K+ across the plasma membrane of eukaryotic cells. The functional structure of this ATPase consists of α- and β-subunits, which play an important role in ATP hydrolysis and ion pumping, and the FXYD subunit, which regulates the pump activity (for review, see Cereijido et al. 2012). The α-subunit is the catalytic subunit and contains binding sites for ATP, cardiotonic steroids and cations (Morth et al. 2009).
The relationship between Na,K-ATPase and the development of cancer has been investigated. In breast and lung cancer cells, treatment with cardiac glycosides, which are well-known Na,K-ATPase inhibitors, caused decreased levels of the p53 protein alongside an increased activation of the Src and MAPK proteins (Kometiani et al. 2005; Wang et al. 2009). Furthermore, reduced expression of the β1-subunit and low activity of Na,K-ATPase have been reported in samples from renal cell carcinoma patients (Rajasekaran et al. 1999). Also, treatment of renal epithelial cells with TGFβ1 reduced β1 Na,K-ATPase expression at the cell surface, and this event was associated with the epithelial-to-mesenchymal transition and increased malignant potential (Rajasekaran et al. 2010).
Some studies have demonstrated that E-cadherin interacts with the β1-subunit of Na,K-ATPase and that this binding is necessary to support AJ stability (Rajasekaran et al. 2005). In this context, disassembly of the E-cadherin/catenin complex has been associated with downregulation of β1 Na,K-ATPase, facilitating the invasion process of non–small cell lung cancer (Mijatovic et al. 2007). Moreover, ouabain, a cardiotonic steroid known to inhibit Na,K-ATPase, induced nuclear accumulation of β-catenin in MDCK cells through an event that requires MAPK activation, suggesting that nuclear β-catenin may lead to the activation of genes related to E-cadherin downregulation (Contreras et al. 2004). On the other hand, ouabain treatment increased the transepithelial electrical resistance (TER) and changed the cell distribution and protein levels of claudins 1, 2 and 4 in MDCK-II cells (Larre et al. 2010). Various studies have suggested that the enzyme-linked signaling function of Na,K-ATPase in renal epithelial cells occurs through a caveolae-associated portion of the enzyme and that ouabain-induced signaling occurs through specific interactions between the α-subunit of Na,K-ATPase, Src kinase and caveolin-1 (Wang et al. 2004; Liang et al. 2007; Li and Xie 2009). However, a recent study revealed that ouabain can activate cell signaling pathways in membranes that do not contain caveolae (Liu et al. 2011). Therefore, more studies are necessary to understand the regulated events for this ATPase in the context of cell–cell adhesion loss and cancer progression.
In the present study, we investigated the effects of ouabain on AJC assembly and downstream signaling pathways in Caco-2, which are caveolae-free colorectal cancer cells. We demonstrated that ouabain treatment triggered disassembly of the AJs, increased claudin-3 protein levels, impaired the TJ barrier function and increased cell viability and proliferation at the shortest treatment times. These events were accompanied by decreased α1 and β1 Na,K-ATPase levels and were dependent on activation of the ERK1/2 pathway. This is the first study that examines the regulation of AJC assembly by Na,K-ATPase, an event modulated by ERK1/2 signaling, in response to ouabain treatment in a cell line that does not contain caveolae.
Materials and Methods
Materials
Anti-E-cadherin mouse monoclonal antibody (clone 36) was purchased from BD Biosciences (San Diego, CA). Anti-α1 and anti-β1 Na,K-ATPase mouse monoclonal antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-p-ERK mouse monoclonal and anti-uvomorulin/E-cadherin rat monoclonal antibodies were purchased from Sigma-Aldrich (St. Louis, MO). Anti-ERK mouse monoclonal, anti-β-catenin rabbit monoclonal and anti-α-tubulin rabbit polyclonal antibodies were obtained from Cell Signaling (Beverly, MA). Anti-claudin-1 and anti-claudin-3 rabbit polyclonal antibodies were purchased from Invitrogen (Carlsbad, CA). Alex-fluor-488 anti-mouse and anti-rabbit and Alex-fluor-546 anti-rat and anti-rabbit were obtained from Invitrogen. Horseradish peroxidase–conjugated anti-rabbit and anti-mouse IgG were purchased from GE Healthcare (Little Chalfont, UK). Ouabain was purchased from Sigma-Aldrich, and 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98059, MEK1 inhibitor) was obtained from Cell Signaling.
Cell Culture and Pharmacological Inhibition
Caco-2 cells (ATCC, Manassas, VA; HTB-37), a cell line derived from human colon adenocarcinoma that does not contain caveolin-1 and caveolae (Breuza et al. 2002), were cultured in Dulbecco’s modified Eagle medium supplemented with 10 % heat-inactivated fetal bovine serum (Invitrogen), 60 mg/ml streptomycin and 100 mg/ml penicillin in a humidified atmosphere of 5 % CO2 and air. The culture medium was changed every 24 h to avoid nutrient depletion. Cell monolayers were treated with 100 nM or 10 or 100 μM ouabain for 6, 8, 12 or 24 h. When indicated, cells were pretreated for 1 h with PD98059 (50 μM final concentration), a specific inhibitor of the MAPK pathway, before ouabain treatment.
Cell Extraction and Immunoblotting
Cells were rinsed in PBS and lysed in lysis buffer (1 % Triton X-100, 0.5 % sodium deoxycholate, 0.2 % SDS, 150 mM NaCl, 10 mM HEPES and 2 mM EDTA; pH 7.3) containing 20 mM sodium fluoride, 1 mM sodium orthovanadate and 1 % protease inhibitor cocktail (Sigma-Aldrich). Equal amounts of protein (30 μg/lane) from the total cell lysates were resolved by SDS-PAGE in 10–12 % gels and transferred to nitrocellulose sheets using a semidry transfer cell at 10 V for 60 min. Then, the membranes were blocked with TBS-T buffer (20 mM Tris–HCl [pH 7.6], 137 mM NaCl and 0.1 % [v/v] Tween 20) containing 5 % low-fat dried milk and incubated for 2 h with the primary antibodies anti-E-cadherin, anti-β-catenin, anti-α1-subunit and anti-β1-subunit of Na,K-ATPase, anti-p-ERK1/2, anti-ERK1/2, anti-claudin-1, anti-claudin-3 and anti-α-tubulin. Membranes were washed and incubated for 1 h with horseradish peroxidase–conjugated secondary antibodies, and the proteins were visualized using a chemiluminescence kit obtained from Amersham Biosciences GE (Little Chalfont, UK). Band images were quantified by optical density using LabWorks 4.6 software (Bio-Rad, Upland, CA).
Electron Microscopic Analysis
Cells were cultured on Transwell polycarbonate filters with a 0.4-μm pore size and 0.33-cm2 surface area (Corning Life Science, Lowell, MA) and fixed for 1 h in a solution containing 2.5 % glutaraldehyde, 1 % paraformaldehyde, 0.2 M sucrose and 2 mM CaCl2 in 0.1 M cacodylate buffer (pH 7.4). Postfixation was carried out in 1 % osmium tetroxide in cacodylate buffer containing 0.8 % potassium ferrocyanide. Subsequently, cells were dehydrated in an acetone series and embedded in Epon resin. Ultrathin sections were obtained, stained with lead citrate and observed in a Zeiss CEM-900 transmission electron microscope (Carl Zeiss, Jena, Germany).
Immunofluorescence
Cells were grown on sterile glass coverslips and, after treatments, rinsed with PBS and fixed with methanol for 10 min at −20 °C. Next, cells were blocked with 0.2 % BSA in PBS for 1 h and permeabilized with 0.1 % Triton X-100. Subsequently, samples were incubated with primary antibodies against E-cadherin, β-catenin, α1-subunit of Na,K-ATPase, claudin-1 or claudin-3 followed by 1 h with their respective secondary antibodies at 37 °C. Coverslips were washed in PBS and mounted using n-propyl-gallate. Cell staining was visualized in an Axio Observe.Z1 microscope equipped with an AxioCam HRc and AxioVision Release 4.8 digital image processing software (Carl Zeiss) or a confocal laser scanning microscope (FV10i-O), in which images were analyzed using FV10-ASW software (Olympus, Tokyo, Japan). Images were captured in the same exposure time and laser intensity. As indicated, fluorescence intensity was quantified in ten areas of cell–cell contact chosen randomly in the captured image, using FV10-ASW software.
TER Measurement
Cells (2 × 104/insert) were grown to confluence on Transwell polycarbonate filters with a 0.4-μm pore size and 0.33-cm2 surface area. After treatments, TER values were determined using a Millicel-ERS system (EMD Millipore, Billerica, MA). All TER values were normalized by the filter area and obtained after background subtraction (filter and bath solution).
Macromolecule Permeability Assay
To further test TJ functionality, macromolecular permeability was assessed by an antibody permeability assay, as described previously (Rajasekaran et al. 2001). Caco-2 cells (105) were seeded on glass coverslips in 24-well plates and incubated until confluence. Afterward, cell monolayers were fixed in 2 % paraformaldehyde and incubated with anti-uvomorulin/E-cadherin antibody for 2 h under nonpermeabilization conditions, followed by 1-h incubation with the respective secondary antibody at 37 °C. Uvomorulin/E-cadherin staining was visualized with the Axio Observer.Z1 microscope.
Cell Viability Analysis
Cells (104/well) were seeded in 96-well plates and treated with ouabain (10 or 100 μM) for 8, 12 and 24 h. Subsequently, 5 % 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was added to the culture medium and incubated for 2 h. Next, dimethyl sulfoxide was used to dissolve the formazan crystals, and the resulting purple solutions were spectrophotometrically measured at 538 nm in a SpectraMax 190 Gemini XS spectrophotometer (Molecular Devices, Sunnyvale, CA).
Cell Proliferation Assay
The crystal violet method was used to measure cell proliferation as described previously (de Freitas et al. 2011). Briefly, cells (2 × 104/well) were seeded in 96-well plates and incubated in culture medium containing ouabain alone or in combination with MEK inhibitor (PD98059) for 8 h before ethanol fixation for 10 min. A crystal violet solution (0.05 % crystal violet and 20 % methanol) was added to the cells for 10 min. Cells were washed and solubilized with methanol, and the absorbances at 595 nm were measured with a Spectra Max 190 spectrophotometer (Molecular Devices).
Statistical Analysis
Statistical analysis was performed using one-way ANOVA in the GraphPad Prism 4.02 software (GraphPad Software, San Diego, CA). All results are expressed as the mean ± standard error of the mean (SEM) of three independent experiments (or ten areas chosen randomly, as in the quantification α1 Na,K-ATPase fluorescence intensity). A difference of p < 0.05 was considered statistically significant.
Results
Ouabain Treatment Alters the Distribution of E-Cadherin and α1 Na,K-ATPase and Induces Decreased α1 and β1 Na,K-ATPase Levels
Previous studies using MDCK cells have demonstrated that Na,K-ATPase plays a critical role in the establishment of the AJC when linked to E-cadherin, the main protein present in AJs (Rajasekaran et al. 2005; Vagin et al. 2007). In the present study, to evaluate the role of Na,K-ATPase in AJC organization of colorectal cancer cells, we used ouabain, a cardiac glycoside known to link to the α1-subunit and inhibit the activity of the Na,K-ATPase. After ouabain treatment, we analyzed the distribution of E-cadherin and α1 Na,K-ATPase in Caco-2 cells by immunofluorescence. In control cells, representative immunofluorescence images show that E-cadherin and α1 Na,K-ATPase were colocalized at cell–cell contacts in normal labeling patterns. After ouabain treatment, E-cadherin and α1 Na,K-ATPase remained colocalized at cell–cell contacts. However, after treatment with 10 μM of the glycoside, a discontinuous staining pattern was observed; and with the higher concentration (100 μM), cytoplasmic projections were also observed (Fig. 1a). Furthermore, we observed that these treatments induced a decrease of α1 Na,K-ATPase staining in cell–cell contacts and of the protein levels of this subunit (Fig. 1b and c, respectively). In addition, treatment with 100 nM of ouabain did not cause an effect on the labeling pattern of E-cadherin. We verified also if the effects observed were due to an ionic imbalance by treating the cells with NaCl (250 mM) and observed that treatment with this salt did not alter the distribution pattern of E-cadherin (Supplementary Fig. S1). With these observations we carried out the subsequent experiments using ouabain concentrations of 10 and 100 μM.

Effect of ouabain treatment on E-cadherin and α1 Na,K-ATPase subcellular distribution, α1 Na,K-ATPase expression and cell morphology. Monolayers of Caco-2 cells were grown on glass coverslips, plates or Transwell polycarbonate filters and treated with ouabain (10 or 100 μM) for 6 h. a Subcellular distribution analysis by immunofluorescence of E-cadherin and α1 Na,K-ATPase. Stained cells were analyzed by confocal microscopy. Bar = 5 μm. b The α1 Na,K-ATPase fluorescence intensity at cell–cell contacts was determined using FV10-ASW software. Intensity is displayed as a ratio intensity comparing treated and untreated cells (control). Error bars indicate the mean ± SEM of ten areas of cell–cell contacts chosen randomly; **p < 0.01, as determined by ANOVA. c Total cell lysates of ouabain-treated or untreated cells were harvested, and the expression of α1 Na,K-ATPase was visualized by immunoblotting. Dye Ponceau staining (Ponc.) was used as a loading control. Numbers represent the ratio of the optical density of ouabain-treated to untreated cells. d Ultrastructural analysis. Representative images of thin sections of control cells and cells treated with ouabain. Arrows indicate tight junctions. AJ adherent junction, α1 α1 Na,K-ATPase, E-cad E-cadherin, Cont control, Ouab ouabain. Bars = 0.35 μm
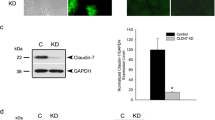
Next, we performed ultrastructural analysis by transmission electron microscopy and observed that ouabain treatment caused a marked disorganization at the AJ region at both concentrations used (10 and 100 μM). However, the TJ structure remained apparently intact (Fig. 1d). Next, as β1 Na,K-ATPase binds directly to E-cadherin and retains it at cell–cell contacts while stabilizing the AJ structure, we analyzed the protein levels of β1 Na,K-ATPase and the AJ proteins E-cadherin and β-catenin after treatment with the glycoside. Ouabain treatment (10 and 100 μM) decreased the protein levels of β1 Na,K-ATPase after both 6 and 8 h (Fig. 2a) but did not alter the levels of the AJ proteins E-cadherin and β-catenin (Fig. 2b). Together, these data indicate that ouabain alters the distribution pattern of both E-cadherin and α1 Na,K-ATPase and decreases the levels of β1 Na,K-ATPase in Caco-2 cells.

Effects of ouabain treatment on expression of AJ proteins and β1 Na,K-ATPase. Total cell lysates of ouabain-treated or untreated cells were harvested, and the expression of (a) β1 Na,K-ATPase and (b) E-cadherin and β-catenin was visualized by immunoblotting. Ponceau staining (Ponc.) or α-tubulin was used as a loading control. Bar graphs show the proportion between optical density of treated cells and untreated cells (Cont) in immunoblotting. Error bars indicate the mean ± SEM (n = 3). **p < 0.01, as determined by ANOVA
Alterations in the Distribution of AJ Proteins Induced by Ouabain Are Mediated by ERK1/2 Signaling
It has been reported that treatment with glycosides might activate the MAPK pathway in MDCK cells (Contreras et al. 2004). We showed also that this pathway is involved in the regulation of E-cadherin–dependent cell–cell adhesion in Caco-2 cells (Barbosa et al. 2003). Based on these studies, we evaluated whether ouabain-induced AJ disorganization may involve activation of the ERK1/2 pathway by specific pharmacological inhibition with PD98059. As shown in Fig. 3a, immunofluorescence analysis revealed that the distribution pattern of E-cadherin and β-catenin was altered in the presence of ouabain and that this alteration was prevented by pretreatment with PD98059. To confirm the involvement of this pathway, we analyzed the phosphorylation status of ERK1/2 by immunoblotting. Ouabain treatment induced ERK1/2 pathway activation, as evidenced by the increased phosphorylation level of these proteins. The increased phosphorylation was prevented by pretreatment with the inhibitor PD98059 (Fig. 3b). Moreover, the inhibition of the ERK1/2 pathway with this inhibitor also prevented the ouabain-induced decrease in protein levels of β1 Na,K-ATPase (Supplementary Fig. S2). Taken together, these results show that the ERK1/2 pathway modulates the ouabain-induced redistribution of AJ proteins.

Impact of ERK1/2 pathway inhibition on ouabain-induced AJ protein distribution. a Monolayers of Caco-2 cells were grown on glass coverslips, pretreated with an inhibitor of the ERK1/2 pathway (PD98059) for 1 h (when indicated) and incubated with ouabain for 6 h. After treatment, cells were processed for E-cadherin immunofluorescence and β-catenin distribution and analyzed by confocal microscopy. Bar = 5 μm. b Total cell lysates were harvested, and the expression of p-ERK1/2 and total ERK1/2 was visualized by immunoblotting. Bar graphs show the proportion between optical density of treated cells and untreated cells (Cont) in immunoblotting. Error bars indicate the mean ± SEM (n = 3). *p < 0.05, **p < 0.01, as determined by ANOVA. β-cat β-catenin, PD98 PD98059 (MEK1 inhibitor)
Ouabain Increases the Levels of Claudin-3, Impairing TJ Barrier Function, and These Events Are Mediated by the ERK1/2 Pathway
MDCK-II cells treated with ouabain exhibit altered cell distribution and protein levels of claudins 1, 2 and 4 alongside increased TER (Larre et al. 2010). Thus, we verified the cell distribution and protein levels of claudins 1 and 3, which are upregulated in colorectal cancer, impairing the barrier function of the TJ as described by us in a previous study (Oliveira et al. 2005). As observed in Fig. 4a, ouabain treatment apparently did not alter the intercellular distribution patterns of claudins 1 and 3, which is in agreement with the electron microscopic observations (Fig. 1b) where an apparently intact TJ structure was noted. However, ouabain increased the protein levels of claudin-3 but not of claudin-1. The increased claudin-3 protein level was prevented by treatment with the inhibitor PD98059, which decreased the protein levels to values below those observed in the control group (Fig. 4b). The claudin proteins are known regulators of the paracellular flux of ions. In this regard, we measured the TER to assess whether ouabain treatment impairs the TJ barrier function along with increased claudin-3 levels. We observed that ouabain decreased the TER values and that this effect was attenuated by ERK1/2 inhibition (Fig. 5a). To further assess the TJ barrier function, we evaluated the paracellular flux to macromolecules, using the macromolecule permeability assay (Rajasekaran et al. 2001). In this assay, using an antibody that recognizes the extracellular region of E-cadherin (anti-uvomorulin/E-cadherin), it is possible to evaluate the TJ ability to regulate the paracellular permeability to macromolecules under nonpermeabilizing conditions. In Fig. 5b, we observed an absence of uvomorulin/E-cadherin staining in untreated cells (control), indicating high regulation of the paracellular flux to macromolecules. On the other hand, after ouabain treatment, we observed a normal pattern of uvomorulin/E-cadherin staining on the plasma membrane, indicating impaired regulation of the macromolecular flux. A positive control to uvomorulin/E-cadherin staining was obtained after calcium depletion, which is known to induce disruption of cell–cell adhesion. Together, these data show that ouabain increases the levels of claudin-3 and impairs the barrier function of TJs, and both events are modulated by the ERK1/2 pathway.

Impact of ERK1/2 pathway inhibition on the ouabain-induced effects in localization and expression of claudins 1 and 3. Monolayers of Caco-2 cells were grown, pretreated (when indicated) with PD98059 for 1 h and treated or not with ouabain (10 μM). a Subcellular distribution analysis of claudins 1 and 3 by immunofluorescence after PD98059 pretreatment and 6-h ouabain treatment. Bar = 5 μm. b Protein level analysis by immunoblotting of claudins 1 and 3 after the same treatment as in a. α-Tubulin was used as a loading control. Bar graphs show the proportion between optical density of treated and untreated cells (Cont) in immunoblotting. Error bars indicate the mean ± SEM (n = 3). **p < 0.01, ***p < 0.001, as determined by ANOVA. Cld-1 claudin-1, Cld-3 claudin-3

Impact of ERK1/2 pathway inhibition on the ouabain-induced effects in TJ barrier function. a Monolayers of Caco-2 cells were grown on Transwell polycarbonate filters, pretreated with PD98059 (as indicated) and treated (3 or 6 h) with ouabain; then, transepithelial electrical resistance (TER) values were determined using a Millicel-ERS system. Error bars indicate the mean ± SEM (n = 3). **p < 0.01, ***p < 0.001, as determined by ANOVA. b Caco-2 cells were seeded on glass coverslips, pretreated with PD98059 (as indicated), treated with ouabain for 6 h and processed to immunofluorescence using nonpermeabilizing conditions to evaluate macromolecular permeability. Uvomorulin/E-cadherin staining was visualized by optical microscopy. A positive control to uvomorulin/E-cadherin staining was performed by incubating the cell monolayers for 30 min in culture medium containing a low concentration (LC) of calcium (4 μM). Bar = 5 μm
ERK1/2 Pathway Mediates the Increase in Cell Proliferation Induced by Early Treatment with Ouabain
It has been reported that ouabain can prompt changes in retinal cell survival (de Rezende Corrêa et al. 2005), proliferation in cardiomyocytes (Mohammadi et al. 2001) and cell death in peripheral blood lymphocytes (Esteves et al. 2005). Thus, we evaluated if ouabain could promote alterations in cell viability and proliferation in Caco-2 cells. Using the MTT method, we showed that ouabain was able to increase cell viability with shorter treatment times (8 h) and only led to a significant decrease in viability after prolonged treatment (12–24 h) (Fig. 6a). Moreover, ouabain increased cell proliferation after treatment for 8 h as observed by the violet crystal method. Pretreatment with PD98059 abolished this effect (Fig. 6b), which suggests that it was modulated by ERK1/2. These results indicate that ouabain increases cell viability and proliferation with early treatment times, and the latter is mediated by ERK1/2 signaling.

Impact of ERK1/2 pathway inhibition on ouabain-induced cell viability and proliferation. a Monolayers of Caco-2 cells were grown and treated with ouabain (10 or 100 μM) for 8, 12 or 24 h; and cell viability was evaluated using the MTT assay. Bar graph shows the optical density (538 nm) relative to control. b Monolayers of Caco-2 cells were grown, pretreated with PD98059 as indicated and treated or not with ouabain (10 or 100 μM) for 8 h. Proliferation was evaluated using the violet crystal method. Bar graph shows the optical density (595 nm) relative to control. Error bars indicate mean ± SEM (n = 3). *p < 0.05, **p < 0.01, as determined by ANOVA
Discussion
The discovery that Na,K-ATPase can modulate the AJC in epithelial cells led to efforts to understand whether this enzyme also participates in regulating cell–cell adhesion in cancer. In particular, the role of this enzyme in colorectal cancer is completely unknown. In the present study, we showed that inhibition of Na,K-ATPase with ouabain caused structural and functional alterations of the AJC and decreased levels of α1 and β1 Na,K-ATPase, which were accompanied by increased cell viability and proliferation at short treatment times. Furthermore, we demonstrated that the ouabain-induced events were mediated by ERK1/2 signaling.
Our results revealed that ouabain treatment caused disorganization of AJs, as evidenced by cell redistribution of E-cadherin and α1 Na,K-ATPase (Fig. 1a), followed by a decrease of α1 Na,K-ATPase in intercellular contacts and its protein levels (Fig. 1b and c, respectively). Furthermore, wide spaces surrounding the AJ area were observed by electron microscopy (Fig. 1d). We also observed decreased protein levels of β1 Na,K-ATPase but no changes in the protein levels of E-cadherin and β-catenin (Fig. 2a, b). It has been reported that the interaction between E-cadherin and the β1-subunit of Na,K-ATPase is necessary to maintain AJ stability (Rajasekaran et al. 2005; Vagin et al. 2007). The observation that ouabain can induce β1 Na,K-ATPase downregulation indicates that this glycoside can destabilize AJs, leading to cell–cell adhesion loss in Caco-2 cells. Other studies have reported that ouabain treatment can lead to internalization of the pump (Tian et al. 2009; Liu and Shapiro 2007), an event that would facilitate the output of E-cadherin and β-catenin from the AJs to the cytoplasm, causing AJ disassembly. We did not observe pump internalization, but we do not preclude that this event may occur at later times given that cytoplasmic staining projections of α1 Na,K-ATPase were observed with a higher ouabain dose (Fig. 1a). However, more studies are necessary to confirm this possibility.
Downregulation of the β1-subunit and inhibition of Na,K-ATPase through activation of the MAPK pathway have been reported in MDCK cells (Contreras et al. 2004; Inge et al. 2008), but this relationship has not been described in colorectal cancer cells. Here, we observed that pharmacologic inhibition of ERK1/2 with the specific inhibitor PD98059 prevented the redistribution of E-cadherin and β-catenin caused by ouabain treatment (Fig. 3a). Furthermore, we showed that ouabain caused a significant increase in the levels of ERK1/2 phosphorylation, indicating activation of this pathway, and that this effect was prevented using PD98059 (Fig. 3b). We did not observe β-catenin nuclear translocation following treatment with ouabain, which does not agree with the findings reported by Contreras et al. (2004). The differences in the cell models and the concentration of ouabain used in both studies may explain these discrepant observations. MAPK pathway activation regulating AJ disassembly has also been described in MCF10A cells transformed with H-Ras (Li and Mattingly 2008) and in Caco-2 cells treated with phorbol ester (Barbosa et al. 2003). In addition, treatment of LLC-PK1 cells with TGFβ1 caused a decrease in protein levels of β1 Na,K-ATPase, which was associated with sustained ERK1/2 activation and induction of epithelial-to-mesenchymal transition, an event commonly associated with tumorigenesis (Rajasekaran et al. 2010). Additionally, a study showed that treatment of colon cancer cells with other cardiac glycosides resulted in tumor suppression (Felth et al. 2009). However, these controversial findings might be explained by the use of these drugs in combination with chemotherapeutic agents and in various cell lines that differ in their malignant potential. Based on these observations and our own, it is reasonable to suggest that inhibition of Na,K-ATPase by ouabain may contribute to the progression of colorectal cancer through AJ disassembly, an initial event of this process.
Studies in MDCK cells have reported that ouabain increases the TER and decreases the dextran flux in addition to upregulation and cell redistribution of claudins 1, 2 and 4 (Larre et al. 2010, 2011). To determine whether ouabain may alter TJ function in our model, we first analyzed the distribution and protein expression of claudins 1 and 3 after treatment with the glycoside. We observed that ouabain did not alter the cell distribution pattern of claudins 1 and 3 (Fig. 4a) or the protein levels of claudin-1. However, ouabain treatment increased claudin-3 protein levels, which were also regulated by the ERK1/2 pathway (Fig. 4b). It has been reported that paracellular flux of ions and small molecules is regulated by claudins, but this regulation is differential, depends on the cell type and is specific to claudin (Krug et al. 2012). Additionally, the participation of other TJ transmembrane proteins such as occludin or tricellulin in the regulation of the macromolecular flux has been reported (Weber 2012). We have reported that patients with colorectal cancer presented increased levels of claudins 1, 3 and 4, which were associated with significant disorganization of the TJ strands and increased paracellular permeability (Oliveira et al. 2005). Claudin-3 upregulation decreased barrier function, while the overexpression of claudin-4 improved the alveolar TER (Mitchell et al. 2011). Additionally, the participation of the ERK1/2 pathway in modulating the expression of claudins 1, 2, 3 and 4 in MDCK cells has been reported (Ikari et al. 2011; Singh and Harris 2004). Here, we showed that ouabain decreases the TER and increases the paracellular flux of macromolecules, impairing the TJ barrier function in Caco-2 cells, which were modulated by the ERK1/2 pathway (Fig. 5a, b). Taken together, our findings suggest that the impairment in TJ function induced by ouabain is a consequence of increased claudin-3 expression and AJ disassembly. However, despite the fact that claudins regulate directly the paracellular flux of ions and small molecules, we do not discard the possibility that increased expression of claudin-3 may cause a redistribution of occludin and/or tricellulin, impairing also the macromolecular flux. Thus, impaired barrier function may facilitate the access of ouabain to Na,K-ATPase, which is mainly present in the basolateral region, and inhibit its activity promoting a sustained loss of cell–cell adhesion in colorectal cancer cells. However, more studies are necessary to confirm this hypothesis.
Note that ouabain is toxic at certain concentrations and treatment times. We observed that ouabain treatment increased viability and proliferation in Caco-2 cells with a short treatment time (8 h). Cell proliferation was also modulated by the ERK1/2 pathway. Decreased cell viability was only observed in long-term ouabain treatments (12–24 h) (Fig. 6a, b). Differential effects on cell proliferation or viability depending on ouabain concentration have been described (Bagrov et al. 2009). The ouabain-induced effects described in the present study showing ERK1/2 signaling activation and increased cell proliferation are in agreement with previously published studies (Xie and Askari 2002; Mohammadi et al. 2001). These authors demonstrated that pretreatment with 50–100 μM ouabain activated MAPK pathways leading to cell proliferation. Importantly, our study describes ouabain-induced ERK1/2 pathway activation in a cell line that does not possess caveolae. The consensus in the literature is that caveolae are necessary for Na,K-ATPase to act as a hormone receptor (Liu et al. 2003; Wang et al. 2004; Li and Xie 2009). However, recently it was demonstrated that a noncaveolar kidney membrane preparation also displayed signaling properties (Liu et al. 2011). This conclusion followed the observation that the noncaveolar model expressed some kinase proteins related with the Na,K-ATPase signaling function more highly than in the caveolar membrane. Accordingly, our study is the first to report that Na,K-ATPase can be modulated to activate cell signaling pathways independently from the membrane domain in a cell culture system.
In conclusion, we showed that ouabain treatment induced α1 and β1 Na,K-ATPase downregulation in addition to structural and functional changes in the AJC. These events led to cell–cell adhesion loss, impaired TJ barrier function and increased viability and proliferation of colorectal cancer cells. These actions were mediated by ERK1/2 signaling but occurred in a caveolae-independent manner. Although more experimental evidence is required to confirm the molecular mechanisms regulating these events, our findings provide a solid foundation to better understand the mechanisms underlying the AJC disassembly mediated by Na,K-ATPase during colorectal cancer progression.
References
Bagrov AY, Shapiro JI, Fedorova OV (2009) Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Am J Hypertens 22(5):559–563
Barbosa LA, Goto-Silva L, Redondo PA et al (2003) TPA-induced signal transduction: a link between PKC and EGFR signaling modulates the assembly of intercellular junctions in Caco-2 cells. Cell Tissue Res 312:319–331
Breuza L, Corby S, Arsanto JP et al (2002) The scaffolding domain of caveolin 2 is responsible for its Golgi localization in Caco-2 cells. J Cell Sci 115(Pt 23):4457–4467
Cereijido M, Contreras RG, Shoshani L, Larre I (2012) The Na+-K+-ATPase as self-adhesion molecule and hormone receptor. Am J Physiol Cell Physiol 302:C473–C481
Contreras RG, Flores-Maldonado C, Lazaro A et al (2004) Ouabain binding to Na+, K+-ATPase relaxes cell attachment and sends a specific signal (NACos) to the nucleus. J Membr Biol 198:147–158
de Freitas JC Jr, Silva Bdu R, de Souza WF et al (2011) Inhibition of N-linked glycosylation by tunicamycin induces E-cadherin-mediated cell–cell adhesion and inhibits cell proliferation in undifferentiated human colon cancer cells. Cancer Chemother Pharmacol 68(1):227–238
de Rezende Corrêa G, Araujo dos Santos A, Frederico Leite Fontes C, Giestal de Araujo E (2005) Ouabain induces an increase of retinal ganglion cell survival in vitro: the involvement of protein kinase C. Brain Res 1049(1):89–94
Esteves MB, Marques-Santos LF, Affonso-Mitidieri OR, Rumjanek VM (2005) Ouabain exacerbates activation-induced cell death in human peripheral blood lymphocytes. An Acad Bras Cienc 77(2):281–292
Felth J, Rickardson L, Rosén J et al (2009) Cytotoxic effects of cardiac glycosides in colon cancer cells, alone and in combination with standard chemotherapeutic drugs. J Nat Prod 72:1969–1974
Ikari A, Takiguchi A, Atomi K, Sugatani J (2011) Epidermal growth factor increases clathrin-dependent endocytosis and degradation of claudin-2 protein in MDCK II cells. J Cell Physiol 226:2448–2456
Inge LJ, Rajasekaran SA, Yoshimoto K et al (2008) Evidence for a potential tumor suppressor role for the Na, K-ATPase beta1-subunit. Histol Histopathol 23:459–467
Kometiani P, Liu L, Askari A (2005) Digitalis-induced signaling by Na+/K+-ATPase in human breast cancer cells. Mol Pharmacol 67:929–936
Krug SM, Günzel D, Conrad MP et al (2012) Charge-selective claudin channels. Ann N Y Acad Sci 1257:20–28
Larre I, Lazaro A, Contreras RG et al (2010) Ouabain modulates epithelial cell tight junction. Proc Natl Acad Sci USA 107(25):11387–11392
Larre I, Contreras RG, Cereijido M (2011) Ouabain modulates cell contacts as well as functions that depend on cell adhesion. Methods Mol Biol 763:155–168
Li Q, Mattingly RR (2008) Restoration of E-cadherin cell–cell junctions requires both expression of E-cadherin and suppression of ERK MAP kinase activation in Ras-transformed breast epithelial cells. Neoplasia 10:1444–1458
Li Z, Xie Z (2009) The Na/K-ATPase/Src complex and cardiotonic steroid-activated protein kinase cascades. Pflugers Arch 457:635–644
Liang M, Tian J, Liu L et al (2007) Identification of a pool of non-pumping Na/K-ATPase. J Biol Chem 282(14):10585–10593
Liu J, Shapiro JI (2007) Regulation of sodium pump endocytosis by cardiotonic steroids: molecular mechanisms and physiological implications. Pathophysiology 14:171–181
Liu L, Mohammadi K, Aynafshar B et al (2003) Role of caveolae in signal-transducing function of cardiac Na+/K+-ATPase. Am J Physiol Cell Physiol 284:C1550–C1560
Liu L, Ivanov AV, Gable ME et al (2011) Comparative properties of caveolar and noncaveolar preparations of kidney Na+/K+-ATPase. Biochemistry 50(40):8664–8673
Mijatovic T, Van Quaquebeke E, Delest B et al (2007) Cardiotonic steroids on the road to anti-cancer therapy. Biochim Biophys Acta 1776:32–57
Mineta K, Yamamoto Y, Yamazaki Y et al (2011) Predicted expansion of the claudin multigene family. FEBS Lett 585(4):606–612
Mitchell LA, Overgaard CE, Ward C et al (2011) Differential effects of claudin-3 and claudin-4 on alveolar epithelial barrier function. Am J Physiol Lung Cell Mol Physiol 301:L40–L49
Miyoshi J, Takai Y (2008) Structural and functional associations of apical junctions with cytoskeleton. Biochim Biophys Acta 1778:670–691
Mohammadi K, Kometiani P, Xie Z, Askari A (2001) Role of protein kinase C in the signal pathways that link Na+/K+-ATPase to ERK1/2. J Biol Chem 276(45):42050–42056
Morth JP, Poulsen H, Toustrup-Jensen MS et al (2009) The structure of the Na+, K+-ATPase and mapping of isoform differences and disease-related mutations. Philos Trans R Soc Lond B 364(1514):217–227
Oliveira SS, Oliveira IM, Souza W, Morgado-Díaz JA (2005) Claudins upregulation in human colorectal cancer. FEBS Lett 579:6179–6185
Rajasekaran SA, Ball WJ Jr, Bander NH et al (1999) Reduced expression of β-subunit of Na, K-ATPase in human clear-cell renal cell carcinoma. J Urol 162:574–580
Rajasekaran SA, Palmer LG, Quan K et al (2001) Na, K-ATPase beta-subunit is required for epithelial polarization, suppression of invasion, and cell motility. Mol Biol Cell 12:279–295
Rajasekaran SA, Barwe SP, Rajasekaran AK (2005) Multiple functions of Na, K-ATPase in epithelial cells. Semin Nephrol 25:328–334
Rajasekaran SA, Huynh TP, Wolle DG et al (2010) Na, K-ATPase subunits as markers for epithelial–mesenchymal transition in cancer and fibrosis. Mol Cancer Ther 9:1515–1524
Singh AB, Harris RC (2004) Epidermal growth factor receptor activation differentially regulates claudin expression and enhances transepithelial resistance in Madin-Darby canine kidney cells. J Biol Chem 279(5):3543–3552
Takizawa Y, Kishimoto H, Kitazato T et al (2012) Changes in protein and mRNA expression levels of claudin family after mucosal lesion by intestinal ischemia/reperfusion. Int J Pharm 426(1–2):82–89
Tian J, Li X, Liang M et al (2009) Changes in sodium pump expression dictate the effects of ouabain on cell growth. J Biol Chem 284:14921–14929
Vagin O, Sachs G, Tokhtaeva E (2007) The roles of the Na, K-ATPase beta 1 subunit in pump sorting and epithelial integrity. J Bioenerg Biomembr 39:367–372
Wang H, Haas M, Liang M et al (2004) Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J Biol Chem 279(17):17250–17259
Wang Z, Zheng M, Li Z et al (2009) Cardiac glycosides inhibit p53 synthesis by a mechanism relieved by Src or MAPK inhibition. Cancer Res 69:6556–6564
Weber CR (2012) Dynamic properties of the tight junction barrier. Ann N Y Acad Sci 1257:77–84
Xie Z, Askari A (2002) Na+/K+-ATPase as a signal transducer. Eur J Biochem 269(10):2434–2439
Yap AS, Crampton MS, Hardin J (2007) Making and breaking contacts: the cellular biology of cadherin regulation. Curr Opin Cell Biol 19:508–514
Acknowledgments
This research was supported by Ministério da Saúde (Brazil), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (Grant E26/170.026/2008), Conselho Nacional de Desenvolvimento Científico e Tecnológico (Grant 573806/2008-0) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior. The text was reviewed by American journal experts.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Fig. S1
Effect of low dose of ouabain and ionic imbalance on cell distribution of E-cadherin.Monolayers of Caco-2 cells were grown on glass coverslips. Cells were treated with a low concentration of ouabain (100 nM) for 6 h or with a molar excess of NaCl (250 mM) for 2.5 h. After, cells were processed for immunofluorescence analysis of E-cadherin distribution. Bar = 10 μm (TIFF 629 kb)
Fig. S2
Impact of ERK1/2 pathway inhibition on the ouabain-induced effects in β1 Na,K-ATPase levels. Monolayers of Caco-2 cells were grown, pretreated with an inhibitor of the ERK1/2 pathway (PD98059) for 1 h (when indicated) and incubated with ouabain for 6 h. After treatment, total cell lysates were harvested and the expression of β1 Na,K-ATPase was visualized by immunoblotting. Ponceau staining (Ponc.) was used as a loading control. Numbers represent the ratio of the optical density of treated to untreated cells (TIFF 59 kb)
Rights and permissions
About this article
Cite this article
de Souza, W.F., Barbosa, L.A., Liu, L. et al. Ouabain-Induced Alterations of the Apical Junctional Complex Involve α1 and β1 Na,K-ATPase Downregulation and ERK1/2 Activation Independent of Caveolae in Colorectal Cancer Cells. J Membrane Biol 247, 23–33 (2014). https://doi.org/10.1007/s00232-013-9607-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00232-013-9607-y