
Abstract
Predicting membrane protein type is a meaningful task because this kind of information is very useful to explain the function of membrane proteins. Due to the explosion of new protein sequences discovered, it is highly desired to develop efficient computation tools for quickly and accurately predicting the membrane type for a given protein sequence. Even though several membrane predictors have been developed, they can only deal with the membrane proteins which belong to the single membrane type. The fact is that there are membrane proteins belonging to two or more than two types. To solve this problem, a system for predicting membrane protein sequences with single or multiple types is proposed. Pseudo–amino acid composition, which has proven to be a very efficient tool in representing protein sequences, and a multilabel KNN algorithm are used to compose this prediction engine. The results of this initial study are encouraging.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Almost all living cells are enclosed by membranes which are composed of mainly lipids and proteins that play various roles (Chou and Elrod 1999); for instance, membranes define cell boundaries, maintaining the essential differences between the cytosol and the extracellular environment, and some of them offer the skeleton for the lipid bilayer membrane. Membrane proteins can be mainly divided into eight types (Chou and Shen 2007b): (1) single-pass type I membrane, (2) single-pass type II membrane, (3) single-pass type III membrane, (4) single-pass type IV membrane, (5) multipass membrane, (6) lipid-anchor membrane, (7) GPI-anchor membrane and (8) peripheral membrane.
Knowledge about the type of a particular membrane protein is very helpful because this kind of information is highly correlated with its function (Nanni and Lumini 2008). Between 20 and 35 % of genes encode membranes, whereas only 1 % of proteins are membrane proteins whose 3D structures are known (Nanni and Lumini 2008). Determining the membrane protein type through multifarious biochemical experiments is not only resource-intensive but time-consuming, so developing automated methods for efficiently and accurately identifying the types of given proteins is highly desired.
The prediction of membrane protein type is similar to the problem of subcellular localization. Several different types of subcellular localization predictors have been proposed in the last decade or so (Chou and Shen 2006, 2007a, 2008, 2010b; Chou et al. 2011, 2012; Shen and Chou 2007, 2009, 2010a, 2010b; Wu et al. 2011, 2012; Xiao et al. 2011b, 2011c). Also, a series of classifiers and methods have been developed to identify membrane protein sequences (Chen and Li 2013; Chou and Cai 2005; Chou and Shen 2007b; Nanni and Lumini 2008). All of them can only deal with membrane protein sequences in which one sequence belongs to only one type. To the best of our knowledge, no predictor can handle the problem that one membrane sequence belongs to two or more than two types. But these types of protein are also very important because they may have some special biological significance. Considering the fact that several predictors which can deal with protein subcellular localization with both single and multiple sites have been established, it is urgent and meaningful to develop methods or predictors which are able to handle membrane protein sequences with single and multiple types.
In this article, we adopt a multilabel algorithm named ML_KNN, whose basic consideration is a multilabel-based K-nearest neighbor algorithm (KNN) derived from the common K-nearest neighbor algorithm (Zhang and Zhou 2007) and pseudo–amino acid composition which has proven to be a very efficient tool in representing protein sequences to compose this new classifier. Application to a rigorous benchmark data set shows that this prediction model performs well, as an initial study on this new topic.
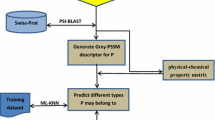
According to a comprehensive review (Chou 2011) and as demonstrated by a series of recent publications (Chen et al. 2012, 2013; Lin et al. 2012; Wang et al. 2011; Xiao et al. 2011a, 2012), to establish a really useful statistical predictor for a protein system, we need to consider the following procedures: (1) construct or select a valid benchmark data set to train and test the predictor, (2) formulate the protein samples with an effective mathematical expression that can truly reflect their intrinsic correlation with the attribute to be predicted, (3) introduce or develop a powerful algorithm (or engine) to operate the prediction and (4) properly perform cross-validation tests to objectively evaluate the anticipated accuracy of the predictor. Below, we describe how to deal with these steps.
Materials and Methods
Data Set
The protein data set was taken from the UniprotKB/Swiss-Prot database at (http://www.ebi.ac.uk/uniprot/) released in June 2012. The detailed procedures are as follows: (1) open the Web site at http://www.uniprot.org/; (2) click the button “Advanced,” select “Subcellular Location” for “Fields,” type in “single-pass type I membrane” for “Term” and select “Experimental” for “Confidence”; (3) click the button “Add&Search,” select “or” and repeat step 2 with the only difference being that each of the following terms is typed in once in order until all of them are used: “single-pass type II membrane,” “single-pass type III membrane,” “single-pass type IV membrane,” “multipass membrane protein,” “lipid-anchor,” “GPI-anchor,” “peripheral membrane protein”; (4) click the button “Add&Search,” choose “and,” select “Fragment(yes/no)” for “Field” and choose “no”; (5) click the button “Add&Search,” choose “and,” select “Sequence Length” for “Field” and choose the sequence length “≥50.”
CD-HIT software (Huang et al. 2010; Li and Godzik 2006; Niu et al. 2010) was used to exclude those sequences which have more than 80 % sequence identity to any others in the same membrane type group.
The information of this benchmark data set is listed in Table 1.
Feature Extraction
Chou’s Pseudo–Amino Acid–Based Features
One of the most efficient methods to engender the sample of a query protein P is the pseudo–amino acid composition (Chou 2001, 2011; Shen and Chou 2008), which has been widely applied to predict a myriad of protein attributes (Esmaeili et al. 2010; Fan and Li 2012; Georgiou et al. 2009; Hayat and Khan 2012; Jiang et al. 2008; Khosravian et al. 2013; Mei 2012; Mohabatkar 2010; Mohabatkar et al. 2011, 2013; Mohammad Beigi et al. 2011; Nanni et al. 2012a, 2012b; Niu et al. 2012; Xiao et al. 2006a, 2006b; Zhang et al. 2008; Zia Ur and Khan 2012). In this study, we also adapted this method to construct the query proteins.
The model can be divided into two parts. One part is just its amino acid composition, which includes 20 discrete numbers, each of them representing the normalized occurrence frequencies of one of the native amino acids in protein P. The other part is the pseudo–amino acid composition part, which takes advantage of the information from the sequence order effect. The steps are shown below.
Suppose a protein including l amino acid residues:
The sequence-order information can be indirectly represented by the following equations
In Eq. (2) the correlation function is defined by
where F(Q i ), G(Q i ) and H(Q j ) are the values of hydrophobicity, hydrophilicity and mass, respectively. There are also three types of value that can be used. Before we use these values, a standard conversion described by the following should be conducted:
where F 0(i) is the original hydrophobicity value of the ith amino acid, G 0(i) is the original hydrophilicity value of the ith amino acid and H 0(i) is the original mass value of the ith amino acid side chain. These data were achieved from the Web server PseACC (Shen and Chou 2008). We use numbers 1–20 to denote the 20 native amino acids according to the order of their three-letter names: Ala (A), Arg (R), Asn (N), Asp (D), Cys (C), Gln (Q), Glu (E), Gly (G), His (H), Ile (I), Leu (L), Lys (K), Met (M), Phe (F), Pro (P), Ser (S), Thr (T), Trp (W), Tyr (Y), Val (V).
Then, a sample protein P can be represented as
where
where μ is the weight factor, which was set at 0.5 (Chou 2005; Chou and Cai 2005) and 0.05 (Chou 2001); \( t_{i} (i = 1,2, \ldots ,20) \) represents the normalized occurrence frequencies of the 20 amino acids in the sample protein P; and δ j is the j-tier sequence-correlation factor, computed according to Eq. (2). In this article, we chose μ = 0.05, η = 20 after careful consideration of easy handling; they can be assigned other values, of course, but the impact on the result would be small.
Algorithms for Classification
ML_KNN
In this article, we follow the notations used by Zhang and Zhou (2007). Define ℑ = Hd as the input vector space, \( {\mathbb{C}} = \{ 1,2,3, \ldots ,C\} \) as the set of C possible labels and \( {\mathbb{Z}} = \{ (q_{i} ,t_{i} ),1 \le i \le N\} \) as a train set in which \( q_{i} \in \Im ,t_{i} \subseteq {\mathbb{C}} \), using \( {\mathbb{Z}} \) to train the multilabel classifier. Usually, the learning model will output a real valued vector based on the function \( g:\;\Im \times {\mathbb{C}} \Rightarrow {\rm H} \). Considering q i and its corresponding label set t i , g(*,*) has the character of \( g(q_{i} ,c_{1} ) < g(q_{i} ,c_{2} ) \) when any \( c_{1} \notin t_{1} \;and\;c_{2} \in t_{2} \). Apparently, the result yields larger values for labels belonging to t i rather than those not belonging to t i . On the side, we use rank g (q i ,c) to represent the ranking function derived from g(q i ,c) which outputs the rank of c(\( {c \in {\mathbb{C}}} \)). Clearly, the larger value of g(q i ,c) corresponds with the higher rank of c. The multilabel classifier t(*) can also be computed by g(*,*) as \( t(q_{i} ) = \{ c|g(q_{i} ,c) > u(q_{i} ),c \in {\mathbb{C}}\} \), where u(*) is a threshold function usually set to 0 for easy handling.
The KNN used here is considered in the multilabel way (Zhang and Zhou 2007). Considering an instance q and its corresponding label set \( C^{\prime} \subseteq {\mathbb{C}} \), let \( \overrightarrow {{f_{q} }} \) be the full category vector for q, in which its jth component, \( \overrightarrow {{f_{q} (j)}} (j \in {\mathbb{C}}) \), takes the value of 0 if \( j \notin C^{\prime} \) and 1 otherwise. Moreover, we use L(q) to represent the set of K-nearest neighbors of q computed in the training set, so an element counting vector can be set as
where \( \overrightarrow {{N_{q} (j)}} \) computes the number of neighbors of q associated with the jth label.
As for each test vector p, its K-nearest neighbor, L(p), is computed first. Let G 0 j represent the fact that p has no label j and G 1 j otherwise. In addition, let \( F_{i}^{j} (i \in \{ 0,1,2,3, \cdots ,K\} ) \) express the fact that there are exactly i examples that have label j among the K-nearest neighbors of p. Thus, given the element counting vector \( \overrightarrow {{N_{p} }} \), the category vector \( \overrightarrow {{f_{p} (j)}} \) can be determined as follows:
According to the Bayesian rule, Eq. (8) can be represented by
The prior probabilities, \( P(G_{j}^{a} )\,(j \in {\mathbb{C}},a \in \{ 0,1\} ) \), and the posterior probabilities, \( P(F_{i}^{j} |G_{j}^{a} )\,(i \in \{ 0,1, \ldots ,K\} ) \), are needed to determine the category vector; and these data can be taken directly from the training set.
Pseudocode and more details of KNN can be viewed in Zhang and Zhou (2007). The source code of ML_KNN can be downloaded at http://cse.seu.edu.cn/people/zhangml/Resources.htm#codes.
Evaluation measures
Given a multilabel test data set, \( \chi = \left\{ {(x_{i} ,X_{i} )\,1 \le i \le n} \right\} \), based on the definition in the previous section, the following popular multilabel evaluation metrics (Schapire and Singer 2000; Zhang 2006, 2009; Zhang et al. 2009; Zhang and Zhou 2007) are used:
-
(1)
Hamming Loss:
\( \Updelta \) represents the symmetric difference between two data sets
-
(2)
One-Error:
-
(3)
Coverage:
-
(4)
Ranking Loss:
-
(5)
Average Precision:
In all, Hamming loss evaluates the times that an instance-label pair is misclassified; one-error evaluates the times that the top-ranked label is not in the set of proper labels of the instance; coverage evaluates the number of steps needed, on the average, to move down the label list in order to cover all the proper labels attached to an instance; ranking loss examines the average fraction of label pairs that are reversely ordered for the instance; average precision evaluates the average fraction of labels which are ranked above a particular label \( h \in X \) and really are in X. Note that, for the first four metrics, the smaller the better and, for the last one, the larger the better performance.
Results and Discussion
In statistical prediction, the following three cross-validation methods are often used to examine a predictor for its effectiveness in practical application: independent data set test, subsampling test and jackknife test. However, of the three test methods, the jackknife test is deemed the least arbitrary that can always yield a unique result for a given benchmark data set, as elaborated in Chou and Shen (2010a) and demonstrated by equations 28–30 in Chou (2011). Accordingly, the jackknife test has been widely recognized and increasingly used by investigators to examine the quality of various predictors (Chen and Li 2013; Esmaeili et al. 2010; Mohabatkar 2010; Sahu and Panda 2010; Sun et al. 2012; Zhao et al. 2012; Zia Ur and Khan 2012). However, to reduce the computational time, we adopted the fivefold cross-validation in this study as done by many investigators with SVM as the prediction engine.
Tables 2, 3, 4, 5 and 6 provide the test results based on different K numbers. For each K number, fivefold cross-validation is performed on the data set, and the performances (mean ± standard deviation) out of five independent runs are presented. As the tables show, for each evaluation measurement, ↓ represents the smaller the better and ↑ represents the larger the better. The K number of the multilabel-based KNN classifier is the most important parameter that may directly affect the predicted result. Thus, it is meaningful to see how prediction is influenced by the parameter K on the membrane data set used. The parameter K was increased from 1 to 20 with a step of 1. The overall values are estimated by the method of fivefold cross-validation. It is quite clear that the differences between results of each multilabel evaluation metric are negligible (Fig. 1). This fact indicates that no matter which K number is chosen, the prediction result is highly robust and dependable. Considering the fact that it is a new study focusing on this very topic, the performance is quite encouraging in comparison with the performance of similar methods applied in other biometric data sets (Zhang 2006, 2009).

Prediction results on the membrane data set using fivefold cross-validation under different K: (a) Hamming loss, (b) one-error, (c) coverage, (d) ranking loss, (e) average precision
Conclusions
Prediction of membrane protein type is a meaningful and challenging task. Even though several models have been proposed, to the best of our knowledge, there is no algorithm to deal with proteins with multiple membrane types. However, those proteins are still important owing to the fact that they may represent some special biological significance worth our attention.
In this study, a new model for predicting membrane proteins with single or multiple types was proposed. The predictor is applicable in annotating membrane protein types. The prediction results are listed in Tables 2, 3, 4, 5 and 6, which are sufficiently good for initial research. We also presented the performances of the algorithm using different K numbers in order to investigate the impact of parameter K on the prediction performance. In the future, we will investigate other types of algorithm for the sake of improving the performance of the prediction.
Since user-friendly and publicly accessible web-servers represent the future direction for developing practically more useful models, simulated methods or predictors (Chou and Shen 2009), we shall make efforts in our future work to provide a web-server for the method presented in this article.
References
Chen YK, Li KB (2013) Predicting membrane protein types by incorporating protein topology, domains, signal peptides, and physicochemical properties into the general form of Chou’s pseudo amino acid composition. J Theor Biol 318:1–12
Chen W, Lin H, Feng PM, Ding C, Zuo YC, Chou KC (2012) iNuc-PhysChem: a sequence-based predictor for identifying nucleosomes via physicochemical properties. PLoS One 7:e47843
Chen W, Feng PM, Lin H, Chou KC (2013) iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. doi:101093/nar/gks1450
Chou KC (2001) Prediction of protein cellular attributes using pseudo–amino acid composition. Proteins 43:246–255
Chou KC (2005) Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 21:10–19
Chou KC (2011) Some remarks on protein attribute prediction and pseudo amino acid composition. J Theor Biol 273:236–247
Chou KC, Cai YD (2005) Prediction of membrane protein types by incorporating amphipathic effects. J Chem Inf Model 45:407–413
Chou KC, Elrod DW (1999) Prediction of membrane protein types and subcellular locations. Proteins 34:137–153
Chou KC, Shen HB (2006) Predicting protein subcellular location by fusing multiple classifiers. J Cell Biochem 99:517–527
Chou KC, Shen HB (2007a) Euk-mPLoc: a fusion classifier for large-scale eukaryotic protein subcellular location prediction by incorporating multiple sites. J Proteome Res 6:1728–1734
Chou KC, Shen HB (2007b) MemType-2L: a web server for predicting membrane proteins and their types by incorporating evolution information through Pse-PSSM. Biochem Biophys Res Commun 360:339–345
Chou KC, Shen HB (2008) Cell-PLoc: a package of web servers for predicting subcellular localization of proteins in various organisms. Nat Protoc 3:153–162
Chou KC, Shen HB (2009) Recent advances in developing Web-servers for predicting protein attributes. Nat Sci 1:63–92
Chou KC, Shen HB (2010a) Cell-PLoc 2.0: an improved package of web-servers for predicting subcellular localization of proteins in various organisms. Nat Sci 2:1090–1103
Chou KC, Shen HB (2010b) Plant-mPLoc: a top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS One 5:e11335
Chou KC, Wu ZC, Xiao X (2011) iLoc-Euk: a multi-label classifier for predicting the subcellular localization of singleplex and multiplex eukaryotic proteins. PLoS One 6:e18258
Chou KC, Wu ZC, Xiao X (2012) iLoc-Hum: using the accumulation-label scale to predict subcellular locations of human proteins with both single and multiple sites. Mol Biosyst 8:629–641
Esmaeili M, Mohabatkar H, Mohsenzadeh S (2010) Using the concept of Chou’s pseudo amino acid composition for risk type prediction of human papilloma viruses. J Theor Biol 263:203–209
Fan GL, Li QZ (2012) Predict mycobacterial proteins subcellular locations by incorporating pseudo-average chemical shift into the general form of Chou’s pseudo amino acid composition. J Theor Biol 304:88–95
Georgiou DN, Karakasidis TE, Nieto JJ, Torres A (2009) Use of fuzzy clustering technique and matrices to classify amino acids and its impact to Chou’s pseudo amino acid composition. J Theor Biol 257:17–26
Hayat M, Khan A (2012) Discriminating outer membrane proteins with fuzzy K-nearest neighbor algorithms based on the general form of Chou’s PseAAC. Protein Pept Lett 19:411–421
Huang Y, Niu B, Gao Y, Fu L, Li W (2010) CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics 26:680–682
Jiang X, Wei R, Zhang T, Gu Q (2008) Using the concept of Chou’s pseudo amino acid composition to predict apoptosis proteins subcellular location: an approach by approximate entropy. Protein Pept Lett 15:392–396
Khosravian M, Faramarzi FK, Beigi MM, Behbahani M, Mohabatkar H (2013) Predicting antibacterial peptides by the concept of Chou’s pseudo–amino acid composition and machine learning methods. Protein Pept Lett 20:180–186
Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659
Lin WZ, Fang JA, Xiao X, Chou KC (2012) Predicting secretory proteins of malaria parasite by incorporating sequence evolution information into pseudo amino acid composition via grey system model. PLoS One 7:e49040
Mei S (2012) Multi-kernel transfer learning based on Chou’s PseAAC formulation for protein submitochondria localization. J Theor Biol 293:121–130
Mohabatkar H (2010) Prediction of cyclin proteins using Chou’s pseudo amino acid composition. Protein Pept Lett 17:1207–1214
Mohabatkar H, Mohammad Beigi M, Esmaeili A (2011) Prediction of GABAA receptor proteins using the concept of Chou’s pseudo-amino acid composition and support vector machine. J Theor Biol 281:18–23
Mohabatkar H, Beigi MM, Abdolahi K, Mohsenzadeh S (2013) Prediction of allergenic proteins by means of the concept of Chou’s pseudo amino acid composition and a machine learning approach. Med Chem 9:133–137
Mohammad Beigi M, Behjati M, Mohabatkar H (2011) Prediction of metalloproteinase family based on the concept of Chou’s pseudo amino acid composition using a machine learning approach. J Struct Funct Genomics 12:191–197
Nanni L, Lumini A (2008) An ensemble of support vector machines for predicting the membrane protein type directly from the amino acid sequence. Amino Acids 35:573–580
Nanni L, Brahnam S, Lumini A (2012a) Wavelet images and Chou’s pseudo amino acid composition for protein classification. Amino Acids 43:657–665
Nanni L, Lumini A, Gupta D, Garg A (2012b) Identifying bacterial virulent proteins by fusing a set of classifiers based on variants of Chou’s pseudo amino acid composition and on evolutionary information. IEEE/ACM Trans Comput Biol Bioinform 9:467–475
Niu B, Fu L, Sun S, Li W (2010) Artificial and natural duplicates in pyrosequencing reads of metagenomic data. BMC Bioinformatics 11:187
Niu XH, Hu XH, Shi F, Xia JB (2012) Predicting protein solubility by the general form of Chou’s pseudo amino acid composition: approached from chaos game representation and fractal dimension. Protein Pept Lett 19:940–948
Sahu SS, Panda G (2010) A novel feature representation method based on Chou’s pseudo amino acid composition for protein structural class prediction. Comput Biol Chem 34:320–327
Schapire RE, Singer Y (2000) BoosTexter: a boosting-based system for text categorization. Mach Learn 39:135–168
Shen HB, Chou KC (2007) Hum-mPLoc: an ensemble classifier for large-scale human protein subcellular location prediction by incorporating samples with multiple sites. Biochem Biophys Res Commun 355:1006–1011
Shen HB, Chou KC (2008) PseAAC: a flexible Web server for generating various kinds of protein pseudo amino acid composition. Anal Biochem 373:386–388
Shen HB, Chou KC (2009) Gpos-mPLoc: a top-down approach to improve the quality of predicting subcellular localization of gram-positive bacterial proteins. Protein Pept Lett 16:1478–1484
Shen HB, Chou KC (2010a) Gneg-mPLoc: a top-down strategy to enhance the quality of predicting subcellular localization of gram-negative bacterial proteins. J Theor Biol 264:326–333
Shen HB, Chou KC (2010b) Virus-mPLoc: a fusion classifier for viral protein subcellular location prediction by incorporating multiple sites. J Biomol Struct Dyn 28:175–186
Sun XY, Shi SP, Qiu JD, Suo SB, Huang SY, Liang RP (2012) Identifying protein quaternary structural attributes by incorporating physicochemical properties into the general form of Chou’s PseAAC via discrete wavelet transform. Mol Biosyst 8:3178–3184
Wang P, Xiao X, Chou KC (2011) NR-2L: a two-level predictor for identifying nuclear receptor subfamilies based on sequence-derived features. PLoS One 6:e23505
Wu ZC, Xiao X, Chou KC (2011) iLoc-Plant: a multi-label classifier for predicting the subcellular localization of plant proteins with both single and multiple sites. Mol Biosyst 7:3287–3297
Wu ZC, Xiao X, Chou KC (2012) iLoc-Gpos: a multi-layer classifier for predicting the subcellular localization of singleplex and multiplex gram-positive bacterial proteins. Protein Pept Lett 19:4–14
Xiao X, Shao S, Ding Y, Huang Z, Chou KC (2006a) Using cellular automata images and pseudo amino acid composition to predict protein subcellular location. Amino Acids 30:49–54
Xiao X, Shao SH, Huang ZD, Chou KC (2006b) Using pseudo amino acid composition to predict protein structural classes: approached with complexity measure factor. J Comput Chem 27:478–482
Xiao X, Wang P, Chou KC (2011a) GPCR-2L: predicting G protein–coupled receptors and their types by hybridizing two different modes of pseudo amino acid compositions. Mol Biosyst 7:911–919
Xiao X, Wu ZC, Chou KC (2011b) iLoc-Virus: a multi-label learning classifier for identifying the subcellular localization of virus proteins with both single and multiple sites. J Theor Biol 284:42–51
Xiao X, Wu ZC, Chou KC (2011c) A multi-label classifier for predicting the subcellular localization of gram-negative bacterial proteins with both single and multiple sites. PLoS One 6:e20592
Xiao X, Wang P, Chou KC (2012) iNR-PhysChem: a sequence-based predictor for identifying nuclear receptors and their subfamilies via physical–chemical property matrix. PLoS One 7:e30869
Zhang ML (2006) Multilabel neural networks with applications to functional genomics and text categorization. IEEE Trans Knowl Data Eng 18:1338–1351
Zhang ML (2009) ML-RBF: RBF neural networks for multi-label learning. Neural Process Lett 29:61–74
Zhang ML, Zhou Z-H (2007) ML-KNN: a lazy learning approach to multi-label learning. Pattern Recognit 40:2038–2048
Zhang SW, Zhang YL, Yang HF, Zhao CH, Pan Q (2008) Using the concept of Chou’s pseudo amino acid composition to predict protein subcellular localization: an approach by incorporating evolutionary information and von Neumann entropies. Amino Acids 34:565–572
Zhang ML, Peña JM, Robles V (2009) Feature selection for multi-label naive Bayes classification. Inf Sci 179:3218–3229
Zhao XW, Ma ZQ, Yin MH (2012) Predicting protein–protein interactions by combing various sequence-derived features into the general form of Chou’s pseudo amino acid composition. Protein Pept Lett 19:492–500
Zia Ur R, Khan A (2012) Identifying GPCRs and their types with Chou’s pseudo amino acid composition: an approach from multi-scale energy representation and position specific scoring matrix. Protein Pept Lett 19:890–903
Acknowledgements
C. H. thanks Dr. H. B. Shen at Shanghai Jiaotong University for useful discussion.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Huang, C., Yuan, JQ. A Multilabel Model Based on Chou’s Pseudo–Amino Acid Composition for Identifying Membrane Proteins with Both Single and Multiple Functional Types. J Membrane Biol 246, 327–334 (2013). https://doi.org/10.1007/s00232-013-9536-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00232-013-9536-9