
Abstract
Posttranslational modification is a common cellular process that is used by cells to ensure a particular protein function. This can happen in a variety of ways, e.g., from the addition of phosphates or sugar residues to a particular amino acid, ensuring proper protein life cycle and function. In this review, we assess the evidence for ubiquitination, glycosylation, phosphorylation, S-nitrosylation as well as other modifications in connexins and pannexin proteins. Based on the literature, we find that posttranslational modifications are an important component of connexin and pannexin regulation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Posttranslational modification is a common method by which proteins can be modulated by intrinsic or extrinsic factors to potentiate or initiate a specific function. It is now well accepted that these modifications are a key way in which a protein can become useful in the context of cellular physiology. This is not different when it comes to the connexin and pannexin family of proteins, where a fairly broad range of modifications has now been described that can alter the function of these proteins, from the dramatic (e.g., opening or closing of the channel/gap junction/hemichannel) to the subtle (e.g., insertion into specialized lipid rafts).
Connexin and pannexin proteins are transmembrane proteins that allow for the passive diffusion of signaling molecules through their pores. Connexins are the key components to gap junctions, linking the cytoplasms of two opposing cells and allowing for rapid electrical or chemical integration among cells in a tissue. These proteins, when composed as a gap junction, allow for several functions, including electrical coordination of cardiac myocytes or ciliary beat frequency between tracheal epithelium, with several mutations in connexins associated with disease states (Boitano and Evans 2000; Johnson and Koval 2009; Kelsell et al. 2001; Lai et al. 2006; Palatinus and Gourdie 2007). Undocked connexin hemichannels have also been hypothesized to play a more paracrine role in cellular communication in more pathological states (De Vuyst et al. 2007; Pearson et al. 2005). While membrane topology is similar to that of connexins, pannexins share no sequence homologies with connexins and are a more recently identified class of transmembrane proteins (Panchin et al. 2000). Pannexins can be found in three different isoforms (Panx1, Panx2 and Panx3) encoded by three different genes, with Panx1 and Panx3 sharing more similarities to each other than to Panx2 (Penuela et al. 2007). In the past decade, a growing body of literature has revealed that pannexins play multiple roles as they have been shown to release ATP and participate in calcium wave propagation and are likely a component of the inflammasome (Dando and Roper 2009; Locovei et al. 2006; Silverman et al. 2009; Sridharan et al. 2010). Thus, both connexins and pannexins are critical for cells to coordinate direct and paracrine communication (Fig. 1). In this review, we aim to bring together data focusing on the roles for glycosylation, phosphorylation, S-nitrosylation, ubiquitination and other posttranslational modifications in regulating the functions of both connexin and pannexin proteins.

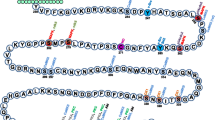
Schematic list of posttranslational modifications throughout the life span of connexins and pannexins. Numbers are the amino acids in the proteins as indicated in the references. Note that only when amino acids or specific regions on the protein could be identified were they included in the figure; e.g., although palmitoylation has been indicated for Panx2 (Swayne et al. 2010), the exact region on the protein remains unknown. *Shown indirectly through PKA; #this cysteine is assumed to be S-nitrosylated in a hemichannel (based on Retamal et al. 2006) but has not been directly shown; +data are now in dispute according to Dunn et al. (2012). 1 Paulson et al. (2000), TenBroek et al. (2001); 2 Locke et al. (2006); 3 Cooper and Lampe (2002); 4 Cottrell et al. (2003), Kanemitsu et al. (1998), Lampe et al. (1998), Norris et al. (2008), Warn-Cramer et al. (1998); 5 Doble et al. (2004), Ek-Vitorin et al. (2006), Lampe et al. (2000); 6 Solan and Lampe (2008); 7 Straub et al. (2011); 8 Kjenseth et al. (2012); 9 Bao et al. (2004b, 2007); 10 Toyofuku et al. (2001); 11 Lampe et al. (2000), Solan et al. (2003); 12 Kanemitsu et al. (1998), Lampe et al. (1998), Xie et al. (1997); 13 Beardslee et al. (2000), Solan et al. (2007); 14 Hertlein et al. (1998); 15 Yin et al. (2001), Yin et al. (2008); 16 Wagner et al. (2011); 17 Boassa et al. (2007), Penuela et al. (2007); 18 Penuela et al. (2007); 19 Chekeni et al. (2010); 20 Kim et al. (2011)
Glycosylation
Glycosylation is a form of posttranslational modification which consists of the enzymatic addition of glycans to form glycosylated proteins. The enzyme glycosyltransferase catalyzes the attachment of glycans to a nitrogen of asparagine (N-linked glycosylation) or to a hydroxyl oxygen of threonine or serine (O-linked glycosylation) residues (Freeze and Sharma 2010; Reis et al. 2010). Glycosylation mostly occurs in the endoplasmic reticulum (ER) or Golgi apparatus and can affect protein folding and stability, influence protein trafficking and interfere with protein function (Pinho et al. 2011; Reis et al. 2010; Roth et al. 2010).
Connexin proteins are not glycosylated despite identification of the N-glycosylation consensus sequence in Cx32 (Martin and Evans 2004; Rahman et al. 1993; Saez et al. 2003). However, it should be noted that a few reports have shown that inhibition of protein glycosylation in cells transfected with Cx43 can increase trafficking of Cx43 to the plasma membrane, its phosphorylation and its opening indirectly via a cAMP pathway (Wang and Mehta 1995; Wang et al. 1995; Wang and Rose 1995).
Currently, the primary identified posttranslational modification in pannexins is glycosylation. Glycosylation of the Panx1 isoforms leads to a migration shift on SDS gel with bands representing three different glycosylation states, whereas the multiple band pattern of Cx43 has been demonstrated to represent different phosphorylated states (Boassa et al. 2007, 2008; Penuela et al. 2007, 2009; Solan and Lampe 2009). The different glycosylation states of Panx1 result in a multiple banding pattern on a Western blot, with the Gly2 species migrating slower than Gly1 as an intermediate band and the Gly0 species being the fastest-migrating form (Boassa et al. 2007, 2008; Penuela et al. 2007, 2009). Of the three pannexin isoforms, Panx1 and Panx3 have been shown to be glycosylated and their glycosylation sites have been defined, whereas the glycosylation site of Panx2 has only been predicted (Boassa et al. 2007, 2008; Penuela et al. 2007, 2009, 2012).
Panx1 and Panx3 are N-glycosylated on the amino acids asparagine (N) 254 and N71, respectively, and can be found in three different states: a core unglycosylated protein (Gly0), a high mannose-glycosylated protein (Gly1) and an extensively glycosylated species (Gly2) (Boassa et al. 2007; Penuela et al. 2007, 2009). The role of N-glycosylation of Panx1 was explored using site-directed mutagenesis of N254 targeted by N-glycosylation (Panx1N254Q) (Boassa et al. 2007, 2008; Penuela et al. 2007, 2008). Similarly, the role of Panx3 N-glycosylation was investigated using the mutant Panx3N71Q (Penuela et al. 2007, 2008). These glycosylation mutants exhibit reduced trafficking to the plasma membrane when transfected in different cell lines, suggesting an important role for N-glycosylation in Panx1 and Panx3 trafficking (Boassa et al. 2007, 2008; Penuela et al. 2007, 2008). However, despite the low amount of Panx1N254Q and Panx3N71Q at the plasma membrane, the mutated proteins still form channels, as evidenced by dye uptake measurements (Penuela et al. 2007, 2009). Additionally, when wild-type Panx1 and Panx1N254Q are cotransfected, the trafficking of the glycosylation mutant to the plasma membrane is rescued, showing that they co-oligomerize and suggesting that N-glycosylation does not play a role in oligomerization of Panx1 (Boassa et al. 2008). Altogether, these studies demonstrate that the Gly0 species appears primarily after protein synthesis, whereas Gly1 is associated within the ER and Gly2 is modified in the Golgi apparatus prior to plasma membrane insertion. However, it should be noted that all Panx1 glycosylated species are capable of trafficking to the plasma membrane but the Gly2 form is preferentially trafficked (Gehi et al. 2011).
Phosphorylation
Posttranslational modification through phosphorylation acts as a major regulatory pathway in normal protein life cycles and can be further affected in pathological states by upregulation of kinase pathways. These pathways regulate protein functions through addition of a phosphate group by kinases to serine (S), threonine (T) and tyrosine (Y) sites, which may lead to alterations in the hydrophobicity, charge and potentially structural reorganization of proteins, either promoting or inhibiting normal functions (Davis 2011; Huttlin et al. 2010; Nishi et al. 2011).
Phosphorylation of connexins has been extensively demonstrated and reviewed (particularly for Cx43) and is integral in their life cycle–altering protein oligomerization, trafficking, membrane insertion and aggregation, gap junction communication and internalization from the membrane (Laird 2005; Laird et al. 1995; Marquez-Rosado et al. 2011; Saez et al. 1990, 1998; Solan and Lampe 2005, 2009). Phosphorylation has been demonstrated for multiple S/T/Y sites through c- and v-SRC; mitogen-activated protein kinase (MAPK), protein kinase C (PKC), protein kinase A (PKA), P34cdc (CDC2), casein kinase 1 (CK1) and calmodulin-dependant protein kinase pathways (Lampe and Lau 2004; Saez et al. 1998; Solan and Lampe 2005, 2009).
Posttranslational modification through phosphorylation of cytoplasmic residues primarily occurs within the connexin carboxyl terminus. Additionally, phosphorylation sites in the Cx43 amino terminus (S5) and the Cx56 intracellular loop have been identified as phosphorylated (Berthoud et al. 1997; Chen et al. 2012; Wisniewski et al. 2010). Cx43 does not contain serines within its intracellular loop and is therefore not modified directly by phosphorylation within this region (Solan and Lampe 2005). As a result of a short 19-amino acid carboxyl terminus containing only two serines, Cx26 is the only isoform that is currently considered not to be posttranslationally modified through phosphorylation (Traub et al. 1989). Conversely, Cx43 contains a total of 66 S/T/Y sites (32 in the carboxyl terminus) and has been demonstrated to be highly regulated through phosphorylation primarily at serine residues (Chen et al. 2012; Crow et al. 1990; Marquez-Rosado et al. 2011).
Phosphoproteomic analyses used to determine the incidence of protein phosphorylation in vitro and across multiple tissues, ranging from brain, kidney, lung, spleen and more, derived from mice, rats and humans have demonstrated that Cx43 as well as Cx47, Cx32 and Cx29 are phosphorylated at multiple sites at their carboxyl terminus as well as the amino terminus (Brill et al. 2009; Cooper et al. 2000; Huttlin et al. 2010; Rikova et al. 2007; Wisniewski et al. 2010). These studies and others further suggest that connexins are regulated by complex multisite phosphorylation, which plays an important role in the rapid generation and turnover of connexin proteins in a multitude of tissues and organ systems (Chen et al. 2012; Johnson and Vaillancourt 1994). This area of study will become increasingly important in terms of understanding how multiple phosphorylation events can “tip the balance” one way or another toward a particular protein function.
Connexins are posttranslationally oligomerized to hexameric hemichannels prior to membrane insertion either within the ER (e.g., Cx26, Cx32) (Falk et al. 1997; Falk and Gilula 1998) or in the trans-Golgi network (e.g., Cx43, Cx46) (Das Sarma et al. 2001; del Castillo et al. 2009; Koval 2006; Koval et al. 1997; Musil and Goodenough 1993; Puranam et al. 1993). A role for connexin phosphorylation in channel oligomerization has not currently been demonstrated (Solan and Lampe 2005). Pulse-chase studies have shown that newly synthesized Cx43 can be rapidly phosphorylated and dephosphorylated (within 15–30 min) (Crow et al. 1990) in the ER and Golgi compartments (Laird et al. 1995). In addition, monomeric Cx43 phosphoisoforms have been identified, which suggests that Cx43 is phosphorylated prior to insertion into the plasma membrane (Musil and Goodenough 1993). Further, increases in PKA activity can promote Cx43 integration to the membrane and are associated with Cx43-S364 phosphorylation, but Cx43 is a poor substrate for PKA and does not appear to directly increase Cx43 phosphorylation, suggesting that other intermediary pathways mediate the response (Paulson et al. 2000; TenBroek et al. 2001). Data demonstrating nonphosphorylated isoforms of connexins at the membrane suggest that they can be inserted to the membrane without phosphorylation (although transient phosphorylations could occur prior to entry to the membrane (Musil et al. 1990; Musil and Goodenough 1991; Solan and Lampe 2005). This is also evident with truncated mutations of Cx43 (Δ252) oligomerizing and trafficking to the plasma membrane. These truncated channels still form functional channels, suggesting that phosphorylation on the carboxyl tail is not a critical requirement (Johnstone et al. 2009; Martinez et al. 2003). Regardless, deletion mutagenesis studies have demonstrated that a portion of the Cx43 carboxyl terminus is required for trafficking. By creating a carboxyl terminus deletion at amino acid 236, Cx43 cannot traffic to the membrane, while deletions after amino acid 239 (Δ243/Δ239) traffic to the membrane and form gap junctions (De Vuyst et al. 2007; Wayakanon et al. 2012). As with Cx43, Cx45 requires its carboxyl terminus to target the membrane. Phosphorylation does not significantly alter Cx45 trafficking to the membrane but may affect protein half-life (Hertlein et al. 1998). Current data therefore suggest that phosphorylation of connexins is not a critical determinant of oligomerization and membrane insertion but could be involved in efficient gap junction assembly and trafficking.
The open and closed states of connexin hemichannels exist as a balance between inhibition and stimulation of carboxyl terminal residues. Mutagenesis studies of Cx43 have shown that mutations after amino acid 239 in Cx43 can still form functional gap junctions but inhibit hemichannel opening, suggesting that regulatory sites within the carboxyl terminus are required for hemichannel opening (De Vuyst et al. 2007). In general, connexin phosphorylation through PKC (e.g., S368) and MAPK activity maintains a closed hemichannel state (Bao et al. 2004a, 2007; Chandrasekhar and Bera 2012; Contreras et al. 2002; Ek-Vitorin et al. 2006; Kwak and Jongsma 1996; Schulz and Heusch 2004; Srisakuldee et al. 2009). Indeed, it has been shown that direct phosphorylation of Cx43-S368 by PKC reduces hemichannel opening (Bao et al. 2004b). Similarly, treatments with lipopolysaccharide and basic fibroblast growth factor decrease hemichannel activity in Cx32-, Cx43- and Cx26-expressing HeLa cells (De Vuyst et al. 2007). However, the same treatments enhance hemichannel opening in C6-Cx43 cells (De Vuyst et al. 2007). This diversity in hemichannel signaling has been attributed to a balance between channel inhibition (phosphorylation) and stimulation (dephosphorylation) modifications on the connexin carboxyl terminus (Contreras et al. 2002; De Vuyst et al. 2007).
As described above, the carboxyl terminus of connexins (particularly Cx43) may act to promote efficient gap junction assembly from hemichannels. Once inserted to the membrane, hemichannels aggregate in “formation plaques” prior to incorporation into the gap junctional plaque (Johnson et al. 2012; Segretain and Falk 2004). Truncation mutants of Cx43 result in reduced hemichannel aggregation and gap junction plaque formation, suggesting that phosphorylation of carboxyl-terminal residues may be involved in efficient delivery of hemichannels to the gap junction plaque (Johnson et al. 2012; Palatinus et al. 2011a). Increased formation and stability of gap junctions have been associated with PKA and CK1 activity. Increases in intracellular cAMP levels promote PKA activity, which promotes (indirectly) Cx43-S364 phosphorylation and increases in gap junction assembly and coupling (TenBroek et al. 2001). CK1 phosphorylation of Cx43-S325/S328/S330 also appears to promote gap junction assembly and stability (Cooper and Lampe 2002). Gap junction aggregation occurs through directed trafficking by chaperone proteins, e.g., zonula occludens-1/2 (ZO-1/ZO-2) (Rhett et al. 2011; Singh et al. 2005). Associations between the Cx43 carboxyl terminus and ZO-1 promote targeting to gap junction plaques (Hunter et al. 2005; Jin et al. 2004; Segretain et al. 2004; Toyofuku et al. 1998). This can be disrupted through c-SRC phosphorylation of Cx43-Y265, which appears to be a critical site in the binding of ZO-1 (Toyofuku et al. 2001). Therefore, phosphorylation may allow for efficient and directed movement of connexins within the membrane to sites of gap junction plaques.
In general, for Cx43, CK1 (and PKA) pathways increase and PKC, MAPK, v-SRC, and CDC2 reduce gap junctional communication (Pahujaa et al. 2007; Solan and Lampe 2005, 2008, 2009). Phosphorylation-induced increases in gap junctional communication occur through CK1 phosphorylation of Cx43-S325/S328/S330 (Cooper and Lampe 2002) and by PKA (Cx43-S364, indirectly) (TenBroek et al. 2001). However, as mentioned, these sites are also associated with increased gap junction assembly; and it has been suggested that they may enhance plaque stability, leading to a prolonged life cycle (Remo et al. 2011). In addition, phosphorylation of Cx40 by PKA pathways leads to altered (higher) conductance states (van Rijen et al. 2000).
PKC activity reduces Cx43 gap junctional communication. Reduced unitary conductance occurs following Cx43-S368 phosphorylation (Ek-Vitorin et al. 2006; Lampe et al. 2000), and site-directed mutagenesis demonstrates that Cx43-S262 phosphorylation can reduce gap junctional communication, which in turn may act to promote DNA synthesis and cell cycle progression (Doble et al. 2004). The impact of reduced communication through PKC phosphorylation of Cx43-S368 ranges from promoting wound closure (potentially by isolating cells from surrounding signals and promoting differentiation) (Richards et al. 2004), causing decreased cell-to-cell communication in the vascular wall (Straub et al. 2009) and reducing ischemia–reperfusion injury in hearts (Palatinus et al. 2011b). MAPK phosphorylation of Cx43-S255/S279/S282 leads to reductions in gap junctional communication by inhibiting the ability of the channels to open (Cottrell et al. 2003; Warn-Cramer et al. 1998). It should also be noted that several sites in the Cx43 tail may be phosphorylated through multiple pathways as is demonstrated for PKC induction by phorbol 12-myristate 13-acetate, which promotes Cx43 S279/S282 phosphorylation (Solan and Lampe 2007, 2009). In addition to the classically accepted MAPK sites, Norris et al. (2008) demonstrated that Cx43-S262 sites were phosphorylated (but not S368) through MAPK pathways, leading to reduced gap junctional communication and meiotic resumption in mouse oocytes. Increases in CDC2 promote phosphorylation of Cx43-S255 and reductions in gap junctional communication, which are associated with progression through the cell cycle (Kanemitsu et al. 1998; Lampe et al. 1998). Activation of v-SRC pathways leads to phosphorylation of Cx43-Y247 and Cx43-Y265, which appears to occur in formed gap junctions, leading to a reduction in gap junctional communication (Solan and Lampe 2008). In addition to these sites, active v-SRC can cause phosphorylation at sites associated with PKC and MAPK in Cx43 (Solan and Lampe 2008).
Less is known for the other connexins. Sequence predictions and homology suggest that Cx37 may be similarly phosphorylated through MAPK pathways, potentially at Cx37-S275 and Cx37-S282, which may affect gap junctional assembly and communication; but this has not been directly demonstrated (Burt et al. 2008). Phosphorylation of Cx45 through PKC pathways alters the occupancy time and conductive state of the channel (Kwak et al. 1995; van Veen et al. 2000). Although Cx45 is primarily phosphorylated at serines, substitution of all carboxyl-terminus serines does not alter gap junctional communication in HeLa cells (Hertlein et al. 1998).
Phosphorylation also plays a role in the internalization of connexins from the gap junctional plaque and has been shown to alter the protein turnover rates for several connexins (Laird 2005). During the cell cycle Cx43 appears to be dynamically regulated at the S, G1, G2 and M phases, all represented by alterations in Cx43 phosphorylation and internalization. As the cell cycle progresses, Cx43 appears at more intracellular locations and is highly phosphorylated at CDC2, PKC and MAPK sites, suggesting that phosphorylation may be involved in the removal of the connexins from the membrane gap junction plaques. However, it has not been conclusively demonstrated whether this results from removal of connexins from gap junction plaques or from reduced assembly to plaques (Solan and Lampe 2009). In mitosis, phosphorylation of Cx43 by CDC2 at the S255 and S262 regions is associated with an internalization of the protein (Kanemitsu et al. 1998; Lampe et al. 1998; Xie et al. 1997). During the cell cycle Cx43-S368 phosphorylation is also associated with a reduction in gap junction plaque size and protein internalization (Lampe et al. 2000; Solan et al. 2003). Conversely, it has been noted that dephosphorylation of Cx43 (S365) promotes an intracellular redistribution of Cx43 (Beardslee et al. 2000; Solan et al. 2007) or removal of Cx43 plaques (Laird et al. 1995). Substitution of Cx45-S381/S382/S384/S385 residues, but not the remaining serines from the carboxyl terminus of Cx45, significantly reduces the protein half-life by approximately 50 %, suggesting that these sites are involved in protein degradation (Hertlein et al. 1998). More recently, the MAPK phosphorylation of Cx43 at S255/262/279/282 was identified as critical to binding with cyclin E and promotion through the cell cycle (Johnstone et al. 2012).
While phosphorylation of connexins is integral to their functionality, much less has been demonstrated for modulation of pannexin channels. Despite this, all mammalian forms of pannexins contain multiple S/T/Y sites (Penuela et al. 2007). Currently, there are limited structural data for the pannexins and their carboxyl-terminus regions (Ambrosi et al. 2010). The membrane topology of pannexins appears to resemble that of the connexins with a cytoplasmic amino terminus, intracellular loop and carboxyl terminus, which shows the greatest sequence variability in the Panx2 isoform (Penuela et al. 2012). As with the connexins, the carboxyl-terminus regions are rich in S/T/Y sites. Consensus sequences for S/Y/T phosphorylation have been predicted based on sequence and membrane topology for pannexins 1–3 to occur primarily in the carboxyl terminus with fewer intracellular loop motifs (Barbe et al. 2006; Penuela et al. 2007). It has been shown that Panx1 ATP release can be induced by P2X7R activation through tyrosine kinase pathways, but it is not clear whether Panx1 is directly phosphorylated or regulated through intermediary pathways (Iglesias et al. 2009). While data on phosphorylation of pannexins are currently in their infancy, there is clearly the potential for this posttranslational modification in the regulation of the protein life cycle or function.
Nitrosylation
Posttranslational modification can also occur through S-nitrosylation, in which nitric oxide (NO) can bind to a reactive cysteine (C) thiol, producing an S-nitrosothiol (SNO) (Stamler et al. 1992). While proteins often contain multiple cysteine residues, the majority of the observed biological effects imparted by NO occur on single or only a few cysteine residues within a protein. In addition to direct modification of proteins by S-nitrosylation, this posttranslational modification can indirectly regulate other protein modifications including acetylation, phosphorylation and ubiquitination (Hess and Stamler 2012; Park et al. 2004; Whalen et al. 2007; Yasukawa et al. 2005).
The effects of NO on hemichannels and gap junctions have primarily been investigated in vascular cell types including vascular endothelial and smooth muscle cells. Of particular note, it was initially shown that exogenous application of the NO donor S-nitroso-N-acetylpenicillamine to cultured human umbilical vein endothelial cells significantly decreased dye transfer between coupled cells in a Cx37-dependent manner (Kameritsch et al. 2005). The observed decrease in gap junction permeability was independent of the effects of NO on guanylate cyclase and cGMP levels, indicating a separate effect of NO on the channels. In a separate study, NO application significantly reduced electrical coupling between cultured human microvascular endothelial cells mediated by effects on gap junctions composed of Cx37, which was similarly independent of cGMP (McKinnon et al. 2009). Conducted vasoconstriction in cremasteric arterioles has been shown to be dependent on Cx37 gap junctions and, importantly, is impaired during sepsis, a pathology resulting in upregulation of nNOS and increased NO production (McKinnon et al. 2006).
While S-nitrosylation of gap junctions composed of Cx37 has not been directly observed, evidence has indicated the ability of Cx43 to be S-nitrosylated; and this modification has been suggested to regulate gap junction permeability at the myoendothelial junction (MEJ) in the blood vessel wall (Straub et al. 2011). In this study, Cx43 was found to be locally enriched along with eNOS at the MEJ. Importantly, Cx43 was constitutively S-nitrosylated at C271. The S-nitrosylation of C271 on Cx43 was directly correlated with an open channel probability as denitrosylation significantly reduced heterocellular communication at the MEJ, namely, the movement of inositol triphosphate from smooth muscle cells to endothelial cells through Cx43 gap junctions. In agreement, pharmacological inhibition or genetic depletion of the denitrosylase GSNOR, which indirectly affects the extent of S-nitrosylation in these cells by reducing denitrosylation of the NO donor S-nitrosogluathione (GSNO), significantly increased gap junctional communication at the MEJ. Altogether this study suggests the potential for connexin proteins to become S-nitrosylated at specific cysteine residues, with the modification affecting channel permeability.
S-nitrosylation has also been proposed to regulate the permeability of undocked Cx43 hemichannels at the plasma membrane. During prolonged periods of ischemia or hypoxia, oxygen deprivation induces the opening of Cx43 hemichannels in cortical astrocytes and cardiomyocytes, leading to cell death (Contreras et al. 2002; John et al. 1999; Kondo et al. 2000). The mechanisms regulating Cx43 hemichannel permeability under these conditions have been suggested to involve dephosphorylation of Cx43 hemichannels (a known stimulus for hemichannel activation (Bao et al. 2004b; Kim et al. 1999) or oxidative modification due to increased production of reactive oxygen species, including NO. A novel study by Retamal et al. (2006) found that metabolic inhibition of cortical astrocytes resulted in both dephosphorylation of Cx43 hemichannels as well as an increase in S-nitrosylation, ultimately leading to an increase in cellular permeability as determined by dye uptake. Interestingly, application of reducing agents to these cells did not affect Cx43 phosphorylation but significantly reduced both cell permeability and Cx43 S-nitrosylation. In agreement with this, application of exogenous NO donors results in increased cell permeability. Together, these observations indicate a functional role for Cx43 S-nitrosylation in regulating hemichannel permeability at the plasma membrane of astrocytes.
The potential regulation of hemichannels composed of Cx46 by NO has also been suggested. When expressed in Xenopus oocytes, Cx46 hemichannels exhibited increased voltage sensitivity and current amplitude upon application of GSNO, as assessed by patch-clamp electrophysiology (Retamal et al. 2009). Treatment of these cells with the reducing agent dithiothreitol (DTT) reversed the effects of GSNO on hemichannel currents, and mutation of the two endogenous carboxyl-terminus cysteine residues inhibited the effect of GSNO application. These observations suggest that critical cysteine residues in the carboxyl terminus of Cx46, like C271 in Cx43, may be modified by S-nitrosylation to regulate channel permeability.
Of the three pannexin isoforms characterized to date, Panx1 is the most widely expressed and is highly expressed in vascular smooth muscle and endothelial cells in the arterial vasculature (Billaud et al. 2011; Godecke et al. 2012; Lohman et al. 2012) and in multiple cell types in the central and peripheral nervous systems (Ray et al. 2005; Vogt et al. 2005; Xia et al. 2012). Importantly, all these tissues have enriched NOS expression and actively utilize NO for signaling. As Panx1 contains multiple cysteine residues and has been shown to be expressed in tissues with enhanced NO production, it is possible that Panx1 channels in these cells may be regulated posttranslationally by S-nitrosylation. Further, it has been shown that mutation of C346 in the carboxyl terminus of Panx1 results in a constitutively leaky channel (Bunse et al. 2010). Also, mutation of C40 in the pore-lining region of the first transmembrane domain of Panx1 produces a constitutively open channel (Bunse et al. 2011). These results indicate the importance of Panx1 cysteine residues in regulating channel gating and permeability. While there are currently no published reports indicating direct modulation of Panx1 channels by S-nitrosylation, studies have indicated a role for Panx1 (as well as Panx2) channels in ischemia-induced neuronal cell death, a condition in which NO production is markedly enhanced (Bargiotas et al. 2011). Similarly, oxygen glucose deprivation and metabolic inhibition, conditions similar to ischemia, both induced excessive NO production and neuronal cell death mediated by opening of Panx channels, which was attenuated by pharmacological blockade of nNOS or reduction of oxidized cysteine thiols with the reducing agent DTT (Zhang et al. 2008). Importantly, inhibition of guanylate cyclase in these experiments had little effect on Panx channel permeability, suggesting potential effects of NO by S-nitrosylation of Panx channels in these cells. Future studies will be essential to directly evaluate the potential for pannexin proteins to be posttranslationally modified by S-nitrosylation and how modification of Panx channels in this manner affects channel gating and permeability.
Ubiquitination
The ubiquitin proteasome system regulates the degradation of many proteins. This can occur at the absolute N-terminal primary amine of proteins to induce autophagy (e.g., Lichtenstein et al. 2011). Although this is a relatively new area of connexin research, it could be quite important in multiple disease states. More traditional ubiquitination is comprised of several components, including E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes and E3 ubiquitin ligases, that together mediate the transfer of the 76-amino acid protein ubiquitin to specific lysine (K) residues on target proteins (Leithe and Rivedal 2007; Willis et al. 2010). Multiple ubiquitins can be covalently linked via K48 to form polyubiquitin chains, which target the ubiquitinated protein for degradation by the multisubunit 26S proteasome (Leithe and Rivedal 2007; Voges et al. 1999; Willis et al. 2010). Both connexins and pannexins have multiple lysine residues to which ubiquitin may be conjugated. Connexins can be ubiquitinated on K9 and K303 (Wagner et al. 2011), while pannexins can have ubiquitin conjugated to K409 (Kim et al. 2011). Thus, the possible ubiquitination of connexins and pannexins implies that these proteins may be regulated by proteasomal degradation.
Several studies have suggested that connexins and pannexins are ubiquitinated and degraded by the proteasome. The proteasomal inhibitor N-acetyl-l-leucyl-l-leucinyl-norleucinal increased levels of Cx43 (Laing and Beyer 1995) and Cx31 (He et al. 2005) and prolonged Cx43 half-life. Furthermore, immunoprecipitation with antibodies against Cx43 and ubiquitin suggested that Cx43 might be polyubiquitinated (Laing and Beyer 1995), indicating that disrupting the ubiquitin proteasome system could alter connexin turnover. The tumorigenic compound phorbol ester 12-O-tetradecanoylphorbol 13-acetate initiated hyperphosphorylation and ubiquitin-mediated degradation of Cx43, which was prevented by proteasomal inhibitors (Leithe and Rivedal 2004), demonstrating the importance of phosphorylation in ubiquitin-mediated connexin degradation. Phosphorylation of connexins facilitates interaction with proteins of the ubiquitin proteasome system, such as the ubiquitin ligase Nedd4 (Leykauf et al. 2006). Disrupting the interaction between connexins and ubiquitin proteasome proteins via proteasomal inhibitors alters the interaction between Cx43 and ZO-1, leading to Cx43 accumulation into enlarged gap junctions at plasma membranes (Girao and Pereira 2007). Therefore, it has been suggested that the ubiquitin proteasome system most likely contributes to the internalization and degradation of connexins.
Despite these findings, there is mounting evidence against ubiquitination and proteasomal degradation of connexins. Degradation of ER-localized Cx43 by the proteasome is partly controlled by Cx43 interactions with the ubiquitin-like protein CIP75, which binds to ubiquitinated substrates. However, immunoprecipitation studies demonstrated that the CIP75-bound Cx43 was not directly ubiquitinated (Su et al. 2010). In addition, a mutant Cx43 construct in which all lysine residues were converted to arginines mimicked the response of wild-type Cx43 to proteasomal inhibitors (Dunn et al. 2012). Taken together, these results indicate that direct ubiquitination may not be required for proteasomal degradation of Cx43. Instead, ubiquitination of connexins may contribute to other pathways regulating turnover. Indeed, recent studies have linked Cx43 monoubiquitination to lysosomal degradation. Following internalization from the plasma membrane, Cx43 can associate with the ubiquitin-binding proteins Hrs (hepatocyte growth factor–regulated tyrosine kinase substrate) and Tsg101 (tumor susceptibility gene 101) and the ubiquitin-binding endocytic adaptor protein Eps15, which shuttle Cx43 along the endocytic pathway for subsequent degradation in lysosomal compartments (Girao et al. 2009; Leithe et al. 2009). Thus, although ubiquitination mediates connexin trafficking and other degradative pathways, it is not clear whether direct ubiquitination is an important step during proteasomal degradation. Connexin turnover by this pathway most likely involves interaction between ubiquitinated substrates, even if connexins themselves are not tagged with ubiquitin.
Perturbation of connexin degradation by the ubiquitin proteasome has physiological and pathological consequences. Connexins are dynamically regulated with a short half-life of 1–6 h (He et al. 2005; Leithe and Rivedal 2004; Musil et al. 2000; Su et al. 2010). Proper connexin turnover rate is vital to maintain normal physiological functions (Li and Wang 2010). Dysregulation of dynamic gap junction protein turnover via inhibition of the ubiquitin proteasome degradation pathway (e.g., from oxidative stress) increases gap junction plaque size and enhances gap junction communication (Girao and Pereira 2007). Inhibition of proteasome-mediated degradation of gap junction proteins and enhanced gap junction communication is associated with several cardiovascular pathologies, such as ischemia–reperfusion injury, cardiac proteinopathy (i.e., irregular protein aggregation), post-myocardial infarct arrhythmias and heart failure (Beardslee et al. 1998; Li and Wang 2010). Thus, the ubiquitin proteasome pathway may provide novel therapeutic targets to treat cardiovascular diseases by restoring proper turnover of cardiac proteins, including connexins.
Much less is known about the involvement of the ubiquitin proteasome system in pannexin degradation. Mass spectrometric screens have identified K409 as a probable site of ubiquitination (Kim et al. 2011). However, biochemical evidence for pannexin ubiquitination and proteasomal degradation is lacking. A recent study has shown that Panx1 internalization and lysosomal degradation are controlled by its carboxyl-terminus region, but Panx1 does not appear to be degraded by traditional endocytic pathways (Gehi et al. 2011). Thus, further studies are needed to investigate the role ubiquitination plays in pannexin turnover (Schalper et al. 2012).
Other Modifications
SUMOylation
It has now been demonstrated that connexins (i.e., Cx43) can be targeted for posttranslation modification by less characterized pathways, e.g., SUMOylation. The small ubiquitin-like modifier (SUMO) family of proteins acts in most cellular compartments to adapt proteins in multiple processes including transcription, translation, cellular transport, protein interactions, cell growth and programmed cell death (Geiss-Friedlander and Melchior 2007). In Cx43, L144 (intracellular loop) and L237 (carboxyl terminus) can be SUMOylated, leading to a reduction in protein expression and gap junction formation (Kjenseth et al. 2012). Consensus motifs for SUMOylation have been identified in the Panx2 sequence; however, studies of immunoprecipitated Panx2 from mouse neuronal stem cells differentiated to neurospheres showed a lack of detection of SUMO1/2/3, suggesting that Panx2 is not SUMOylated (Swayne et al. 2010). Given the diversity of activity of SUMO proteins, this posttranslational modification could potentially play a significant role in connexins and pannexin protein regulation and channel formation.
Lipid Modification, e.g., Palmitoylation
The correct insertion of transmembrane proteins into the plasma membrane is a delicate process that involves the coordination of protein modifications for the binding of specific phospholipids (termed “palmitoylation”). It has been well described that connexins are associated with lipid rafts, especially those enriched with caveoli (e.g., Langlois et al. 2008; Locke et al. 2005). However, only the carboxyl terminus of Cx32 (amino acids 277–283) has been identified to have a palmitoylation site (Locke et al. 2006). Regardless, this concept was further elucidated by a series of experiments designed to identify all the phospholipids that could be associated with either Cx26 or Cx32 gap junctions. The authors found that the lipids associated with the two homotypic gap junctions were dramatically different, indicating these connexins may reside in very distinct parts of the plasma membrane (Locke and Harris 2009), possibly placed there by palmitoylation modifications.
Thus far, there is no indication that pannexin isoforms associate with caveolin (Gehi et al. 2011). However, it has been demonstrated that Panx2 has distinct depalmitoylated and palmitoylated species that migrate at different molecular weights (~60 and ~85 kDa, respectively) as determined by treatment of Western blots with hydroxylamine or metabolically labeling cells with BODIPY FL hexadecanoic acid. Swayne et al. (2010) further suggested that the palmitoylated form of Panx2 resides in the Golgi and/or ER, whereas the depalmitoylated form resides primarily at the plasma membrane and indicates these changes are associated with differentiation of neuronal cells. Elucidation of the exact site where Panx2 may be palmitoylated could prove quite interesting.
Caspase Cleavage
The posttranslational modification of proteins is generally considered to be a reversible process. However, a new type of terminal posttranslational modification was recently described where the Panx1 protein was cleaved by caspase 3 during apoptosis, inducing a constitutive open state of the channel for the release of ATP (Chekeni et al. 2010; Dunn et al. 2012). The caspase 3 cleavage site was identified in human Panx1 at amino acids 376–379 (DVVD) (Chekeni et al. 2010). The implications from the work were that the carboxyl terminus of Panx1 could regulate the pore open or closed state. This concept was recently demonstrated in a novel experimental model (Sandilos et al. 2012). In this work the authors inserted a TEV sequence into the Panx1 caspase cleavage site, allowing for controlled enzymatic cleavage of the carboxyl terminus. Next, a disulfide bond between the carboxyl terminus (C426) and an engineered cysteine within the pore of the Panx1 channel prevented removal of the carboxyl terminus from the pore (Sandilos et al. 2012). Finally, cleavage of the carboxyl terminus by TEV protease failed to produce current, and only after disulfide bond removal was current through the Panx1 channel restored (Sandilos et al. 2012). When summed with the caspase cleavage, the data indicate a role for the carboxyl terminus in regulating an open or closed pore and that cleavage of the carboxyl terminus as the specific caspase cleavage site induces a constitutive open channel during apoptosis. Although multiple connexins have been implicated throughout the apoptotic pathway (e.g., Andrade-Rozental et al. 2000; Minogue et al. 2009; Seul et al. 2004), there are no direct links or functional results like that described for the action of caspase 3 on Panx1.
Conclusion
Although much has been learned about how posttranslational modifications can have dramatic or subtle effects on connexin and pannexin function, most of these discoveries have happened only in the last decade. The creation of more specific antibodies toward modified amino acids (e.g., Solan and Lampe 2008) and accessibility of proteomic arrays to more researchers will enable an even greater study of posttranslational modifications associated with connexins and pannexins and their functional effect.
References
Ambrosi C, Gassmann O, Pranskevich JN, Boassa D, Smock A, Wang J, Dahl G, Steinem C, Sosinsky GE (2010) Pannexin1 and pannexin2 channels show quaternary similarities to connexons and different oligomerization numbers from each other. J Biol Chem 285:24420–24431
Andrade-Rozental AF, Rozental R, Hopperstad MG, Wu JK, Vrionis FD, Spray DC (2000) Gap junctions: the “kiss of death” and the “kiss of life”. Brain Res Brain Res Rev 32:308–315
Bao L, Locovei S, Dahl G (2004a) Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett 572:65–68
Bao X, Reuss L, Altenberg GA (2004b) Regulation of purified and reconstituted connexin 43 hemichannels by protein kinase C-mediated phosphorylation of serine 368. J Biol Chem 279:20058–20066
Bao X, Lee SC, Reuss L, Altenberg GA (2007) Change in permeant size selectivity by phosphorylation of connexin 43 gap-junctional hemichannels by PKC. Proc Natl Acad Sci USA 104:4919–4924
Barbe MT, Monyer H, Bruzzone R (2006) Cell–cell communication beyond connexins: the pannexin channels. Physiology (Bethesda) 21:103–114
Bargiotas P, Krenz A, Hormuzdi SG, Ridder DA, Herb A, Barakat W, Penuela S, von Engelhardt J, Monyer H, Schwaninger M (2011) Pannexins in ischemia-induced neurodegeneration. Proc Natl Acad Sci USA 108:20772–20777
Beardslee MA, Laing JG, Beyer EC, Saffitz JE (1998) Rapid turnover of connexin43 in the adult rat heart. Circ Res 83:629–635
Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, Kleber AG, Schuessler RB, Saffitz JE (2000) Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res 87:656–662
Berthoud VM, Beyer EC, Kurata WE, Lau AF, Lampe PD (1997) The gap-junction protein connexin 56 is phosphorylated in the intracellular loop and the carboxy-terminal region. Eur J Biochem 244:89–97
Billaud M, Lohman AW, Straub AC, Looft-Wilson R, Johnstone SR, Araj CA, Best AK, Chekeni FB, Ravichandran KS, Penuela S, Laird DW, Isakson BE (2011) Pannexin1 regulates alpha1-adrenergic receptor- mediated vasoconstriction. Circ Res 109:80–85
Boassa D, Ambrosi C, Qiu F, Dahl G, Gaietta G, Sosinsky G (2007) Pannexin1 channels contain a glycosylation site that targets the hexamer to the plasma membrane. J Biol Chem 282:31733–31743
Boassa D, Qiu F, Dahl G, Sosinsky G (2008) Trafficking dynamics of glycosylated pannexin 1 proteins. Cell Commun Adhes 15:119–132
Boitano S, Evans WH (2000) Connexin mimetic peptides reversibly inhibit Ca2+ signaling through gap junctions in airway cells. Am J Physiol Lung Cell Mol Physiol 279:L623–L630
Brill LM, Xiong W, Lee KB, Ficarro SB, Crain A, Xu Y, Terskikh A, Snyder EY, Ding S (2009) Phosphoproteomic analysis of human embryonic stem cells. Cell Stem Cell 5:204–213
Bunse S, Schmidt M, Prochnow N, Zoidl G, Dermietzel R (2010) Intracellular cysteine 346 is essentially involved in regulating Panx1 channel activity. J Biol Chem 285:38444–38452
Bunse S, Schmidt M, Hoffmann S, Engelhardt K, Zoidl G, Dermietzel R (2011) Single cysteines in the extracellular and transmembrane regions modulate pannexin 1 channel function. J Membr Biol 244:21–33
Burt JM, Nelson TK, Simon AM, Fang JS (2008) Connexin 37 profoundly slows cell cycle progression in rat insulinoma cells. Am J Physiol Cell Physiol 295:C1103–C1112
Chandrasekhar A, Bera AK (2012) Hemichannels: permeants and their effect on development, physiology and death. Cell Biochem Funct 30:89–100
Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS (2010) Pannexin 1 channels mediate “find-me” signal release and membrane permeability during apoptosis. Nature 467:863–867
Chen VC, Gouw JW, Naus CC, Foster LJ (2012) Connexin multi-site phosphorylation: mass spectrometry-based proteomics fills the gap. Biochim Biophys Acta. doi:10.1016/j.bbamem.2012.02.028
Contreras JE, Sanchez HA, Eugenin EA, Speidel D, Theis M, Willecke K, Bukauskas FF, Bennett MV, Saez JC (2002) Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc Natl Acad Sci USA 99:495–500
Cooper CD, Lampe PD (2002) Casein kinase 1 regulates connexin-43 gap junction assembly. J Biol Chem 277:44962–44968
Cooper CD, Solan JL, Dolejsi MK, Lampe PD (2000) Analysis of connexin phosphorylation sites. Methods 20:196–204
Cottrell GT, Lin R, Warn-Cramer BJ, Lau AF, Burt JM (2003) Mechanism of v-Src- and mitogen-activated protein kinase-induced reduction of gap junction communication. Am J Physiol Cell Physiol 284:C511–C520
Crow DS, Beyer EC, Paul DL, Kobe SS, Lau AF (1990) Phosphorylation of connexin43 gap junction protein in uninfected and Rous sarcoma virus-transformed mammalian fibroblasts. Mol Cell Biol 10:1754–1763
Dando R, Roper SD (2009) Cell-to-cell communication in intact taste buds through ATP signalling from pannexin 1 gap junction hemichannels. J Physiol 587:5899–5906
Das Sarma J, Meyer RA, Wang F, Abraham V, Lo CW, Koval M (2001) Multimeric connexin interactions prior to the trans-Golgi network. J Cell Sci 114:4013–4024
Davis FP (2011) Phosphorylation at the interface. Structure 19:1726–1727
De Vuyst E, Decrock E, De Bock M, Yamasaki H, Naus CC, Evans WH, Leybaert L (2007) Connexin hemichannels and gap junction channels are differentially influenced by lipopolysaccharide and basic fibroblast growth factor. Mol Biol Cell 18:34–46
del Castillo FJ, Cohen-Salmon M, Charollais A, Caille D, Lampe PD, Chavrier P, Meda P, Petit C (2009) Consortin, a trans-Golgi network cargo receptor for the plasma membrane targeting and recycling of connexins. Hum Mol Genet 19:262–275
Doble BW, Dang X, Ping P, Fandrich RR, Nickel BE, Jin Y, Cattini PA, Kardami E (2004) Phosphorylation of serine 262 in the gap junction protein connexin-43 regulates DNA synthesis in cell–cell contact forming cardiomyocytes. J Cell Sci 117:507–514
Dunn CA, Su V, Lau AF, Lampe PD (2012) Activation of Akt, not connexin 43 protein ubiquitination, regulates gap junction stability. J Biol Chem 287:2600–2607
Ek-Vitorin JF, King TJ, Heyman NS, Lampe PD, Burt JM (2006) Selectivity of connexin 43 channels is regulated through protein kinase C-dependent phosphorylation. Circ Res 98:1498–1505
Falk MM, Gilula NB (1998) Connexin membrane protein biosynthesis is influenced by polypeptide positioning within the translocon and signal peptidase access. J Biol Chem 273:7856–7864
Falk MM, Buehler LK, Kumar NM, Gilula NB (1997) Cell-free synthesis and assembly of connexins into functional gap junction membrane channels. EMBO J 16:2703–2716
Freeze HH, Sharma V (2010) Metabolic manipulation of glycosylation disorders in humans and animal models. Semin Cell Dev Biol 21:655–662
Gehi R, Shao Q, Laird DW (2011) Pathways regulating the trafficking and turnover of pannexin1 protein and the role of the C-terminal domain. J Biol Chem 286:27639–27653
Geiss-Friedlander R, Melchior F (2007) Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol 8:947–956
Girao H, Pereira P (2007) The proteasome regulates the interaction between Cx43 and ZO-1. J Cell Biochem 102:719–728
Girao H, Catarino S, Pereira P (2009) Eps15 interacts with ubiquitinated Cx43 and mediates its internalization. Exp Cell Res 315:3587–3597
Godecke S, Roderigo C, Rose CR, Rauch BH, Godecke A, Schrader J (2012) Thrombin-induced ATP release from human umbilical vein endothelial cells. Am J Physiol Cell Physiol 302:C915–C923
He LQ, Cai F, Liu Y, Liu MJ, Tan ZP, Pan Q, Fang FY, de Liang S, Wu LQ, Long ZG, Dai HP, Xia K, Xia JH, Zhang ZH (2005) Cx31 is assembled and trafficked to cell surface by ER–Golgi pathway and degraded by proteasomal or lysosomal pathways. Cell Res 15:455–464
Hertlein B, Butterweck A, Haubrich S, Willecke K, Traub O (1998) Phosphorylated carboxy terminal serine residues stabilize the mouse gap junction protein connexin45 against degradation. J Membr Biol 162:247–257
Hess DT, Stamler JS (2012) Regulation by S-nitrosylation of protein posttranslational modification. J Biol Chem 287:4411–4418
Hunter AW, Barker RJ, Zhu C, Gourdie RG (2005) Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell 16:5686–5698
Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143:1174–1189
Iglesias R, Dahl G, Qiu F, Spray DC, Scemes E (2009) Pannexin 1: the molecular substrate of astrocyte “hemichannels”. J Neurosci 29:7092–7097
Jin C, Martyn KD, Kurata WE, Warn-Cramer BJ, Lau AF (2004) Connexin43 PDZ2 binding domain mutants create functional gap junctions and exhibit altered phosphorylation. Cell Commun Adhes 11:67–87
John SA, Kondo R, Wang SY, Goldhaber JI, Weiss JN (1999) Connexin-43 hemichannels opened by metabolic inhibition. J Biol Chem 274:236–240
Johnson LN, Koval M (2009) Cross-talk between pulmonary injury, oxidant stress, and gap junctional communication. Antioxid Redox Signal 11:355–367
Johnson GL, Vaillancourt RR (1994) Sequential protein kinase reactions controlling cell growth and differentiation. Curr Opin Cell Biol 6:230–238
Johnson RG, Reynhout JK, TenBroek EM, Quade BJ, Yasumura T, Davidson KG, Sheridan JD, Rash JE (2012) Gap junction assembly: roles for the formation plaque and regulation by the C-terminus of connexin43. Mol Biol Cell 23:71–86
Johnstone SR, Ross J, Rizzo MJ, Straub AC, Lampe PD, Leitinger N, Isakson BE (2009) Oxidized phospholipid species promote in vivo differential cx43 phosphorylation and vascular smooth muscle cell proliferation. Am J Pathol 175:916–924
Johnstone SR, Kroncke BM, Straub AC, Best AK, Dunn CA, Mitchell LA, Peskova Y, Nakamoto RK, Koval M, Lo CW, Lampe PD, Columbus L, Isakson BE (2012) MAPK phosphorylation of connexin 43 promotes binding of cyclin E and smooth muscle cell proliferation. Circ Res
Kameritsch P, Khandoga N, Nagel W, Hundhausen C, Lidington D, Pohl U (2005) Nitric oxide specifically reduces the permeability of Cx37-containing gap junctions to small molecules. J Cell Physiol 203:233–242
Kanemitsu MY, Jiang W, Eckhart W (1998) Cdc2-mediated phosphorylation of the gap junction protein, connexin43, during mitosis. Cell Growth Differ 9:13–21
Kelsell DP, Di WL, Houseman MJ (2001) Connexin mutations in skin disease and hearing loss. Am J Hum Genet 68:559–568
Kim DY, Kam Y, Koo SK, Joe CO (1999) Gating connexin 43 channels reconstituted in lipid vesicles by mitogen-activated protein kinase phosphorylation. J Biol Chem 274:5581–5587
Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, Gygi SP (2011) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 44:325–340
Kjenseth A, Fykerud TA, Sirnes S, Bruun J, Kolberg M, Yohannes Z, Omori Y, Rivedal E, Leithe E (2012) The gap junction channel protein connexin43 is covalently modified and regulated by SUMOylation. J Biol Chem 287:15851–15861
Kondo RP, Wang SY, John SA, Weiss JN, Goldhaber JI (2000) Metabolic inhibition activates a non-selective current through connexin hemichannels in isolated ventricular myocytes. J Mol Cell Cardiol 32:1859–1872
Koval M (2006) Pathways and control of connexin oligomerization. Trends Cell Biol 16:159–166
Koval M, Harley JE, Hick E, Steinberg TH (1997) Connexin46 is retained as monomers in a trans-Golgi compartment of osteoblastic cells. J Cell Biol 137:847–857
Kwak BR, Jongsma HJ (1996) Regulation of cardiac gap junction channel permeability and conductance by several phosphorylating conditions. Mol Cell Biochem 157:93–99
Kwak BR, Hermans MM, De Jonge HR, Lohmann SM, Jongsma HJ, Chanson M (1995) Differential regulation of distinct types of gap junction channels by similar phosphorylating conditions. Mol Biol Cell 6:1707–1719
Lai A, Le DN, Paznekas WA, Gifford WD, Jabs EW, Charles AC (2006) Oculodentodigital dysplasia connexin43 mutations result in non-functional connexin hemichannels and gap junctions in C6 glioma cells. J Cell Sci 119:532–541
Laing JG, Beyer EC (1995) The gap junction protein connexin43 is degraded via the ubiquitin proteasome pathway. J Biol Chem 270:26399–26403
Laird DW (2005) Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim Biophys Acta 1711:172–182
Laird DW, Castillo M, Kasprzak L (1995) Gap junction turnover, intracellular trafficking, and phosphorylation of connexin43 in brefeldin A-treated rat mammary tumor cells. J Cell Biol 131:1193–1203
Lampe PD, Lau AF (2004) The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol 36:1171–1186
Lampe PD, Kurata WE, Warn-Cramer BJ, Lau AF (1998) Formation of a distinct connexin43 phosphoisoform in mitotic cells is dependent upon p34cdc2 kinase. J Cell Sci 111(pt 6):833–841
Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF (2000) Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol 149:1503–1512
Langlois S, Cowan KN, Shao Q, Cowan BJ, Laird DW (2008) Caveolin-1 and -2 interact with connexin43 and regulate gap junctional intercellular communication in keratinocytes. Mol Biol Cell 19:912–928
Leithe E, Rivedal E (2004) Ubiquitination and down-regulation of gap junction protein connexin-43 in response to 12-O-tetradecanoylphorbol 13-acetate treatment. J Biol Chem 279:50089–50096
Leithe E, Rivedal E (2007) Ubiquitination of gap junction proteins. J Membr Biol 217:43–51
Leithe E, Kjenseth A, Sirnes S, Stenmark H, Brech A, Rivedal E (2009) Ubiquitylation of the gap junction protein connexin-43 signals its trafficking from early endosomes to lysosomes in a process mediated by Hrs and Tsg101. J Cell Sci 122:3883–3893
Leykauf K, Salek M, Bomke J, Frech M, Lehmann WD, Durst M, Alonso A (2006) Ubiquitin protein ligase Nedd4 binds to connexin43 by a phosphorylation-modulated process. J Cell Sci 119:3634–3642
Li YF, Wang X (2010) The role of the proteasome in heart disease. Biochim Biophys Acta 1809:141–149
Lichtenstein A, Minogue PJ, Beyer EC, Berthoud VM (2011) Autophagy: a pathway that contributes to connexin degradation. J Cell Sci 124:910–920
Locke D, Harris AL (2009) Connexin channels and phospholipids: association and modulation. BMC Biol 7:52
Locke D, Liu J, Harris AL (2005) Lipid rafts prepared by different methods contain different connexin channels, but gap junctions are not lipid rafts. Biochemistry 44:13027–13042
Locke D, Koreen IV, Harris AL (2006) Isoelectric points and posttranslational modifications of connexin26 and connexin32. FASEB J 20:1221–1223
Locovei S, Bao L, Dahl G (2006) Pannexin 1 in erythrocytes: function without a gap. Proc Natl Acad Sci USA 103:7655–7659
Lohman AW, Billaud M, Straub AC, Johnstone SR, Best AK, Lee MY, Barr K, Penuela S, Laird DW, Isakson BE (2012) Expression of pannexin isoforms in the systemic murine arterial network. J Vasc Res (in press)
Marquez-Rosado L, Solan JL, Dunn CA, Norris RP, Lampe PD (2011) Connexin43 phosphorylation in brain, cardiac, endothelial and epithelial tissues. Biochim Biophys Acta
Martin PE, Evans WH (2004) Incorporation of connexins into plasma membranes and gap junctions. Cardiovasc Res 62:378–387
Martinez AD, Hayrapetyan V, Moreno AP, Beyer EC (2003) A carboxyl terminal domain of connexin43 is critical for gap junction plaque formation but not for homo- or hetero-oligomerization. Cell Commun Adhes 10:323–328
McKinnon RL, Lidington D, Bolon M, Ouellette Y, Kidder GM, Tyml K (2006) Reduced arteriolar conducted vasoconstriction in septic mouse cremaster muscle is mediated by nNOS-derived NO. Cardiovasc Res 69:236–244
McKinnon RL, Bolon ML, Wang HX, Swarbreck S, Kidder GM, Simon AM, Tyml K (2009) Reduction of electrical coupling between microvascular endothelial cells by NO depends on connexin37. Am J Physiol Heart Circ Physiol 297:H93–H101
Minogue PJ, Tong JJ, Arora A, Russell-Eggitt I, Hunt DM, Moore AT, Ebihara L, Beyer EC, Berthoud VM (2009) A mutant connexin50 with enhanced hemichannel function leads to cell death. Invest Ophthalmol Vis Sci 50:5837–5845
Musil LS, Goodenough DA (1991) Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol 115:1357–1374
Musil LS, Goodenough DA (1993) Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell 74:1065–1077
Musil LS, Cunningham BA, Edelman GM, Goodenough DA (1990) Differential phosphorylation of the gap junction protein connexin43 in junctional communication-competent and -deficient cell lines. J Cell Biol 111:2077–2088
Musil LS, Le AC, VanSlyke JK, Roberts LM (2000) Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J Biol Chem 275:25207–25215
Nishi H, Hashimoto K, Panchenko AR (2011) Phosphorylation in protein–protein binding: effect on stability and function. Structure 19:1807–1815
Norris RP, Freudzon M, Mehlmann LM, Cowan AE, Simon AM, Paul DL, Lampe PD, Jaffe LA (2008) Luteinizing hormone causes MAP kinase-dependent phosphorylation and closure of connexin 43 gap junctions in mouse ovarian follicles: one of two paths to meiotic resumption. Development 135:3229–3238
Pahujaa M, Anikin M, Goldberg GS (2007) Phosphorylation of connexin43 induced by Src: regulation of gap junctional communication between transformed cells. Exp Cell Res 313:4083–4090
Palatinus JA, Gourdie RG (2007) Xin and the art of intercalated disk maintenance. Am J Physiol Heart Circ Physiol 293:H2626–H2628
Palatinus JA, Rhett JM, Gourdie RG (2011a) The connexin43 carboxyl terminus and cardiac gap junction organization. Biochim Biophys Acta 1818:1831–1843
Palatinus JA, Rhett JM, Gourdie RG (2011b) Enhanced PKCepsilon mediated phosphorylation of connexin43 at serine 368 by a carboxyl-terminal mimetic peptide is dependent on injury. Channels (Austin) 5:236–240
Panchin Y, Kelmanson I, Matz M, Lukyanov K, Usman N, Lukyanov S (2000) A ubiquitous family of putative gap junction molecules. Curr Biol 10:R473–R474
Park HS, Yu JW, Cho JH, Kim MS, Huh SH, Ryoo K, Choi EJ (2004) Inhibition of apoptosis signal-regulating kinase 1 by nitric oxide through a thiol redox mechanism. J Biol Chem 279:7584–7590
Paulson AF, Lampe PD, Meyer RA, TenBroek E, Atkinson MM, Walseth TF, Johnson RG (2000) Cyclic AMP and LDL trigger a rapid enhancement in gap junction assembly through a stimulation of connexin trafficking. J Cell Sci 113(pt 17):3037–3049
Pearson RA, Dale N, Llaudet E, Mobbs P (2005) ATP released via gap junction hemichannels from the pigment epithelium regulates neural retinal progenitor proliferation. Neuron 46:731–744
Penuela S, Bhalla R, Gong XQ, Cowan KN, Celetti SJ, Cowan BJ, Bai D, Shao Q, Laird DW (2007) Pannexin 1 and pannexin 3 are glycoproteins that exhibit many distinct characteristics from the connexin family of gap junction proteins. J Cell Sci 120:3772–3783
Penuela S, Celetti SJ, Bhalla R, Shao Q, Laird DW (2008) Diverse subcellular distribution profiles of pannexin 1 and pannexin 3. Cell Commun Adhes 15:133–142
Penuela S, Bhalla R, Nag K, Laird DW (2009) Glycosylation regulates pannexin intermixing and cellular localization. Mol Biol Cell 20:4313–4323
Penuela S, Gehi R, Laird DW (2012) The biochemistry and function of pannexin channels. Biochim Biophys Acta (in press)
Pinho SS, Seruca R, Gartner F, Yamaguchi Y, Gu J, Taniguchi N, Reis CA (2011) Modulation of E-cadherin function and dysfunction by N-glycosylation. Cell Mol Life Sci 68:1011–1020
Puranam KL, Laird DW, Revel JP (1993) Trapping an intermediate form of connexin43 in the Golgi. Exp Cell Res 206:85–92
Rahman S, Carlile G, Evans WH (1993) Assembly of hepatic gap junctions. Topography and distribution of connexin 32 in intracellular and plasma membranes determined using sequence-specific antibodies. J Biol Chem 268:1260–1265
Ray A, Zoidl G, Weickert S, Wahle P, Dermietzel R (2005) Site-specific and developmental expression of pannexin1 in the mouse nervous system. Eur J Neurosci 21:3277–3290
Reis CA, Osorio H, Silva L, Gomes C, David L (2010) Alterations in glycosylation as biomarkers for cancer detection. J Clin Pathol 63:322–329
Remo BF, Qu J, Volpicelli FM, Giovannone S, Shin D, Lader J, Liu FY, Zhang J, Lent DS, Morley GE, Fishman GI (2011) Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ Res 108:1459–1466
Retamal MA, Cortes CJ, Reuss L, Bennett MV, Saez JC (2006) S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: induction by oxidant stress and reversal by reducing agents. Proc Natl Acad Sci USA 103:4475–4480
Retamal MA, Yin S, Altenberg GA, Reuss L (2009) Modulation of Cx46 hemichannels by nitric oxide. Am J Physiol Cell Physiol 296:C1356–C1363
Rhett JM, Jourdan J, Gourdie RG (2011) Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol Biol Cell 22:1516–1528
Richards TS, Dunn CA, Carter WG, Usui ML, Olerud JE, Lampe PD (2004) Protein kinase C spatially and temporally regulates gap junctional communication during human wound repair via phosphorylation of connexin43 on serine368. J Cell Biol 167:555–562
Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, Hu Y, Tan Z, Stokes M, Sullivan L, Mitchell J, Wetzel R, Macneill J, Ren JM, Yuan J, Bakalarski CE, Villen J, Kornhauser JM, Smith B, Li D, Zhou X, Gygi SP, Gu TL, Polakiewicz RD, Rush J, Comb MJ (2007) Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131:1190–1203
Roth J, Zuber C, Park S, Jang I, Lee Y, Kysela KG, Le Fourn V, Santimaria R, Guhl B, Cho JW (2010) Protein N-glycosylation, protein folding, and protein quality control. Mol Cells 30:497–506
Saez JC, Nairn AC, Czernik AJ, Spray DC, Hertzberg EL, Greengard P, Bennett MV (1990) Phosphorylation of connexin 32, a hepatocyte gap-junction protein, by cAMP-dependent protein kinase, protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Eur J Biochem 192:263–273
Saez JC, Martinez AD, Branes MC, Gonzalez HE (1998) Regulation of gap junctions by protein phosphorylation. Braz J Med Biol Res 31:593–600
Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC (2003) Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev 83:1359–1400
Sandilos JK, Chiu YH, Chekeni FB, Armstrong AJ, Walk SF, Ravichandran KS, Bayliss DA (2012) Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C terminal autoinhibitory region. J Biol Chem (in press)
Schalper KA, Riquelme MA, Branes MC, Martinez AD, Vega JL, Berthoud VM, Bennett MV, Saez JC (2012) Modulation of gap junction channels and hemichannels by growth factors. Mol BioSyst 8:685–698
Schulz R, Heusch G (2004) Connexin 43 and ischemic preconditioning. Cardiovasc Res 62:335–344
Segretain D, Falk MM (2004) Regulation of connexin biosynthesis, assembly, gap junction formation, and removal. Biochim Biophys Acta 1662:3–21
Segretain D, Fiorini C, Decrouy X, Defamie N, Prat JR, Pointis G (2004) A proposed role for ZO-1 in targeting connexin 43 gap junctions to the endocytic pathway. Biochimie 86:241–244
Seul KH, Kang KY, Lee KS, Kim SH, Beyer EC (2004) Adenoviral delivery of human connexin37 induces endothelial cell death through apoptosis. Biochem Biophys Res Commun 319:1144–1151
Silverman WR, de Rivero Vaccari JP, Locovei S, Qiu F, Carlsson SK, Scemes E, Keane RW, Dahl G (2009) The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J Biol Chem 284:18143–18151
Singh D, Solan JL, Taffet SM, Javier R, Lampe PD (2005) Connexin 43 interacts with zona occludens-1 and -2 proteins in a cell cycle stage-specific manner. J Biol Chem 280:30416–30421
Solan JL, Lampe PD (2005) Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta 1711:154–163
Solan JL, Lampe PD (2007) Key connexin 43 phosphorylation events regulate the gap junction life cycle. J Membr Biol 217:35–41
Solan JL, Lampe PD (2008) Connexin 43 in LA-25 cells with active v-src is phosphorylated on Y247, Y265, S262, S279/282, and S368 via multiple signaling pathways. Cell Commun Adhes 15:75–84
Solan JL, Lampe PD (2009) Connexin43 phosphorylation: structural changes and biological effects. Biochem J 419:261–272
Solan JL, Fry MD, TenBroek EM, Lampe PD (2003) Connexin43 phosphorylation at S368 is acute during S and G2/M and in response to protein kinase C activation. J Cell Sci 116:2203–2211
Solan JL, Marquez-Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD (2007) Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J Cell Biol 179:1301–1309
Sridharan M, Adderley SP, Bowles EA, Egan TM, Stephenson AH, Ellsworth ML, Sprague RS (2010) Pannexin 1 is the conduit for low oxygen tension-induced ATP release from human erythrocytes. Am J Physiol Heart Circ Physiol 299:H1146–H1152
Srisakuldee W, Jeyaraman MM, Nickel BE, Tanguy S, Jiang ZS, Kardami E (2009) Phosphorylation of connexin-43 at serine 262 promotes a cardiac injury-resistant state. Cardiovasc Res 83:672–681
Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J (1992) S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci USA 89:444–448
Straub AC, Johnstone SR, Heberlein KR, Rizzo MJ, Best AK, Boitano S, Isakson BE (2009) Site-specific connexin phosphorylation is associated with reduced heterocellular communication between smooth muscle and endothelium. J Vasc Res 47:277–286
Straub AC, Billaud M, Johnstone SR, Best AK, Yemen S, Dwyer ST, Looft-Wilson R, Lysiak JJ, Gaston B, Palmer L, Isakson BE (2011) Compartmentalized connexin 43 S-nitrosylation/denitrosylation regulates heterocellular communication in the vessel wall. Arterioscler Thromb Vasc Biol 31:399–407
Su V, Nakagawa R, Koval M, Lau AF (2010) Ubiquitin-independent proteasomal degradation of endoplasmic reticulum-localized connexin43 mediated by CIP75. J Biol Chem 285:40979–40990
Swayne LA, Sorbara CD, Bennett SA (2010) Pannexin 2 is expressed by postnatal hippocampal neural progenitors and modulates neuronal commitment. J Biol Chem 285:24977–24986
TenBroek EM, Lampe PD, Solan JL, Reynhout JK, Johnson RG (2001) Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J Cell Biol 155:1307–1318
Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Hori M, Tada M (1998) Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J Biol Chem 273:12725–12731
Toyofuku T, Akamatsu Y, Zhang H, Kuzuya T, Tada M, Hori M (2001) c-Src regulates the interaction between connexin-43 and ZO-1 in cardiac myocytes. J Biol Chem 276:1780–1788
Traub O, Look J, Dermietzel R, Brummer F, Hulser D, Willecke K (1989) Comparative characterization of the 21-kD and 26-kD gap junction proteins in murine liver and cultured hepatocytes. J Cell Biol 108:1039–1051
van Rijen HV, van Veen TA, Hermans MM, Jongsma HJ (2000) Human connexin40 gap junction channels are modulated by cAMP. Cardiovasc Res 45:941–951
van Veen TA, van Rijen HV, Jongsma HJ (2000) Electrical conductance of mouse connexin45 gap junction channels is modulated by phosphorylation. Cardiovasc Res 46:496–510
Voges D, Zwickl P, Baumeister W (1999) The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem 68:1015–1068
Vogt A, Hormuzdi SG, Monyer H (2005) Pannexin1 and pannexin2 expression in the developing and mature rat brain. Brain Res Mol Brain Res 141:113–120
Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C (2011) A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics 10:M111.013284
Wang Y, Mehta PP (1995) Facilitation of gap-junctional communication and gap-junction formation in mammalian cells by inhibition of glycosylation. Eur J Cell Biol 67:285–296
Wang Y, Rose B (1995) Clustering of Cx43 cell-to-cell channels into gap junction plaques: regulation by cAMP and microfilaments. J Cell Sci 108(pt 11):3501–3508
Wang Y, Mehta PP, Rose B (1995) Inhibition of glycosylation induces formation of open connexin-43 cell-to-cell channels and phosphorylation and triton X-100 insolubility of connexin-43. J Biol Chem 270:26581–26585
Warn-Cramer BJ, Cottrell GT, Burt JM, Lau AF (1998) Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J Biol Chem 273:9188–9196
Wayakanon P, Bhattacharjee R, Nakahama KI, Morita I (2012) The role of the Cx43 C-terminus in GJ plaque formation and internalization. Biochem Biophys Res Commun 420:456–461
Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, Nelson CD, Benhar M, Keys JR, Rockman HA, Koch WJ, Daaka Y, Lefkowitz RJ, Stamler JS (2007) Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell 129:511–522
Willis MS, Townley-Tilson WH, Kang EY, Homeister JW, Patterson C (2010) Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ Res 106:463–478
Wisniewski JR, Nagaraj N, Zougman A, Gnad F, Mann M (2010) Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J Proteome Res 9:3280–3289
Xia J, Lim JC, Lu W, Beckel JM, Macarak EJ, Laties AM, Mitchell CH (2012) Neurons respond directly to mechanical deformation with pannexin-mediated ATP release and autostimulation of P2X7 receptors. J Physiol 590(pt 10):2285–2304
Xie H, Laird DW, Chang TH, Hu VW (1997) A mitosis-specific phosphorylation of the gap junction protein connexin43 in human vascular cells: biochemical characterization and localization. J Cell Biol 137:203–210
Yasukawa T, Tokunaga E, Ota H, Sugita H, Martyn JA, Kaneki M (2005) S-nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. J Biol Chem 280:7511–7518
Yin X, Gu S, Jiang JX (2001) The development-associated cleavage of lens connexin 45.6 by caspase-3-like protease is regulated by casein kinase II-mediated phosphorylation. J Biol Chem 276:34567–34572
Yin X, Liu J, Jiang JX (2008) Lens fiber connexin turnover and caspase-3-mediated cleavage are regulated alternately by phosphorylation. Cell Commun Adhes 15:1–11
Zhang L, Deng T, Sun Y, Liu K, Yang Y, Zheng X (2008) Role for nitric oxide in permeability of hippocampal neuronal hemichannels during oxygen glucose deprivation. J Neurosci Res 86:2281–2291
Acknowledgement
This study was supported by National Institutes of Health grants HL088554 and HL107963 (to B. E. I.), an American Heart Association Scientist Development Grant (to B. E. I.), American Heart Association postdoctoral fellowships (to M. B. and S. R. J.) and a National Institutes of Health Cardiovascular Training Grant (to A. W. L.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Johnstone, S.R., Billaud, M., Lohman, A.W. et al. Posttranslational Modifications in Connexins and Pannexins. J Membrane Biol 245, 319–332 (2012). https://doi.org/10.1007/s00232-012-9453-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00232-012-9453-3