
Abstract
In contrast to other organisms, gram-negative bacteria have evolved numerous systems for protein export. Eight types are known that mediate export across or insertion into the cytoplasmic membrane, while eight specifically mediate export across or insertion into the outer membrane. Three of the former secretory pathway (SP) systems, type I SP (ISP, ABC), IIISP (Fla/Path) and IVSP (Conj/Vir), can export proteins across both membranes in a single energy-coupled step. A fourth generalized mechanism for exporting proteins across the two-membrane envelope in two distinct steps (which we here refer to as type II secretory pathways [IISP]) utilizes either the general secretory pathway (GSP or Sec) or the twin-arginine targeting translocase for translocation across the inner membrane, and either the main terminal branch or one of several protein-specific export systems for translocation across the outer membrane. We here survey the various well-characterized protein translocation systems found in living organisms and then focus on the systems present in gram-negative bacteria. Comparisons between these systems suggest specific biogenic, mechanistic and evolutionary similarities as well as major differences.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, our laboratory has been concerned with the phylogenetic characterization and classification of transmembrane transport systems (Saier, 1998). Transporters generally consist of channels and carriers (Saier, 1999), and carriers can function by uniport, cation:solute symport, cation:solute antiport or solute:solute antiport (Saier, 2000a). Additionally, cytoplasmic protein domains and/or subunits can be superimposed upon these integral membrane channel and carrier proteins to allow the direct coupling of chemical energy, as is provided by adenosine triphosphate (ATP) hydrolysis, to transport (Saier, 2000b; Saier & Tseng, 1999). The transporter classification (TC) system (Bush & Saier, 2002) involves transport protein categorization (Table 1) in five steps as follows: first, permeases are grouped according to transporter type (e.g., category 1, channels; category 2, secondary carriers; category 3, primary active transporters; category 4, group translocators; category 5, transmembrane electron flow carriers; category 8, accessory transport proteins; and category 9, transporters or putative transporters of unknown mechanism of action); second, these major divisions of transporters are subdivided according to protein structural type or energy coupling mechanism; third, the resultant permease types are divided into recognizable families; fourth, these families are subdivided into phylogenetic clusters or subfamilies; and fifth, within each phylogenetic cluster, all functionally characterized transporters that catalyze different transport processes and/or exhibit different substrate specificities are separately tabulated (Busch & Saier, 2002). The TC system is therefore based on both function and phylogeny. This classification system has been adopted by the International Union of Biochemistry and Molecular Biology (IUBMB). The system is described briefly on the IUBMB website and in more detail in the transporter classification database (TCDB) (http://www.tcdb.org; Saier et al., 2006). It includes all systems currently recognized to catalyze protein secretion and membrane insertion.
The Diversity of Protein Translocases in Gram-Negative Bacteria and Eukaryotic Organelles
Protein export systems are present in all living organisms. All protein export/membrane insertion systems currently recognized in living organisms fall into TC categories 1A, 1B, 1C, 1E, 2A, 3A, 9A and 9B of the TC system (Table 1; http://www.tcdb.org) (Saier, 2000a,b; Saier et al., 2006). Table 2 lists the organismal distributions and the energy sources of most types of protein secretory pathway (PSP) systems that are currently recognized in living organisms. References cited in this review are reviews and original research articles that, among other things, provide phylogenetic descriptions of the protein families. Currently characterized type I (ATP-binding cassette or ABC-type) protein secretory pathway (ISP) systems are restricted to some bacteria, especially gram-negative bacteria, as well as archaea and a few eukaryotes, although members of the ABC superfamily are found in essentially all living organisms (Schmitt et al., 2003; Yamane et al., 2004). The gram-negative bacterial export systems transport their protein substrates across both membranes of the cell envelope. These systems include ABC efflux pumps (TC 3.A.1.109 and 110) complexed with membrane fusion proteins (MFPs, TC 8.A.1) and outer membrane factors (OMFs, TC 1.B.17). ABC-type protein exporters in gram-positive bacteria often depend upon an MFP for activity but lack an OMF (Harley et al., 2000). This suggests that at least some ABC systems depend on an MFP for activity, but the molecular basis for this observation is not known.
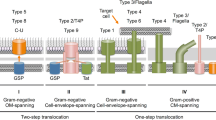
By contrast, general secretory pathway (GSP or Sec) systems for protein export across the cytoplasmic membrane are found ubiquitously in all living organisms (Cao & Saier, 2003). The twin arginine targeting translocases (Tat), also for export specifically across the cytoplasmic membrane, are by no means ubiquitous, but they are widely distributed. By contrast, main terminal branch (MTB) systems, responsible for transport of many proteins across the outer membranes of gram-negative bacteria, are exclusively restricted to these organisms (Sandkvist, 2001). In this article, we refer to the Sec or Tat plus MTB systems as type IISP systems, in parallel with the type ISP, type IIISP and type IVSP systems, which translocate their protein substrates across both membranes.
Well-characterized type III (flagellar [fla]- and pathogenesis [path]-related) and type IV (conjugation [conj]- and virulence [vir]-related) systems are largely restricted to gram-negative bacteria. However, flagellar export systems, related to but distinct from the pathogenesis-type systems, are prevalent in both gram-negative and gram-positive bacteria (Christie, 2001; Plano, Day & Ferracci, 2001). Type IV conjugation systems are also present in gram-positive bacteria (Dubnau, 1999), although these systems are still relatively poorly characterized. All proteins of these secretory systems – ISP, IISP, IIISP and IVSP, as referred to here – export proteins across both membranes of the gram-negative bacterial envelopes. They are probably energized by ATP, although guanosine triphosphate (GTP) and the proton motive force (pmf) may contribute to protein export via the Sec and MTB systems (Economou, 2002). Tat translocases, which usually translocate fully folded redox enzyme and other protein complexes into the periplasm of gram-negative bacteria, are energized by the pmf exclusively. In gram-negative bacteria, when protein substrates of the Sec and Tat systems are exported from the periplasm to the external milieu, they often use the MTB for their final secretion across the outer membrane (Johnson et al., 2006), but other protein-specific systems are also present (Theg et al., 2005; Yen et al., 2002b).
Mitochondrial protein translocases (MPTs, TC 3.A.8), possibly derived from primitive protein translocation systems in α-proteobacteria, appear to have evolved to their current level of complexity after the degeneration of endosymbiotic bacteria into mitochondria. Their evolution probably followed transfer of much of their genetic material to the nucleus of the host eukaryotic cell, requiring extensive import (Wickner & Schekman, 2005). Although there are only a few prokaryotic homologues of constituents of these mitochondrial import systems (see also TCDB, TC 1.B.33) (Lister et al., 2005), several chloroplast envelope protein translocase CEPT family constituents (TC 3.A.9) (Schleiff et al., 2003) have bacterial homologues, suggesting an origin in the primordial cyanobacterial cell (Ertel et al., 2005; Steiner et al., 2005).
The cytochrome oxidase biogenesis (Oxal) family is still poorly characterized. Functional data were available first for mitochondria and subsequently for plant chloroplasts and bacteria (Luirink, Samuelsson & de Gier, 2001; Pohlschroder et al., 2005; Yi & Dalbey, 2005). In both eukaryotic organelles as well as Escherichia coli, these proteins seem to function primarily (but not exclusively) to effect integral membrane protein insertion (Luirink et al., 2005). The large-conductance mechanosensitive channels (MscL) possibly export small proteins such as thioredoxin as well as osmolytes from the cytoplasm of the bacterial cell following osmotic downshift (for review, see Pivetti et al., 2003).
Small holin proteins, of which there are more than two dozen recognized families, form oligomeric pores in membranes. They function to export autolytic enzymes that hydrolyze the peptidoglycan layer of the bacterial cell wall. Export is from the cell cytoplasm to an extracytoplasmic locale, where these enzymes can promote cell death (Ramanculov & Young, 2001; Wang, Smith & Young, 2000; Young, 2002; Young & Bläsi, 1995). The holin/autolysin pair is usually encoded either by phage genes, in which case they promote cell lysis and phage release, or by bacterial chromosomal genes, in which case they mediate programmed cell death. Cell death-inducing systems of animal cells include the Bcl-2 family proteins (TC 1.A.21), which interact with voltage-dependent anion channel proteins (VDAC, TC 1.B.8) in the outer membranes and the ATP/ADP exchanger of the mitochondrial carrier family (MC, TC 2.A.29) in the inner membranes of mitochondria (Adams & Cory, 1998).
Finally, bacteria synthesize and secrete numerous toxins such as diphtheria and tetanus toxins, which insert into a host animal cell membrane, forming pores that transport cytoplasmic toxic proteins into the cell. All such toxins are included in TC classification 1.C. Descriptions of all of these systems as well as references to research concerning them can be found at our TCDB database (http://www.tcdb.org). These proteins will not be the focus of this review.
A number of systems in addition to the MTB cited above are known to function exclusively in export across the lipopolysaccharide (LPS)-containing outer membranes of gram-negative bacteria (Table 2). These systems include (1) the fimbrial usher porins (FUPs) that translocate fimbrial subunits across the outer membranes and assemble these subunits into intact fimbriae (Yen et al., 2002a); (2 and 3) two independently evolving families of autotransporters (AT-1 and AT-2) that exhibit C-terminal, oligomeric, pore-forming, β-barrel domains that are thought to translocate their N-terminal virulence-related protein domains to the external surface of the membrane (Cotter, Surana & St. Geme, 2005; Roggenkamp et al., 2003; Thanassi et al., 2005); (4) the two-partner secretons (TPSs), most of which translocate and sometimes modify toxins and other exported proteins (Jacob-Dubuisson, Locht & Antoine, 2001); (5) secretins, oligomeric pore-forming constituents of types II (MTB) and III (Path) systems (Thanassi, 2002); (6) OMFs that are pore-forming constituents that function with type I (ABC) protein exporters to allow protein transport across the outer gram-negative bacterial membrane in a process coupled to ATP hydrolysis catalyzed by the cytoplasmic membrane ABC exporter (Federici et al., 2005); and (7) the outer membrane protein insertion porin (OmpIP), a multicomponent system that appears to facilitate insertion of periplasmic β-structured outer membrane proteins from the periplasm into this membranous structure (Doerrler & Raetz, 2005; Voulhoux et al., 2003).
There are thus eight independently functioning systems that specifically effect protein export across or insertion into the inner (cytoplasmic) membranes of gram-negative bacteria and eight that effect export across or insertion into the outer (LPS) membranes (Table 2) (Yen et al., 2002a). Interestingly, the inner membrane protein export systems can function by a threading mechanism (ABC and Sec), by translocating fully or partially folded subunits (IIISP, IVSP, MscL and possibly holins) and one (Tat) by translocating fully folded and assembled multisubunit protein complexes. Strictly outer membrane translocases usually (but not always) function by energy-independent diffusion-type mechanisms that may translocate partially or fully folded substrate proteins. The nature of the substrates of and the translocation mechanisms utilized by the outer membrane FUP, AT-1, AT-2 and TPS family channels are still ill-defined. In the following sections, we will discuss the functions and phylogenies of individual protein secretion systems in gram-negative bacteria.
Complex Inner Membrane Secretory Systems
TYPE I (ABC-, MFP-, OMF-TYPE) PROTEIN EXPORTERS
Type I ABC macromolecular export systems are widespread in nature. Of the 64 currently recognized families of these strictly ATP-dependent systems, two are specific for large proteins (TC 3.A.1.109, -110), both from gram-negative bacteria. Members of four other families export peptides or small proteins (TC 3.A.1.111-113, 3.A.1.123), and exporters of six other families are specific for complex carbohydrates. These ABC exporters generally consist of two integral membrane domains and two cytoplasmic “energizer” domains that hydrolyze ATP. The systems may recognize a C-terminal targeting sequence in the transported substrate protein, but transport seems to be limited by the size and ease of unfolding of the substrate protein. ABC-type protein export systems, several of which can be present in a single bacterial cell (Ma et al., 2003), can associate with two auxiliary proteins, the MFPs and the OMFs, that allow transport across both membranes of the gram-negative bacterial envelope in a single step, as noted above (Holland, Schmitt & Young, 2005).
ABC transporters usually exhibit substrate specificities that reflect the phylogenies of these systems. In cases that have been studied, the constituents of these systems seem to have rarely, if at all, undergone shuffling during their evolutionary histories (Kuan et al., 1995; Paulsen, Beness & Saier, 1997a; Tam & Saier, 1993). It is presumed that this restriction reflects a need for strict protein:protein interactions for maximal function (Cao & Saier, 2001; Nguyen et al., 2000; Peabody et al., 2003).
Comparable studies with the two auxiliary constituents, the MFPs and the OMFs, have revealed that while the MFPs have evolved in parallel with their primary permeases (Dinh, Paulsen & Saier, 1994), the OMFs have not (Paulsen et al., 1997b). The discovery that MFP homologues are present in gram-positive bacteria (Harley et al., 2000) and the demonstration that at least some of these proteins are essential for transport activity (Axelsson & Holck, 1995; Quadri et al., 1997; Venema et al., 1996) are in agreement with their close connection with the permeases.
Recently, the high-resolution three-dimensional structures of both uptake- and efflux-type ABC systems have been determined. The solved efflux pump (Chang & Roth, 2001; Reyes & Chang, 2005) is MsbA of E. coli, specific for drugs and lipids, while the solved uptake system is BtuCDF, specific for vitamin B12 (Karpowich et al., 2003; Locher, Lee & Rees, 2002; Oloo & Tieleman, 2004). Their three-dimensional structures revealed marked differences between systems catalyzing uptake and efflux.
TolC of E. coli is an OMF that functions with several types of transporters. Its structure has also been solved (Eswaran et al., 2004; Higgins & Linton, 2004; Higgins et al., 2004a, b; Koronakis, 2003; Koronakis et al., 2000; Koronakis, Eswaran & Hughes, 2004; Touze et al., 2004). The TolC protein exhibits a three-dimensional fold unlike any previously characterized protein. It forms a trimeric, outer membrane β-barrel pore structure (12 β-strands, four per subunit) as well as a transperiplasmic, trimeric, α-helical conduit (12 α-helices: six continuous, six discontinuous; four per subunit) which probably connects the inner membrane permease to the outer membrane pore. The OMF by itself provides the transperiplasmic channel. The MFP probably interlinks the inner and outer membrane transport pathways. However, it may serve other functions. This last postulate is consistent with the occurrence of MFP homologues in gram-positive bacteria that lack outer membranes (Harley et al., 2000). Some of these MFP proteins have been shown to be essential for transport function, as noted above.
GENERAL SECRETORY TRANSLOCASES (SEC SYSTEMS)
Type II GSP (Sec) systems in gram-negative bacteria consist of essential and auxiliary protein subunits (Wickner & Schekman, 2005). Every living organism that has been examined has a Sec system, and most have only one (Cao & Saier, 2003). Nevertheless, only some of the constituents are found universally. The essential E. coli translocase constituents include a heterotrimeric integral inner membrane protein complex (SecYEG), the cytoplasmic ATPase (SecA) and several additional proteins, all of which follow the phylogenies of the host organisms with few exceptions (Cao & Saier, 2003; see Fig. 1). SecA may recruit SecYEG complexes (or vice versa) to form reversible active translocation complexes (Benach et al., 2003). The active assembly includes a SecA homodimer and a SecYEG homodimeric or homotetrameric complex (Scheuring et al., 2005; Tziatzios et al., 2004). SecA is apparently found only in prokaryotes. SecY of E. coli is a 10- Transmembrane Segment (TMS) protein of about 450 amino acyl residues that is believed to form the protein-translocating channel when complexed with the two small integral membrane proteins SecE and SecG, each maximally of about 140 amino acyl residues in length (Ito, 1992). The SecYEG complex is ubiquitous, being present in every bacterium, archaeon and eukaryote with a fully sequenced genome (Kinch, Saier & Grishin, 2002).

Phylogenetic tree of sequenced homologues of the SecY protein of E. coli and the Sec61α protein of Saccharomyces cerevisiae. Reproduced from Cao & Saier (2003), with permission, wherein protein and organismal abbreviations can be found. The tree was generated with the CLUSTAL X and TREEVIEW programs (Thompson et al., 1997; Zhai, Tchieu & Saier, 2002). Note that the tree reveals one eukaryotic and one archaeal cluster (clusters 2 and 1, respectively) plus “splinter” group members from yeast and plants. The other eight phylogenetic clusters are exclusively of bacterial origin, where the proteins cluster according to the phylogenies of their source organisms (16S rRNAs).
Two auxiliary proteins, SecD and SecF in E. coli, are homologous to members of the Resistance/ Nodulation Division (RND) superfamily (TC 2.A.6). They are not present in many organisms. Another protein, YajC of E. coli, forms a complex with SecD-SecF, both independently of and in complexation with SecYEG. The SecDF-YajC complex is not essential for secretion, but it stimulates secretion up to 10-fold, particularly at lower temperatures. The mechanistic role of this auxiliary prokaryote-specific complex is not clearly defined, but it is not required for maintenance of the pmf (Nouwen, van der Laan & Driessen, 2001).
Although Sec-dependent protein export and integral membrane protein insertion are driven by ATP/GTP hydrolysis, the pmf is stimulatory and may function solely in translocating the C-terminal parts of the unfolded substrate proteins. Thus, it is possible that both energy sources are required for efficient translocation, with each acting at different steps (Geller, 1991; Rapoport, Jungnickel & Kutay, 1996). Point mutations in SecY have been described that abolish the pmf dependence of the translocation process, but nucleoside triphosphate hydrolysis appears to be essential under all conditions.
Insertion of integral inner membrane proteins in bacteria is dependent on a complex resembling the eukaryotic signal recognition particle (SRP) protein-RNA complex, which functions as an essential constituent for protein membrane insertion (Müller et al., 2001). It has also been shown to play a role in the export of some secretory proteins, such as DsbA, β-lactamase and some autotransporters in E. coli (Sijbrandi et al., 2003; Takamatsu et al., 1997). The primary protein constituents of the bacterial complex, Ffh (an SRP54-like protein) and FtsY (an SRP receptor [subunit α]-like protein), probably act as GTP-dependent chaperones, feeding the substrate protein into the SecYEG complex (Scotti et al., 1999). Insertion of most polytopic inner membrane proteins shows a dependence on Ffh and FtsY as well as the SecYEG channel complex, although in some cases the Oxa1 homologue in E. coli, YidC, may replace this complex (Fröderberg et al., 2003; van der Laan, Nouwen & Driessen, 2005). How the SecYEG channel may facilitate membrane protein insertion, based on the high-resolution X-ray structure of an archaeal SecYEG channel complex, has been discussed (Rapoport et al., 2004; van den Berg et al., 2004).
TYPE III FLAGELLAR AND PATHOGENICITY-RELATED SYSTEMS
Proteins of the IIISP family are found in gram-negative bacteria and allow secretion of cytoplasmically synthesized proteins across both membranes of the cell envelope (Hueck, 1998). These systems are often concerned with secretion of virulence factors across the envelopes of pathogenic gram-negative bacteria (Yip et al., 2005). Genes encoding these proteins are sometimes in the chromosome, in which case they are most frequently found within “pathogenicity islands,” which are inserted DNA segments derived from foreign organismal sources (Hansen-Wester & Hensel, 2001). They may also be plasmid-encoded. Many of these proteins are homologous to proteins concerned with bacterial flagellar protein export (Nguyen et al., 2000; Saier, 2004), and the flagellar export machinery has been shown to be capable of secreting virulence factors (Young, Schmiel & Miller, 1999). They are thus functionally and structurally equivalent, although the constituents generally cluster separately on a phylogenetic tree (Nguyen et al., 2000).
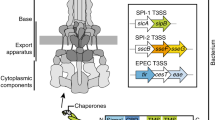
As many as 20 distinct proteins may comprise the type III secretion apparatus. The biochemical functions of most of the individual constituents are not known. The best-characterized systems are derived from Yersinia species, although they occur in many gram-negative bacterial pathogens (Mota & Cornelis, 2005). These protein complexes export Yersinia virulence-related proteins called YOPS. One of the constituents of the IIISP system, YscN, is an ATPase that is believed to couple ATP hydrolysis to protein export. Six integral inner membrane proteins (LcrD and YscD, R, S, T and U) may form a complex that provides the transport pathway. The YscC protein, an outer membrane secretin, forms dodecameric pores (Burghout et al., 2004a, b; Nguyen et al., 2000).
The IIISP systems often secrete proteins directly into the host cell cytoplasm without exposure to the extracellular milieu (Coombes & Finlay, 2005). This fact implies the existence of a pore complex that spans the host cell cytoplasmic membrane and is contiguous with the bacterial secretion apparatus. The Yersinia proteins that are believed to provide this function are YopB and YopD, which span the host cell cytoplasmic membrane and form oligomeric pore complexes (Olsson et al., 2004; Ryndak et al., 2005). These putative host cell membrane pore proteins comprise the bacterial type III-target cell pore (IIITCP) family (TC 1.C.36).
The mechanism by which the substrates are recognized by the IIISP machinery remains questionable. The N-terminal ∼15 amino acyl residues as well as mRNA signals have been proposed to target proteins to the secretory apparatus (Ramamurthi & Schneewind, 2003a,b). The former proposal is now favored by most leading experts in the field (Cornelis, 2002; Karavolos et al., 2005). However, the IIISP-associated chaperones that interact with internal segments of the target proteins may also confer specificity to the secretion pathway (Lee & Galán, 2004).
TYPE IV CONJUGATION- AND VIRULENCE-RELATED (IVSP) SYSTEMS
Protein complexes of the IVSP family consist of multiple subunits that span the two membranes and the peptidoglycan wall of the gram-negative bacterial cell envelope or the single membrane plus wall of the gram-positive bacterial cell envelope. They export proteins and DNA-protein complexes out of the cell and into the cytoplasm of a recipient cell (Christie & Cascales, 2005; Winans, Burns & Christie, 1996). These systems are very promiscuous, being capable of transporting various DNA-protein complexes into other bacteria, yeast and plants or into the external medium (Hamilton et al., 2005; Li et al., 2005). For example, the VirB systems of agrobacterial species are specifically designed to transfer T-DNA into plant cells, causing cancerous growth; but they can also transfer the IncQ plasmid RSF1010 into both plant and bacterial cells (Bohne, Yim & Binns, 1998). Further, the various Inc IVSP systems are designed to mediate plasmid transfer from the donor bacterium to a recipient bacterium, but cross-specificity has been demonstrated (Lybarger & Sandkvist, 2004). C-terminal sequences of proteins covalently linked to the DNA may be recognized as a prelude to nucleoprotein transport (Christie & Cascales, 2005). While proteins in addition to the VirB2-B11 proteins may be involved in the transfer process, the VirB proteins appear to be the primary ones involved in export from the cytoplasm across the two membranes of the agrobacterial envelope. Schematic models of type IVSP systems have been presented (Cao & Saier, 2001; Christie & Cascales, 2005).
The VirB system of Agrobacterium tumefaciens is related to (1) a natural competence (CAG, ComB) system of Helicobacter pylori, which may also be involved in transfer of virulence factors including the CagA antigen into host animal cells by a type IV secretion-related mechanism; (2) the Ptl system, involved in secretion of pertussis toxin from the Bordetella pertussis cell; (3) the TraS/TraB system of the Pseudomonas aeruginosa conjugative plasmid RP1; (4) the Trb system of plasmid pTiC58 of Agrobacterium, one of three genetically encoded systems for conjugal transfer of this Ti plasmid; (5) the Tra system of plasmid F in E. coli; and (6) the Dot conjugative transfer/virulence system of Legionella pneumophila (see TC entry 3.A.7) (Chen, Christie & Dubnau, 2005).
Although members of the type IV secretion family share many characteristics, not all systems contain the same sets of genes. Thus, the virB systems of Ti plasmids and the trb system of RP4 have only six recognized genes in common. The distantly related CAG system of H. pylori contains eight known constituents (Karnholz et al., 2006), and the dot system of L. pneumophila contains only two easily recognizable virB homologues. Homologues of only one VirB protein, VirB10 (TrbI), are demonstrably present in all known type IV secretion systems characterized (Cao & Saier, 2001). A model for the conjugative transfer of DNA protein complexes through the A. tumefaciens VirB-VirD4 system has been presented (Chen et al., 2005).
Transport of proteins and DNA protein complexes has been thought to occur in a single energy-coupled step. However, this hypothesis has been challenged by the observation of periplasmic intermediates of the IVSP substrates (Pantoja et al., 2002). It is possible that, as originally proposed, transport across both membranes is normally linked but that some periplasmic leakage can occur.
TAT SYSTEMS
The TatABCE system of E. coli has been extensively characterized (Sargent, Berks & Palmer, 2006; Yen et al., 2002b). This system forms a large (∼600 kDa) complex, which interacts with fully folded substrate redox proteins that have an N-terminal (S/T)RRXFLK “twin arginine” leader motif (Müller, 2005). It translocates several redox enzymes to the E. coli periplasm including nitrate reductase (NapA), formate dehydrogenase (FdnGHI), dimethylsulfoxide reductase (DmsABC) and trimethylamine N-oxide reductase (TorA), all of which have this leader motif (Gralnick et al., 2006; Sambasivarao et al., 2000, 2001, 2002; Stanley et al., 2002). Hydrogenases, formate dehydrogenases and several other proteins, including nonredox proteins and some integral membrane proteins (several dozen altogether in E. coli), use this pathway (Berks, Palmer & Sargent, 2005). These proteins associate with their cofactors in the cell cytoplasm before translocation.
The Tat system apparently functions independently of other types of protein secretory systems present in E. coli (Palmer, Sargent & Berks, 2005). Only one TatB homologue and TatC are absolutely required for function (Bogsch et al., 1998; Hicks et al., 2005; Sargent et al., 1999). TatA, TatB and TatE are paralogues of each other and exhibit a single TMS each (Muller & Klosgen, 2005). TatA and TatE exhibit much more similarity in sequence to each other than they do to TatB (Yen et al., 2002b). They can functionally substitute for each other. TatA (the major, more conserved constituent) and TatB (the minor, less conserved constituent) together comprise large cylindrical channel-forming complexes of variable diameters that may serve as the channel for protein translocation (Gohlke et al., 2001; Hicks et al., 2005). TatE is absent in many species, such as P. aeruginosa.
Homologues of representative E. coli Tat proteins are found in a variety of gram-negative and gram-positive bacteria, archaea and thylakoid membranes of plant chloroplasts (van Dijl et al., 2002; Yen et al., 2002b). TatC, with six putative TMSs, may serve as a specificity determinant for the complex (Jongbloed et al., 2000). Substrate proteins bind to the receptor complex, inducing formation of the protein-translocating channel (Berks, Palmer & Sargent, 2003; Berks et al., 2005; Dabney-Smith, Mori & Cline, 2006; Gerard & Cline, 2006). An organism may encode within its genome one, two or three TatA homologues and one, two or three TatC homologues; but no organism with a completely sequenced genome encodes one but not the other (Yen et al., 2002b). Homologues are not demonstrable in yeast, in animals or in many prokaryotes, particularly those with small genomes and a fermentative lifestyle. Thus, these systems are not ubiquitous as are the GSP systems (Cao & Saier, 2003). Energy coupling for transport involves the pmf in both chloroplasts and E. coli (Müller, 2005; Theg et al., 2005). A protein:proton antiport mechanism is inferred (Berks et al., 2003, 2005; Theg et al., 2005).
The TatC phylogenetic tree reveals tremendous diversity in the sequences of these proteins (Yen et al., 2002b). All of the low G+C gram-positive bacterial homologues cluster together, as do the high G+C gram-positive bacterial homologues; and most of the gram-negative bacterial proteins form two distinct but adjacent clusters. However, the archaeal homologues are found in multiple clusters, while the plant proteins are found in two clusters. It is possible that a few gene duplication events that occurred early during the evolution of Tat family constituents were responsible for these unexpected phylogenetic characteristics (van Dijl et al., 2002; Yen et al., 2002b).
Outer Membrane Protein Translocases
PROPERTIES OF THE MTB
The MTB is very complex, consisting of at least 14 proteins that somehow function in the energized transport of folded exoproteins from the periplasm across the outer membrane to the external milieu (Peabody et al., 2003). One of the proteins of the Klebsiella MTB complex, the PulD secretin (TC 1.B.22), is homologous to one of the constituents of the IIISP system (TC 3.A.6) (Peabody et al., 2003). PulD and its homologues form dodecameric ring structures with large, central, gated pores (internal diameters of 50–100 Å) (Chami et al., 2005; Collins et al., 2001; Schmidt et al., 2001). Another constituent of the MTB (the PulE ATPase) is homologous to an ATPase (VirB11) of the IVSP complex (TC 3.A.7). Otherwise these distinct protein translocases appear to be nonhomologous. They share few structural and functional features and probably evolved independently of each other. However, they sometimes use common constituents, as noted above, and parallels with type IV protein secretion systems have been noted (Filloux, 2004).
The other constituents of the MTB are either integral constituents of the inner membrane (PulC, F, G, H, I, J, K, L, M, N and O), a peripheral constituent of the inner membrane (PulE) or in one case, a peripheral outer membrane lipoprotein which probably functions as a secretin-specific chaperone/anchor protein (PulS) (Nouwen et al., 1999). One of the inner membrane proteins (PulO) is a peptidase/N-methyl transferase that processes the pilin-like precursors of PulG, H, I, J and K. PulE is an ATP-binding ATPase/kinase that exhibits an essential zinc-finger motif. Another protein, PulL, is required for PulE to associate with the membrane. These proteins probably form a transperiplasmic complex called a “secreton” that (1) recognizes the substrate proteins in the periplasm, (2) energizes transport across the outer membrane and (3) controls opening of the PulD secretin pore. Retraction and extension of the periplasmic pseudopilus, consisting in part of the PulG protein, may energize transport (Burrows, 2005; Vignon et al., 2003). Other proteins may be involved in secreton assembly. Substrate proteins fold in the periplasm prior to transport across the outer membrane. The secretion signal may be contained in the tertiary conformation of the native protein, or multiple signals may be present (Francetic & Pugsley, 2005).
FUP SYSTEMS
FUP systems are responsible for the biogenesis of numerous fimbriae (pili) in gram-negative bacteria, cyanobacteria and Deinococcus radiodurans, a gram-positive bacterium with an unusual dual membrane envelope (Yen et al., 2002a). The operon encoding the structural proteins of each fimbrium also encodes a fimbrium-specific periplasmic chaperone protein and a fimbrium-specific outer membrane usher protein. The chaperone and usher proteins, in general, evolved in parallel from their evolutionary precursor proteins (Van Rosmalen & Saier, 1993). The usher proteins contain a large central domain that spans the outer membrane 24 times as β-strands, presumbaly forming a β-barrel structure (Mol & Oudega, 1996). Following translocation across the inner membrane by the Sec system, the pilus subunits are bound to the chaperone proteins, which prevents the self-assembly of pili in the periplasm. Interactions between the chaperone and usher proteins release the pilus subunits, which are subsequently exported through the usher protein across the outer membrane as a prelude to pilus assembly on the outer surface of the outer membrane (Sauer et al., 2000; Thanassi & Hultgren, 2000). The mechanism by which the assembled fimbrial structure is exported through the usher protein is not well understood.
AT-1 SYSTEMS
The autotransporters consist of a single protein with an N-terminal Sec-type signal peptide, a central passenger domain and a C-terminal β-domain of 250–300 amino acyl residues in each system. Although the β-domains of different autotransporters of the AT-1 family are homologous, they are extremely diverse in sequence. Moreover, the passenger domains, which determine the functions of the exoproteins, vary significantly in sequence and size (Loveless & Saier, 1997). These proteins are found primarily in proteobacteria, but the chlamydial kingdom also has recognizable AT-1-type autotransporters (Yen et al., 2002a). Following secretion across the inner membrane by the Sec system and cleavage of the signal peptide, multiple β-domains form oligomeric ring-shaped complexes of ∼500 kDa in the outer membrane, allowing passage of the folded N-terminal domains through the channel (Veiga et al., 2002). The passenger domains, many of which are virulence factors in gram-negative bacteria, are either released to the environment or remain attached to the cell surface.
AT-2 SYSTEMS
Recently, a novel type of autotransporter was identified in proteobacteria and a few more distantly related gram-negative bacteria. The prototype is the adhesin protein YadA in Yersinia enterocolitica (Roggenkamp et al., 2003). Rather than having 14 or 15 amphipathic β-strands, as is true for the conventional AT-1 autotransporters described above, YadA contains a C-terminal putative transport domain of only four amphipathic β-strands that is joined to the N-terminal passenger domain by a coiled-coil linker. This linker is essential for stability and translocation of the passenger domain through the outer membrane. The structures of the transporter domains and the nature of the passenger domains are not yet fully defined. However, the available evidence suggests that the passenger domains, and possibly the AT-2 domains, arose by intragenic duplication of segments of defined size, creating multiple repeat units in the proteins. The repeat units vary in size from 7 to about 50 amino acyl residues (Kim, Chao & Saier, 2006). A single AT-2 protein may have multiple copies of as many as three different types of repeat units, where the largest repeats are near the N-termini of the passenger domain and the smallest are near the C-termini of this domain, overlapping the “linker” region connecting the passenger domain and the AT-2 domain (Kim et al., 2006).
TPS SYSTEMS
Each TPS system is composed of a substrate protein and a transport protein that are usually encoded by two neighboring genes (Jacob-Dubuisson et al., 2001). Although TPS homologues have not been identified in archeae, they have been found in bacteria and the animal, plant and fungal kingdoms of eukaryotes (Yen et al., 2002a). Most protein substrates of the bacterial TPS systems are large proteins with hemolytic and/or adhesive activities that are linked to bacterial virulence (Thanassi et al., 2005). The transport protein consists of a β-domain with 19 predicted amphipathic β-strands that presumably form a β-barrel channel in the outer membrane (Jacob-Dubuisson et al., 1999; Könninger et al., 1999). Both proteins are secreted to the periplasm by the Sec system, and the passenger domain is further exported across the outer membrane by the transport constituent of the binary system (Newman & Stathopoulos, 2004). The mechanism of protein secretion by the outer membrane transporter is unclear. The substrate protein contains a conserved N-terminal domain of approximately 115 amino acyl residues that is specifically recognized by its cognate transport protein (Jacob-Dubuisson et al., 1997).
THE OMPIP FAMILY
Gram-negative bacterial outer membrane proteins (OMPs) are assembled from the periplasm into the outer membrane in a process that is poorly understood. A large (∼800 aa) outer membrane antigen in Neisseria species, Omp85 (TC 1.B.33.1.1) (Genevrois et al., 2003), is homologous to the protective surface antigen D15 precursor in Haemophilus influenzae (TC 1.B.33.1.2). These bacterial proteins are very distantly related to the chloroplast import-associated β-barrel channel protein IAP75 (TC 1.B.33.2.1), a constituent of the chloroplast envelope protein translocase (CEPT or Tic-Toc) family (TC 3.A.9). IAP75 has been found to be a β-barrel porin in the outer membrane of plant chloroplasts (Ertel et al., 2005; Gentle, Burri & Lithgow, 2005).
Omp85 is also distantly related to the yeast mitochondrial Sorting and Assembly Machinery (SAM) constituent SAM50 (Kozjak et al., 2003). The SAM complex in yeast mitochondria consists of at least three proteins, SAM50, SAM35 and MAS37 (Kozjak et al., 2003; Milenkovic et al., 2004; Wiedemann et al., 2003). It is required for the assembly of outer membrane β-barrel proteins in mitochondria.
The functionally characterized homologue in Neisseria meningitidis, Omp85, is essential for bacterial viability (Gentle et al., 2004). It has a two-domain structure with an N-terminal periplasmic domain rich in POTRA repeats and a C-terminal domain that forms in integral outer membrane β-barrel (Gentle et al., 2005). Unassembled forms of various outer membrane proteins accumulate when Omp85 is depleted (Voulhoux et al., 2003). Moreover, immunofluorescence microscopy showed decreased surface exposure of outer membrane proteins, particularly at the cell division planes. Homologues of Omp85 are present in all gram-negative bacteria examined (Voulhoux et al., 2003).
Generally in gram-negative bacteria, LPS and phospholipids (PLs) destined for the outer membrane are made in the inner membrane. Genevrois et al. (2003) reported that the Omp85 structural gene is cotranscribed with genes involved in lipid biosynthesis. Depletion of Omp85 results in accumulation of LPS and PL in the inner membrane and loss from the outer membrane. The effects on lipids were reported to precede the effects on outer membrane protein (PorA and Opa) insertion, suggesting that the latter effects were secondary to the effects on LPS and PL translocation (Genevrois et al., 2003). However, Doerrler & Raetz (2005) came to the opposite conclusion when studying the effects of mutations in the yaeT gene of E. coli. YaeT is the E. coli Omp85 orthologue. These investigators and others concluded that YaeT functions primarily in protein insertion into the outer membrane (Gentle et al., 2004). A different protein, OstA or Imp (784 aa in E. coli; TC 1.B.42; P31554), may mediate LPS export (Bos et al., 2004).
Normally, OMPs are translocated into the periplasm via the Sec translocase (TC 3.A.5). They are believed to fold in the periplasm before being inserted into the outer membrane. Folding is stimulated by the small periplasmic chaperone protein SurA (P21202) and by a peptidyl prolyl cis-trans isomerase (PPIase) called Rotamase C or parvulin (POA9L5). Two other periplasmic/outer membrane proteins, Skp (OmpH, HlpA, P11457) and another PPIase FkpA (P45523), also function in this capacity (Missiakas, Betton & Raina, 1996). Still other proteins may be involved. It is even possible that an energy source will prove to be required.
In E. coli, a multiprotein complex has been shown to be required for outer membrane biogenesis (Wu et al., 2005). This complex includes the Omp85 homologue YaeT, the lipoprotein YfgL and two other proteins, YfiO and NlpB. It is believed that these proteins function in outer membrane protein assembly. The specific biochemical roles of the individual protein constituents have not been determined. Gentle et al. (2005) provide a current review on possible evolutionary pathways taken by Omp85 homologues.
Comparisons and Overview
Table 3 compares the properties of the four primary secretory pathway systems involved in inner membrane secretion. The Tat system is tabulated separately in Table 3 for clarity. Each of these systems apparently evolved independently of the others, even though they exhibit overlapping properties and occasionally share one or two homologous protein constituents. Type I systems (ABC plus MFP and OMF proteins) consist of three or four protein constituents. Type II systems as defined here include the Sec or Tat system for inner membrane transport plus the MTB or a protein-specific translocase for transport across the outer membrane. The Sec system consists of 7–10 constituents. It transports proteins across or inserts proteins into the cytoplasmic gram-negative or gram-positive bacterial cell membrane (Cao & Saier, 2003). The E. coli Tat system consists of four dissimilar protein constituents, but three of these proteins are homologous and of similar sizes with the same topology. Tat systems in other bacteria can be much simpler with only two nonhomologous constituents (Yen et al., 2002b). However, multiple subunits of TatA/B homologues generate the oligomeric pore structure (Mangels et al., 2005; Müller, 2005; Sargent et al., 2001). The MTB includes 10–12 essential constituents for transport across the outer membrane, while the substrate-specific outer membrane translocases each usually consist of a single protein. Both type III and type IV systems consist minimally of about 10–12 constituents. Although these two systems share certain functional characteristics, none of the protein constituents is recognizably common to the two systems. It is thus clear that all of these systems are distinct, complex, multicomponent systems.
The basis for assuming that these systems arose independently in part involves the fact that homology cannot be demonstrated for constituents of one system with those of any other except for the secretins of the MTB, which are distantly related to the secretins of IIISP systems, and the ATPases of the MTB, homologues of which are found in IVSP systems. These ATPases may be involved in assembly of the complex and/or protein export (Table 2). While type I, III and IV systems span the two membranes of the gram-negative bacterial envelope and translocate protein substrates across both membranes in a single energy-coupled step, type II systems, as defined here, actually consist of two distinct, independently functioning complexes, one (Sec or Tat) for transport across the inner membrane and one (MTB) for transport across the outer membrane. There are also several types of protein-specific outer membrane export systems. Like Sec, Tat systems transport proteins only across the cytoplasmic membrane (Table 2); but unlike Sec, they transport fully folded and assembled enzyme complexes using the pmf instead of ATP to energize transport.
Well-characterized protein-translocating ABC-type (type I) systems are found primarily in gram-negative bacteria, although ABC peptide and small protein export systems are also found in gram-positive bacteria and eukaryotes. While Sec systems are ubiquitous, being found in every living cell in which they have been sought, MTB systems seem to be restricted to gram-negative bacteria. Homologues of Tat systems are widespread, but they are not found in all living organisms, being absent in some bacteria, some archaea and yeast and animals of the eukaryotic domain. However, they are represented in each of the three primary domains of life (Table 2). While type III pathogenesis-related systems are present only in gram-negative bacteria, flagellar type III systems are present in gram-positive bacteria as well and, similarly, the type IV virulence/conjugation-related systems occur in gram-positive as well as gram-negative bacteria.
Table 3 also reveals that while type III and IV systems have undergone frequent lateral transfer, shuffling of constituents during evolution has only been shown to occur in type I systems, not in types II, III and IV systems. In type I systems, OMFs have shuffled relative to the ABC and MFP constituents (Dinh et al., 1994; Paulsen et al., 1997b), but MFPs have not shuffled relative to their cognate ABC transporters. Specific protein-protein interactions, rendering essential the coevolution of all or most protein constituents of a complex, may account for these startling findings.
Finally, the protein species transported in these four primary types of systems are distinguishable in that Sec, ABC and type III systems transport unfolded protein substrates; the MTB and type IV systems transport fully or partially folded protein subunits; and Tat systems transport fully folded and assembled enzyme complexes (Table 3). It is therefore clear that the predominant well-characterized protein export systems in a gram-negative bacterium like E. coli or P. aeruginosa (Ma et al., 2003) use different protein constituents, energy sources and mechanisms to transport their protein substrates, which are transported in different states of assembly depending on the system. The poorly characterized Oxa1/YidC-type systems are believed to be energized by the pmf, as are the Tat systems (Luirink et al., 2001; Yen et al., 2001; Yi & Dalbey, 2005). However, the Oxa1-mediated mode of transport and the conformation of the translocated substrates of this system have not been extensively investigated (Luirink et al., 2005).
It is hoped that this short treatise allows the reader to conceptualize and discriminate between the various types of protein secretory systems found in nature. It is worth briefly marveling at the remarkable functional diversity that the evolutionary process has yielded, even within a single gram-negative bacterium.
References
Adams J.M., Cory S. 1998. The Bcl-2 protein family: arbiters of cell survival. Science 281:1322–1326
Axelsson L., Holck A. 1995. The genes involved in production of and immunity to sakacin A, a bacteriocin from Lactobacillus sake Lb706. J. Bacteriol. 177:2125–2137
Benach J., Chou Y.T., Fak J.J., Itkin A., Nicolae D.D., Smith P.C., Wittrock G., Floyd D.L., Golsaz C.M., Gierasch L.M., Hunt J.F. 2003. Phospholipid-induced monomerization and signal-peptide-induced oligomerization of SecA. J. Biol. Chem. 278:3628–3638
Berks B.C., Palmer T., Sargent F. 2003. The Tat protein translocation pathway and its role in microbial physiology. Adv. Microb. Physiol. 47:187–254
Berks B.C., Palmer T., Sargent F. 2005. Protein targeting by the bacterial twin-arginine translocation (Tat) pathway. Curr. Opin. Microbiol. 8:174–181
Bogsch E.G., Sargent F., Stanley N.R., Berks B.C., Robinson C., Palmer T. 1998. An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J. Biol. Chem. 273:18003–18006
Bohne J., Yim A., Binns A.N. 1998. The Ti plasmid increases the efficiency of Agrobacterium tumefaciens as a recipient in virB-mediated conjugal transfer of an IncQ plasmid. Proc. Natl. Acad. Sci. USA 95:7057–7062
Bos M.P., Tefsen B., Geurtsen J., Tommassen J. 2004. Identification of an outer membrane protein required for the transport of lipopolysaccharide to the bacterial cell surface. Proc. Natl. Acad. Sci. USA 101:9417–9422
Burghout P., Beckers F., de Wit E., van Boxtel R., Cornelis G.R., Tommassen J., Koster M. 2004a. Role of the pilot protein YscW in the biogenesis of the YscC secretin in Yersinia enterocolitica. J. Bacteriol. 186:5366–5375
Burghout P., van Boxtel R., Van Gelder P., Ringler P., Muller S.A., Tommassen J., Koster M. 2004b. Structure and electrophysiological properties of the YscC secretin from the type III secretion system of Yersinia enterocolitica. J. Bacteriol. 186:4645–4654
Burrows L.L. 2005. Weapons of mass retraction. Mol. Microbiol. 57:878–888
Busch W., Saier M.H. Jr. 2002. The transporter classification (TC) system, 2002. CRC Crit. Rev. Biochem. Mol. Biol. 37:287–337
Cao T.B., Saier M.H. Jr. 2001. Conjugal type IV macromolecular transfer systems of gram-negative bacteria: Organismal distribution, structural constraints and evolutionary conclusions. Microbiology 147:3201–3214
Cao T.B., Saier M.H. Jr. 2003. The general protein secretory pathway: Phylogenetic analyses leading to evolutionary conclusions. Biochim. Biophys. Acta 1609:115–125
Chami M., Guilvout I., Gregorini M., Remigy H.W., Muller S.A., Valerio M., Engel A., Pugsley A.P., Bayan N. 2005. Structural insights into the secretin PulD and its trypsin-resistant core. J. Biol. Chem. 280:37732–37741
Chang G., Roth C.B. 2001. Structure of MsbA from E. coli: A homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science 293:1793–1800
Chen I., Christie P.J., Dubnau D. 2005. The ins and outs of DNA transfer in bacteria. Science 310:1456–1460
Christie P.J. 2001. Type IV secretion: Intercellular transfer of macromolecules by systems ancestrally related to conjugation machines. Mol. Microbiol. 40:294–305
Christie P.J., Cascales E. 2005. Structural and dynamic properties of bacterial type IV secretion systems. Mol. Membr. Biol. 22:51–61
Collins R.F., Davidsen L., Derrick J.P., Ford R.C., Tonjum T. 2001. Analysis of the PilQ secretion from Neisseria meningitidis by transmission electron microscopy reveals a dodecameric quaternary structure. J. Bacteriol. 183:3825–3832
Coombes B.K., Finlay B.B. 2005. Insertion of the bacterial type III translocon: Not your average needle stick. Trends Microbiol. 13:92–95
Cornelis G.R. 2002. The Yersinia Ysc-Yop “type III” weaponry. Nat. Rev. Mol. Cell. Biol. 3:742–752
Cotter S.E., Surana N.K., St. Geme J.W. III. 2005. Trimeric autotransporters: A distinct subfamily of autotransporter proteins. Trends Microbiol. 13:199–205
Crompton M., Barksby E., Johnson N., Capano M. 2002. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie 84:143–152
Dabney-Smith C., Mori H., Cline K. 2006. Oligomers of Tha4 organize at the thylakoid Tat translocase during protein transport. J. Biol. Chem. 281:5476–5483
Dinh T., Paulsen I.T., Saier M.H. Jr. 1994. A family of extracytoplasmic proteins that allow transport of large molecules across the outer membranes of gram-negative bacteria. J. Bacteriol. 176:3825–3831
Doerrler W.T., Raetz C.R. 2005. Loss of outer membrane proteins without inhibition of lipid export in an Escherichia coli YaeT mutant. J. Biol. Chem. 280:27679–27687
Dubnau D. 1999. DNA uptake in bacteria. Annu. Rev. Microbiol. 53:217–244
Economou A. 2002. Bacterial secretome: The assembly manual and operating instructions. Mol. Membr. Biol. 19:159–169
Ertel F., Mirus O., Bredemeier R., Moslavac S., Becker T., Schleiff E. 2005. The evolutionarily related β-barrel polypeptide transporters from Pisum sativum and Nostoc PCC7120 contain two distinct functional domains. J. Biol. Chem. 280:28281–28289
Eswaran J., Koronakis E., Higgins M.K., Hughes C., Koronakis V. 2004. Three’s company: Component structures bring a closer view of tripartite drug efflux pumps. Curr. Opin. Struct. Biol. 14:741–747
Federici L., Du D., Walas F., Matsumura H., Fernandez-Recio J., McKeegan K.S., Borges-Walmsley M.I., Luisi B.F., Walmsley A.R. 2005. The crystal structure of the outer membrane protein VceC from the bacterial pathogen Vibrio cholerae at 1.8 Å resolution. J. Biol. Chem. 280:15307–15314
Filloux A. 2004. The underlying mechanisms of type II protein secretion. Biochim. Biophys. Acta 1694:163–179
Francetic O., Pugsley A.P. 2005. Towards the identification of type II secretion signals in a nonacylated variant of pullulanase from Klebsiella oxytoca. J. Bacteriol. 187:7045–7055
Froderberg L., Houben E., Samuelson J.C., Chen M., Park S.K., Phillips G.J., Dalbey R., Luirink J., De Gier J.W. 2003. Versatility of inner membrane protein biogenesis in Escherichia coli. Mol. Microbiol. 47:1015–1027
Geller B.L. 1991. Energy requirements for protein translocation across the Escherichia coli inner membrane. Mol. Microbiol. 5:2093–2098
Genevrois S., Steeghs L., Roholl P., Letesson J.J., van der Ley P. 2003. The Omp85 protein of Neisseria meningitidis is required for lipid export to the outer membrane. EMBO J. 22:1780–1789
Gentle I., Gabriel K., Beech P., Waller R., Lithgow T. 2004. The Omp85 family of proteins is essential for outer membrane biogenesis in mitochondria and bacteria. J. Cell Biol. 164:19–24
Gentle I.E., Burri L., Lithgow T. 2005. Molecular architecture and function of the Omp85 family of proteins. Mol. Microbiol. 58:1216–1225
Gerard F., Cline K. 2006. Efficient twin arginine translocation (Tat) pathway transport of a precursor protein covalently anchored to its initial cpTatC binding site. J. Biol. Chem. 281:6130–6135
Gohlke S.F., De Leeuw E., Stanley N.R., Palmer T., Saibil H.R., Berks B.C. 2001. Purified components of the Escherichia coli Tat protein transport system form a double-layered ring structure. Eur. J. Biochem. 268:3361–3367
Gralnick J.A., Vali H., Lies D.P., Newman D.K. 2006. Extracellular respiration of dimethyl sulfoxide by Shewanella oneidensis strain MR-1. Proc. Natl. Acad. Sci. USA 103:4669–4674
Hamilton H.L., Dominguez N.M., Schwartz K.J., Hackett K.T., Dillard J.P. 2005. Neisseria gonorrhoeae secretes chromosomal DNA via a novel type IV secretion system. Mol. Microbiol. 55:1704–1721
Hansen-Wester I., Hensel M. 2001. Salmonella pathogenicity islands encoding type III secretion systems. Microbes Infect. 3:549–559
Harley K.T., Djordjevic G.M., Tseng T.-T., Saier M.H. Jr. 2000. Membrane-fusion protein homologues in gram-positive bacteria. Mol. Microbiol. 36:516–517
Hicks M.G., Lee P.A., Georgiou G., Berks B.C., Palmer T. 2005. Positive selection for loss-of-function tat mutations identifies critical residues required for TatA activity. J. Bacteriol. 187:2920–2925
Higgins C.F., Linton K.J. 2004. The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 11:918–926
Higgins M.K., Bokma E., Koronakis E., Hughes C., Koronakis V. 2004a. Structure of the periplasmic component of a bacterial drug efflux pump. Proc. Natl. Acad. Sci. USA 101:9994–9999
Higgins M.K., Eswaran J., Edwards P., Schertler G.F., Hughes C., Koronakis V. 2004b. Structure of the ligand-blocked periplasmic entrance of the bacterial multidrug efflux protein TolC. J. Mol. Biol. 342:697–702
Holland I.B., Schmitt L., Young J. 2005. Type 1 protein secretion in bacteria, the ABC-transporter dependent pathway. Mol. Membr. Biol. 22:29–39
Hueck C.J. 1998. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev. 62:379–433
Ito K. 1992. SecY and integral membrane components of the Escherichia coli protein translocation system. Mol. Microbiol. 6:2423–2428
Jacob-Dubuisson F., Buisine C., Willery E., Renauld-Mongenie G., Locht C. 1997. Lack of functional complementation between Bordetella pertussis filamentous hemagglutinin and Proteus mirabilis HpmA hemolysin secretion machineries. J. Bacteriol. 179:775–783
Jacob-Dubuisson F., El-Hamel C., Saint N., Guedin S., Willery E., Molle G., Locht C. 1999. Channel formation by FhaC, the outer membrane protein involved in the secretion of the Bordetella pertussis filamentous hemagglutinin. J. Biol. Chem. 274:37731–37735
Jacob-Dubuisson F., Locht C., Antoine R. 2001. Two-partner secretion in gram-negative bacteria: A thrifty, specific pathway for large virulence proteins. Mol. Microbiol. 40:306–313
Johnson T.L., Abendroth J., Hol W.G., Sandkvist M. 2006. Type II secretion: From structure to function. FEMS Microbiol. Lett. 255:175–186
Jongbloed J.D.H., Martin U., Antelmann H., Hecker M., Tjalsma H., Venema G., Bron S., van Dijl J.M., Müller J. 2000. TatC is a specificity determinant for protein secretion via the twin-arginine translocation pathway. J. Biol. Chem. 275:41350–41357
Karavolos M.H., Roe A.J., Wilson M., Henderson J., Lee J.J., Gally D.L., Khan C.M. 2005. Type III secretion of the Salmonella effector protein SopE is mediated via an N-terminal amino acid signal and not an mRNA sequence. J. Bacteriol. 187:1559–1567
Karnholz A., Hoefler C., Odenbreit S., Fischer W., Hofreuter D., Haas R. 2006. Functional and topological characterization of novel components of the comB DNA transformation competence system in Helicobacter pylori. J. Bacteriol. 188:882–893
Karpowich N.K., Huang H.H., Smith P.C., Hunt J.F. 2003. Crystal structures of the BtuF periplasmic-binding protein for vitamin B12 suggest a functionally important reduction in protein mobility upon ligand binding. J. Biol. Chem. 278:8429–8434
Kim, S.H., Chao, Y. Saier, M.H., Jr. 2006. Protein-translocating trimeric autotransporters of gram-negative bacteria. J. Bacteriol. 188:5655–5667
Kinch L.N., Saier M.H. Jr. Grishin N.V. 2002. Sec61β – a component of the archaeal protein secretory system. Trends Biochem. Sci. 27:170–171
Könninger U.W., Hobbie S., Benz R., Braun V. 1999. The haemolysin-secreting ShlB protein of the outer membrane of Serratia marcescens: Determination of surface-exposed residues and formation of ion-permeable pores by ShlB mutants in artificial lipid bilayer membranes. Mol. Microbiol. 32:1212–1225
Koronakis V. 2003. TolC – the bacterial exit duct for proteins and drugs. FEBS Lett. 555:66–71
Koronakis V., Eswaran J., Hughes C. 2004. Structure and function of TolC: The bacterial exit duct for proteins and drugs. Annu. Rev. Biochem. 73:467–489
Koronakis V., Sharff A., Koronakis E., Luisi B., Hughes C. 2000. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature 405:914–919
Kozjak V., Wiedemann N., Milenkovic D., Lohaus C., Meyer H.E., Guiard B., Meisinger C., Pfanner N. 2003. An essential role of Sam50 in the protein sorting and assembly machinery of the mitochondrial outer membrane. J. Biol. Chem. 278:48520–48523
Kuan G., Dassa E., Saurin W., Hofnung M., Saier M.H. Jr. 1995. Phylogenic analyses of the ATP-binding constituents of bacterial extracytoplasmic receptor-dependent ABC-type nutrient uptake permeases. Res. Microbiol. 146:271–278
Lee S.H., Galán J.E. 2004. Salmonella type III secretion-associated chaperones confer secretion-pathway specificity. Mol. Microbiol. 51:483–495
Li J., Wolf S.G., Elbaum M., Tzfira T. 2005. Exploring cargo transport mechanics in the type IV secretion systems. Trends Microbiol. 13:295–298
Lister R., Hulett J.M., Lithgow T., Whelan J. 2005. Protein import into mitochondria: Origins and functions today. Mol. Membr. Biol. 22:87–100
Locher K.P., Lee A.T., Rees D.C. 2002. The E. coli BtuCD structure: A framework for ABC transporter architecture and mechanism. Science 296:1091–1098
Loveless B.J., Saier M.H. Jr. 1997. A novel family of channel-forming, autotransporting, bacterial virulence factors. Mol. Membr. Biol. 14:113–123
Luirink J., Samuelsson T., de Gier J.-W. 2001. YidC/Oxa1p/Alb3: Evolutionarily conserved mediators of membrane protein assembly. FEBS Lett. 501:1–5
Luirink J., von Heijne G., Houben E., de Gier J.W. 2005. Biogenesis of inner membrane proteins in Escherichia coli. Annu. Rev. Microbiol. 59:329–355
Lybarger S.R., Sandkvist M. 2004. A hitchhiker’s guide to type IV secretion. Science 304:1122–1123
Ma Q., Zhai Y., Schneider C.J., Ramseier T.M., Saier M.H. Jr. 2003. Protein secretion systems of Pseudomonas aeruginosa and P. fluorescens. Biochim. Biophys. Acta 1611:223–233
Mangels D., Mathers J., Bolhuis A., Robinson C. 2005. The core TatABC complex of the twin-arginine translocase in Escherichia coli: TatC drives assembly where TatA is essential for stability. J. Mol. Biol. 345:415–423
Milenkovic D., Kozjak V., Wiedemann N., Lohaus C., Meyer H.E., Guiard B., Pfanner N., Meisinger C. 2004. Sam35 of the mitochondrial protein sorting and assembly machinery is a peripheral outer membrane protein essential for cell viability. J. Biol. Chem. 279:22781–22785
Missiakas D., Betton J.M., Raina S. 1996. New components of protein folding in extracytoplasmic compartments of Escherichia coli, SurA, FkpA and Skp/OmpH. Mol. Microbiol. 21:871–886
Mol O., Oudega B. 1996. Molecular and structural aspects of fimbriae biosynthesis and assembly in Escherichia coli. FEMS Microbiol. Rev. 19:25–52
Mota L.J., Cornelis G.R. 2005. The bacterial injection kit: Type III secretion systems. Ann. Med. 37:234–249
Müller M. 2005. Twin-arginine-specific protein export in Escherichia coli. Res. Microbiol. 156:131–136
Müller M., Klosgen R.B. 2005. The Tat pathway in bacteria and chloroplasts. Mol. Membr. Biol. 22:113–121
Müller M., Koch H.-G., Beck K., Schäfer U. 2001. Protein traffic in bacteria: Multiple routes from the ribosome to and across the membrane. Prog. Nucleic Acid Res. Mol. Biol. 66:107–157
Newman C.L., Stathopoulos C. 2004. Autotransporter and two-partner secretion: Delivery of large-size virulence factors by gram-negative bacterial pathogens. Crit. Rev. Microbiol. 30:275–286
Nguyen L., Paulsen I.T., Tchieu J., Hueck C.J., Saier M.H. Jr. 2000. Phylogenetic analyses of the constituents of type III protein secretion systems. J. Mol. Microbiol. Biotechnol. 2:125–144
Nouwen N., Ranson N., Saibil H., Wolpensinger B., Engel A., Ghazi A., Pugsley A.P. 1999. Secretin PulD: Association with pilot PulS, structure, and ion-conducting channel formation. Proc. Natl. Acad. Sci. USA 96:8173–8177
Nouwen N., van der Laan M., Driessen A.J. 2001. SecDFyajC is not required for the maintenance of the proton motive force. FEBS Lett. 508:103–106
Oloo E.O., Tieleman D.P. 2004. Conformational transitions induced by the binding of MgATP to the vitamin B12 ATP-binding cassette (ABC) transporter BtuCD. J. Biol. Chem. 279:45013–45019
Olsson J., Edqvist P.J., Bröms J.E., Forsberg A., Wolf-Watz H., Francis M.S. 2004. The YopD translocator of Yersinia pseudotuberculosis is a multifunctional protein comprised of discrete domains. J. Bacteriol. 186:4110–4123
Palmer T., Sargent F., Berks B.C. 2005. Export of complex cofactor-containing proteins by the bacterial Tat pathway. Trends Microbiol. 13:175–180
Pantoja M., Chen L., Chen Y., Nester E.W. 2002. Agrobacterium type IV secretion is a two-step process in which export substrates associate with the virulence protein VirJ in the periplasm. Mol. Microbiol. 45:1325–1335
Paulsen I.T., Beness A.M., Saier M.H. Jr. 1997a. Computer-based analyses of the protein constituents of transport systems catalysing export of complex carbohydrates in bacteria. Microbiology 143:2685–2699
Paulsen I.T., Park J.H., Choi P.S., Saier M.H. Jr. 1997b. A family of gram-negative bacterial outer membrane factors that function in the export of proteins, carbohydrates, drugs and heavy metals from gram-negative bacteria. FEMS Microbiol. Lett. 156:1–8
Peabody C.R., Chung Y.-J., Yen M.-R., Vidal-Ingigliardi D., Pugsley A.P., Saier M.H. Jr. 2003. Type II protein secretion and its relationship to bacterial type IV pili and archaeal flagella. Microbiology 149:3051–3072
Pivetti C.D., Yen M.-R., Miller S., Busch W., Tseng Y.-H., Booth I.R., Saier M.H. Jr. 2003. Two families of mechanosensitive channel proteins. Microbiol. Mol. Biol. Rev. 67:66–85
Plano G.V., Day J.B., Ferracci F. 2001. Type III export: New uses for old pathway. Mol. Microbiol. 40:284–293
Pohlschroder M., Hartmann E., Hand N.J., Dilks K., Haddad A. 2005. Diversity and evolution of protein translocation. Annu. Rev. Microbiol. 59:91–111
Quadri L.E., Kleerebezem M., Kuipers O.P., de Vos W.M., Roy K.L., Vederas J.C., Stiles M.E. 1997. Characterization of a locus from Carnobacterium piscicola LV17B involved in bacteriocin production and immunity: Evidence for global inducer-mediated transcriptional regulation. J. Bacteriol. 179:6163–6171
Ramamurthi K.S., Schneewind O. 2003a. Substrate recognition by the Yersinia type III protein secretion machinery. Mol. Microbiol. 50:1095–1102
Ramamurthi K.S., Schneewind O. 2003b. Yersinia yopQ mRNA encodes a bipartite type III secretion signal in the first 15 codons. Mol. Microbiol. 50:1189–1198
Ramanculov E., Young R. 2001. Genetic analysis of the T4 holin: Timing and topology. Gene 265:25–36
Rapoport T.A., Goder V., Heinrich S.U., Matlack K.E. 2004. Membrane-protein integration and the role of the translocation channel. Trends Cell. Biol. 14:568–575
Rapoport T.A., Jungnickel B., Kutay U. 1996. Protein transport across the eukaryotic endoplasmic reticulum and bacterial inner membranes. Annu. Rev. Biochem. 65:271–303
Reyes C.L., Chang G. 2005. Lipopolysaccharide stabilizes the crystal packing of the ABC transporter MsbA. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 61:655–658
Robinson C., Woolhead C., Edwards W. 2000. Transport of proteins into and across the thylakoid membrane. J. Exp. Bot. 51(Special issue):369–374
Roggenkamp, A., Ackermann, N., Jacobi, C.A., Truelzsch, K., Hoffmann, H., Heesemann, J. 2003. Molecular analysis of transport and oligomerization of the Yersinia enterocolitica adhesin YasA. J. Bactorial. 185:3735–3744
Ryndak M.B., Chung H., London E., Bliska J.B. 2005. Role of predicted transmembrane domains for type III translocation, pore formation, and signaling by the Yersinia pseudotuberculosis YopB protein. Infect. Immun. 73:2433–2443
Saier M.H. Jr. 1998. Molecular phylogeny as a basis for the classification of transport proteins from bacteria, archaea and eukarya. In: Poole R.K., editor. Advances in Microbial Physiology, Academic Press, San Diego pp 81–136
Saier M.H. Jr. 1999. A functional-phylogenetic system for the classification of transport proteins. J. Cell. Biochem. Suppl. 32/33:84–94
Saier M.H. Jr. 2000a. A functional/phylogenetic classification system for transmembrane solute transporters. Microbiol. Mol. Biol. Rev. 64:354–411
Saier M.H. Jr. 2000b. Families of proteins forming transmembrane channels. J. Membr. Biol. 175:165–180
Saier M.H. Jr. 2004. Evolution of bacterial type III protein secretion systems. Trends Microbiol. 12:113–115
Saier M.H. Jr., Tran C.V., Barabote R.D. 2006. TCDB: The transporter classification database for membrane transport protein analyses and information. Nucleic Acids Res. 34:D181–D186
Saier M.H., Jr., Tseng T.-T. 1999. Evolutionary origins of transmembrane transport systems. In: Broome-Smith J.K., Baumberg S., Stirling C.J., Ward F.B. editors. Transport of Molecules Across Microbial Membranes. Symposium 58, Society for General Microbiology, Cambridge University Press, Cambridge pp 252–274
Sambasivarao D., Dawson H.A., Zhang G., Shaw G., Hu J., Weiner J.H. 2001. Investigation of Escherichia coli dimethyl sulfoxide reductase assembly and processing in strains defective for the sec-independent protein translocation system membrane targeting and translocation. J. Biol. Chem. 276:20167–20174
Sambasivarao D., Turner R.J., Bilous P.T., Rothery R.A., Shaw G., Weiner J.H. 2002. Differential effects of a molybdopterin synthase sulfurylase (moeB) mutation on Escherichia coli molybdoenzyme maturation. Biochem. Cell. Biol. 80:435–443
Sambasivarao D., Turner R.J., Simala-Grant J.L., Shaw G., Hu J., Weiner J.H. 2000. Multiple roles for the twin arginine leader sequence of dimethyl sulfoxide reductase of Escherichia coli. J. Biol. Chem. 275:22526–22531
Sandkvist M, 2001. Biology of type II secretion. Mol. Microbiol. 40:271–283
Sargent F., Berks B.C., Palmer T. 2006. Pathfinders and trailblazers: a prokaryotic targeting system for transport of folded proteins. FEMS Microbiol. Lett. 254:198–207
Sargent F., Gohlke U., De Leeuw E., Stanley N.R., Palmer T., Saibil H.R., Berks B.C. 2001. Purified components of the Escherichia coli Tat protein transport system form a double-layered ring structure. Eur. J. Biochem. 268:3361–3667
Sargent F., Stanley N.R., Berks B.C., Palmer T. 1999. Sec-independent protein translocation in Escherichia coli. A distinct and pivotal role for the TatB protein. J. Biol. Chem. 274:36073–36082
Sauer F.G., Knight S.D., Waksman G.J., Hultgren S.J. 2000. PapD-like chaperones and pilus biogenesis. Semin. Cell Dev. Biol. 11:27–34
Scheuring J., Braun N., Nothdurft L., Stumpf M., Veenendaal A.K., Kol S., van der Does C., Driessen A.J., Weinkauf S. 2005. The oligomeric distribution of SecYEG is altered by SecA and translocation ligands. J. Mol. Biol. 354:258–271
Schleiff E., Soll J., Küchler M., Kühlbrandt W., Harrer R. 2003. Characterization of the translocon of the outer envelope of chloroplasts. J. Cell Biol. 160:541–551
Schmidt S.A., Bieber D., Ramer S.W., Hwang J., Wu C.Y., Schoolnik G. 2001. Structure-function analysis of BfpB, a secretin-like protein encoded by the bundle-forming-pilus operon of enteropathogenic Escherichia coli. J. Bacteriol. 183:4848–4859
Schmitt L., Benabdelhak H., Blight M.A., Holland I.B., Stubbs M.T. 2003. Crystal structure of the nucleotide-binding domain of the ABC-transporter haemolysin B: Identification of a variable region within ABC helical domains. J. Mol. Biol. 330:333–342
Scotti P.A., Valent Q.A., Manting E.H., Urbanus M.L., Driessen A.J., Oudega B., Luirink J. 1999. SecA is not required for signal recognition particle-mediated targeting and initial membrane insertion of a nascent inner membrane protein. J. Biol. Chem. 274:29883–29888
Sijbrandi R., Urbanus M.L., ten Hagen-Jongman C.M., Bernstein H.D., Oudega B., Otto B.R., Luirink J. 2003. Signal recognition particle (SRP)-mediated targeting and Sec-dependent translocation of an extracellular Escherichia coli protein. J. Biol. Chem. 278:4654–4659
Stanley N.R., Sargent F., Buchanan G., Shi J., Stewart V., Palmer T., Berks B.C. 2002. Behaviour of topological marker proteins targeted to the Tat protein transport pathway. Mol. Microbiol. 43:1005–1021
Steiner J.M., Yusa F., Pompe J.A., Loffelhardt W. 2005. Homologous protein import machineries in chloroplasts and cyanelles. Plant J. 44:646–652
Takamatsu H., Bunai K., Horinaka T., Oguro A., Nakamura K., Watabe K., Yamane K. 1997. Identification of a region required for binding to presecretory protein in Bacillus subtilis Ffh, a homologue of the 54-kDa subunit of mammalian signal recognition particle. Eur. J. Biochem. 248:575–582
Tam R., Saier M.H. Jr. 1993. Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol. Rev. 57:320–346
Thanassi D.G. 2002. Ushers and secretins: Channels for the secretion of folded proteins across the bacterial outer membrane. J. Mol. Microbiol. Biotechnol. 4:11–20
Thanassi D.G., Hultgren S.J. 2000. Assembly of complex organelles: Pilus biogenesis in gram-negative bacteria as a model system. Methods 20:111–126
Thanassi D.G., Stathopoulos C., Karkal A., Li H. 2005. Protein secretion in the absence of ATP: The autotransporter, two-partner secretion and chaperone/usher pathways of gram-negative bacteria. Mol. Membr. Biol. 22:63–72
Theg, S.M., Cline, K., Finazzi, G., Wollman, F.A. 2005. The energetics of the chloroplast Tat protein transport pathway revisited. Trends Plant Sci. 10:153–154
Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F., Higgins D.G. 1997. The CLUSTAL X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876–4882
Touze T., Eswaran J., Bokma E., Koronakis E., Hughes C., Koronakis V. 2004. Interactions underlying assembly of the Escherichia coli AcrAB-TolC multidrug efflux system. Mol. Microbiol. 53:697–706
Tziatzios C., Schubert D., Lotz M., Gundogan D., Betz H., Schagger H., Haase W., Duong F., Collinson I. 2004. The bacterial protein-translocation complex: SecYEG dimers associate with one or two SecA molecules. J. Mol. Biol. 340:513–524
van den Berg B., Clemons W.M. Jr., Collinson I., Modis Y., Hartmann E., Harrison S.C., Rapoport T.A. 2004. X-ray structure of a protein-conducting channel. Nature 427:36–44
van der Laan M., Nouwen N.P., Driessen A.J. 2005. YidC – an evolutionary conserved device for the assembly of energy-transducing membrane protein complexes. Curr. Opin. Microbiol. 8:182–187
van Dijl J.M., Braun P.G., Robinson C., Quax W.J., Antelmann H., Hecker M., Muller J., Tjalsma H., Bron S., Jongbloed J.D. 2002. Functional genomic analysis of the Bacillus subtilis Tat pathway for protein secretion. J. Biotechnol. 98:243–254
Van Rosmalen M., Saier M.H. Jr. 1993. Structural and evolutionary relationships between two families of bacterial extracytoplasmic chaperone proteins which function cooperatively in fimbrial assembly. Res. Microbiol. 144:507–527
Veiga E., Sugawara E., Nikaido H., de Lorenzo V., Fernández L.A. 2002. Export of autotransported proteins proceeds through an oligomeric ring shape by C-terminal domains. EMBO J. 21:2122–2131
Venema K., Dost M.H., Beun P.A., Haandrikman A.J., Venema G., Kok J. 1996. The genes for secretion and maturation of lactococcins are located on the chromosome of Lactococcus lactis IL1403. Appl. Environ. Microbiol. 62:1689–1692
Vignon G., Kohler R., Larquet E., Giroux S., Prevost M.C., Roux P., Pugsley A.P. 2003. Type IV-like pili formed by the type II secreton: Specificity, composition, bundling, polar localization, and surface presentation of peptides. J. Bacteriol. 185:3416–3428
Voulhoux R., Bos M.P., Geurtsen J., Mols M., Tommassen J. 2003. Role of a highly conserved bacterial protein in outer membrane protein assembly. Science 299:262–265
Wang I.-N., Smith D.L., Young R. 2000. Holins: The protein clocks of bacteriophage infections. Annu. Rev. Microbiol. 54:799–825
Wickner W., Schekman R. 2005. Protein translocation across biological membranes. Science 310:1452–1456
Wiedemann N., Kozjak V., Chacinska A., Schönfisch B., Rospert S., Ryan M.T., Pfanner N., Meisinger C. 2003. Machinery for protein sorting and assembly in the mitochondrial outer membrane. Nature 424:565–571
Winans S.C., Burns D.L., Christie P.J. 1996. Adaptation of a conjugal transfer system for the export of pathogenic macromolecules. Trends Microbiol. 4:64–68
Wu T., Malinverni J., Ruiz N., Kim S., Silhavy T.J., Kahne D. 2005. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121:235–245
Yamane K., Bunai K., Kakeshita H. 2004. Protein traffic for secretion and related machinery of Bacillus subtilis. Biosci. Biotechnol. Biochem. 68:2007–2023
Yen M.-R., Harley K.T., Tseng Y.-H., Saier M.H. Jr. 2001. Phylogenetic and structural analyses of the Oxa1 family of protein translocases. FEMS Microbiol Lett. 204:223–231
Yen M.-R., Peabody C.R., Partovi S.M., Zhai Y., Tseng Y.-H., Saier M.H. Jr. 2002a. Protein-translocating outer membrane porins of gram-negative bacteria. Biochim. Biophys. Acta 1562:6–31
Yen M.-R., Tseng Y.-H., Nguyen E.H., Wu L.F., Saier M.H. Jr. 2002b. Sequence and phylogenetic analyses of the twin-arginine targeting (Tat) protein export system. Arch. Microbiol. 177:441–450
Yi L., Dalbey R.E. 2005. Oxa1/Alb3/YidC system for insertion of membrane proteins in mitochondria, chloroplasts and bacteria. Mol. Membr. Biol. 22:101–111
Yip C.K., Kimbrough T.G., Felise H.B., Vuckovic M., Thomas N.A., Pfuetzner R.A., Frey E.A., Finlay B.B., Miller S.I., Strynadka N.C. 2005. Structural characterization of the molecular platform for type III secretion system assembly. Nature 435:702–707
Young G.M., Schmiel D.H., Miller V.L. 1999. A new pathway for the secretion of virulence factors by bacteria, the flagellar export apparatus functions as a protein-secretion system. Proc. Natl. Acad. Sci. USA 96:6456–6461
Young R. 2002. Bacteriophage holins: Deadly diversity. J. Mol. Microbiol. Biotechnol. 4:21–36
Young R., Bläsi U. 1995. Holins: Form and function in bacteriophage lysis. FEMS Microbiol. Rev. 17:191–205
Zhai Y., Tchieu J., Saier M.H. Jr. 2002. A web-based Tree View (TV) program for the visualization of phylogenetic trees. J. Mol. Microbiol. Biotechnol. 4:69–70
Acknowledgement
I thank both Dr. Chin Hong Ma and Ms. Mary Beth Hiller for useful discussions and contributions to the preparation of this manuscript. This work was supported by National Institutes of Health grant GM077402.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Saier, M.H. Protein Secretion and Membrane Insertion Systems in Gram-Negative Bacteria. J Membrane Biol 214, 75–90 (2006). https://doi.org/10.1007/s00232-006-0049-7
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00232-006-0049-7