
Abstract
We previously showed that activation of the cystic fibrosis (CF) transmembrane conductance regulator (CFTR) Cl− conductance (gCFTR) supports parallel activation of amiloride-sensitive epithelial Na+ channel (ENaC) in the native human sweat duct. However, it is not clear whether phosphorylated CFTR, phosphorylated ENaC, or only Cl− -channel function is required for activation. We used basilaterally α-toxin-permeabilized human sweat ducts to test the hypothesis that ENaC activation depends only on Cl− -channel function and not on phosphorylation of either CFTR or ENaC. CFTR is classically activated by PKA plus millimolar ATP, but cytosolic glutamate activation of gCFTR is independent of ATP and phosphorylation. We show here that both phosphorylation-dependent (PKA) and phosphorylation-independent (glutamate) activation of CFTR Cl− channel function support gENaC activation. We tested whether cytosolic application of 5 mM ATP alone, phosphorylation by cAMP, cGMP, G-protein dependent kinases (all in the presence of 100 μM ATP), or glutamate could support ENaC activation in the absence of gCFTR. We found that none of these agonists activated gENaC by themselves when Cl− current ( \( I_{\rm{Cl}^{-}}\) ) through CFTR was blocked by: 1) Cl− removal, 2) DIDS inhibition, 3) lowering the ATP concentration to 100 μM (instead of 5 mM required to support CFTR channel function), or 4) mutant CFTR (homozygous ΔF508 CF ducts). However, Cl− gradients in the direction of absorption supported, while Cl− gradients in the direction of secretion prevented ENaC activation. We conclude that the interaction between CFTR and ENaC is dependent on activated \( I_{\rm{Cl}^{-}}\) through CFTR in the direction of absorption (Cl− gradient from lumen to cell). But such activation of ENaC is independent of phosphorylation and ATP. However, reversing \( I_{\rm{Cl}^{-}}\) through CFTR in the direction of secretion (Cl− gradient from cell to lumen) prevents ENaC activation even in the presence of \( I_{\rm{Cl}^{-}}\) through CFTR.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
CFTR and ENaC are the principal epithelial ion channels that allow transepithelial salt (NaCl) absorption in human sweat duct as well as in other salt-absorptive epithelia, including airways, salivary ducts and kidney. In spite of its immense significance in health and diseases, including pseudohypoaldosteronism, Liddle’s syndrome and hypo- and hypertension and even though we have significant understanding of the regulation of the CFTR Cl− channel [31], little is known about the regulation of ENaC channels in native cells. Knowledge of ENaC regulation mostly comes from studies on heterologous expression of ENaC channels in non-epithelial systems such as Xenopus oocytes, NIH-3T3 fibroblasts or planar lipid bilayers [5, 10, 29, 32]
Our previous studies indicated that activating endogenous kinases (with cAMP or cGMP or G-proteins) in the presence of ATP simultaneously activated CFTR and ENaC [22]. However, it was not clear from these early studies whether phosphorylation and ATP play a direct or indirect role in regulating ENaC. In fact, the inability to activate CFTR without phosphorylation and ATP made it impossible to distinguish between the roles of phosphorylation, ATP and CFTR in enhancing gENaC. However, we recently found that gCFTR can be activated by cytosolic glutamate, apparently completely independently of phosphorylation and ATP [25]. Therefore, we can now evaluate the role of phosphorylation and ATP in the activation of ENaC.
Earlier patch-clamp studies primarily on culture cells and heterologous expression systems suggested that phosphorylation acutely regulates ENaC channels [5, 29, 34]. Reports also suggested that phosphorylation activation of ENaC could be inhibited following phosphorylation activation of CFTR [33, 34]. It was also reported that the intracellular domains of CFTR (including NBD1 that bind and hydrolyze ATP) might interact with ENaC [16, 29]. These results, taken together with the observation that ATP alone may activate ENaC in the presence of actin [11, 29], suggest that in addition to phosphorylation, ATP also might play a direct role in regulating ENaC. Nonetheless, direct evidence indicating a role for acute in vivo phosphorylation and ATP in regulating ENaC activity is missing.
In addition, we and others have indicated that the CFTR Cl− channel function might play a crucial role in the interaction between CFTR and ENaC [19, 22, 29, 34]. Therefore it seems important to know whether Cl− channel function is critically required for ENaC activation regardless of the agonist used (including different kinase activators, ATP and glutamate) in order to determine the role of electrochemical coupling between these two channels. We have addressed this question by studying the effect of different agonists on ENaC in the presence and absence of CFTR Cl− channel function.
It is intriguing that the nature of interaction between CFTR and ENaC appears to be tissue-specific [22, 29]. That is, in the salt-absorbing sweat duct, activating CFTR causes simultaneous activation of ENaC. However, in some other tissues that are either purely secretory or both secretory as well as absorptive, depending on the physiological need, activating CFTR appears to reciprocally inhibit ENaC [29]. The physiological mechanisms underlying this phenomenon are unclear. Hence, in the event that the CFTR Cl− channel function does in fact play a physiological role in this channel interaction, it seems logical to ask whether the direction of this Cl− current could determine the nature of interaction between these two ion channels. We have addressed this question by studying the effect of reversing the direction of Cl− current from absorption to secretion and vice versa on the stimulation of ENaC following activation of CFTR Cl− conductance.
We have addressed whether 1) interaction between CFTR and ENaC is dependent on phosphorylation and ATP, 2) channel interaction is dependent on Cl− channel function, 3) the direction of Cl− current (absorption vs secretion) determines the nature of interaction between CFTR and ENaC. We now show that neither ATP nor phosphorylation conditions directly regulate ENaC in the absence of CFTR Cl− channel function, and that ENaC activation is dependent on the direction of Cl− current across the apical membrane.
Materials and Methods
Sweat ducts were obtained from skin biopsies of volunteer subjects giving informed consent. Sweat ducts were dissected and microperfused as described previously [22, 23, 25]. Segments of ducts usually longer than 1 mm were microperfused with a double-barreled luminal micropipette that served to perfuse and record transepithelial voltage through one barrel and to pass constant current pulses through the other. This arrangement allowed estimation of the specific membrane conductance from the cable equation [22, 23, 25]. After confirming the integrity of the perfused tubule, we applied 1,000–2,000 units/ml of α-toxin from Staphylococcus aureus to the bath solution in order to selectively permeabilize the basilateral membrane [22, 23, 25]. This procedure leaves the epithelium with an intact, functional apical membrane and a non-selective basal membrane, permeable to molecules of up to about 5,000 mwu including cAMP, ATP, other organic and inorganic ions. We then determined the magnitude of CFTR-gCl activation by measuring the trans-apical membrane Cl− diffusion potential and the simultaneous changes in membrane conductance following activation of CFTR with cAMP and ATP in the cytoplasm (see Fig. 1A).

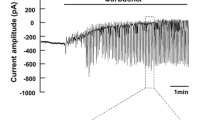
Effect of activation of gCFTR on gENaC. (A) Model of basolaterally α-toxin-permeabilized apical plasma membrane. The apical membrane conductance is predominantly comprised of CFTR and ENaC with no detectable contribution from other ion channels. In these experiments, the basilateral membrane was permeabilized with 1000 U/ml of α-toxin from Staphylococcus aureus. Ion gradients were established for both Na+ and Cl− from lumen to cytoplasmic bath by perfusing with 150 mM NaCl and 140 mM KGlu in lumen and cytoplasmic bath, respectively. Amiloride was always present in the luminal perfusate except when gENaC and Na+ diffusion potentials were measured. gCFTR and gENaC were measured both under unstimulated control conditions and after stimulating gCFTR. CFTR agonists (cAMP, cGMP, GTP-γ-S, glutamate and ATP) and the intracellular ion composition were readily manipulated through α-toxin pores in the basilateral membrane. The magnitudes of gCFTR and gENaC were monitored as their respective ion diffusion potentials and conductances. Transepithelial potential was measured using the perfusion side of the double-barrel microperfusion pipette in the lumen with reference to the cytoplasmic bath ground agar bridge. 50–100 nA/500 ms constant current pulses were injected through the second barrel of the perfusion pipette in order to calculate the transmembrane conductance using the cable equation. (B) A representative electrical trace showing activation of gCFTR and gENaC in the presence of cAMP+ATP in the cytoplasmic bath. Under control conditions, gCFTR was completely deactivated but a small gENaC was detectable after removing amiloride from the luminal perfusate. However, application of cAMP + ATP resulted in a significant coordinated activation of both gCFTR and gENaC. (C) Summary of data on gCFTR and gENaC acquired from experiments similar to that shown in panel (A). n = number of ducts = 12, P < 0.001.
The luminal perfusion Ringer’s solutions contained (in mM) NaCl 150, K 5, PO4 3.5, MgSO4 1.2, Ca2+ 1, and pH. 7.4. Cl−-free luminal Ringer’s solution was prepared by complete substitution of Cl− with the impermeant anion gluconate. The cytoplasmic/bath solution contained K+ 145, gluconate 140, PO4 3.5, MgSO4 1.2, and 260 μM Ca2+ buffered with 2.0 mM EGTA (Sigma) to 80 nM free Ca2+, pH 6.8. ATP 0.01–5 (K+ salt), ATP-γ-S 5, cAMP 0.1 and glutamate 10 or α-ketoglutarate, 1, were added to the cytoplasmic bath as needed.
gCFTR and gENaC were calculated using the following formulae: Gt = gENaC + gX and Gt* = gCFTR + gENaC* + gX, where Gt and Gt* are total transepithelial conductances before and after stimulating gCFTR (with either cAMP + 5 mM ATP or 10 mM glutamate or 1 mM α-ketoglutarate in the cytoplasmic bath) respectively; gX is nonspecific shunt conductance measured after blocking gENaC (with luminal amiloride) and gCFTR (by removing cAMP and ATP from cytoplasmic bath); gENaC and gENaC* represent ENaC conductance before and after activating CFTR, as mentioned above, with cyclic nucleotides or by G-protein agonists or glutamate. Transepithelial electrical conductance was measured by injecting 50–100 nA/500 ms transepithelial constant current pulse as described earlier [22]. Statistical significance was determined with Student’s t-test. A P value of 0.05 was taken to be significantly different.
Results
PHOSPHORYLATION ACTIVATION OF gCFTR ALSO ACTIVATES gENaC
Before application of cAMP and ATP to the cytoplasmic bath, gCFTR was completely deactivated, but a small gENaC persisted. Phosphorylation activation of gCFTR in the presence of 5 mM ATP significantly activated gENaC (Fig.1, n = number of ducts = 12). Even though the effect of activating gCFTR always increased absolute gENaC under these experimental conditions, the absolute magnitude of responses of Na+ diffusion potentials and gENaC (after washing out amiloride) significantly varied between different human subjects, which might reflect individual differences including but not limited to dietary habits and physical activity.
CYTOSOLIC GLUTAMATE METABOLITES ACTIVATE CFTR AND ENaC
To test whether simultaneous activation of CFTR and ENaC involves a phosphorylation process, we have used glutamate or its metabolite α-ketoglutarate to stimulate gCFTR. We have used these agonists because activation of CFTR by these agonists does not involve either ATP or phosphorylation, as previously shown [25]. We showed that application of 10 mM glutamate or 1 mM α-ketoglutarate to the cytoplasmic bath in the complete absence of cAMP and ATP resulted in simultaneous activation of both gCFTR and gENaC. Activation of gCFTR by cytosolic glutamate resulted in a concomitant activation of gENaC (gENaC = 4.5 ± 1.8 mS/cm2 and 28 ± 5 mS/cm2 before and after glutamate activation of gCFTR, respectively) (Fig. 2).

Effect of cytosolic α-ketoglutarate on CFTR and ENaC. (A) Representative electrical trace showing the effect of 1 mM α-ketoglutarate in the cytoplasm on Cl− and Na+ conductance mediated by CFTR and ENaC, respectively. Notice that no Cl− diffusion potential (only anion junction potential between Cl− and gluconate) and a small Na+ diffusion potential were detectable (in the absence of amiloride) before application of α-ketoglutarate. However, stimulating CFTR with α-ketoglutarate almost doubled the Na+ diffusion potential in face of a significantly larger Cl− shunt due to CFTR activation. (B) Summary of the data acquired from the experiments similar to the one shown in Fig. 2A showing the effect of glutamate on the ΔCl− (VCl) and ΔNa+ (ENa) diffusion potentials; n = 8, P < 0.001. (C) Summary of the data acquired from the experiments similar to the one shown in Fig. 2A showing the effect of glutamate on gCFTR and gENaC. n = 8, P < 0.001.
NO EFFECT OF cAMP/ATP ON gENaC AFTER REMOVAL OF Cl−
We have further investigated whether \( I_{\rm{Cl}^{-}}\) through CFTR is required for activating gENaC. We have substituted Cl− by the impermeant anion gluconate in the lumen as well as in the cytosolic bath. Substituting Cl− with gluconate did not significantly affect the magnitude of baseline unstimulated gENaC. However, application of cAMP + ATP under these conditions did not activate gENaC in the absence of \( I_{\rm{Cl}^{-}}\) through CFTR (Fig. 3).

The effect of Cl− removal on cAMP / ATP activation of gENaC. Notice that cAMP and 5 mM ATP had little effect on gENaC when CFTR was not conducting Cl−, indicating that gENaC activation by cAMP / ATP is critically dependent on CFTR conducting Cl−.
DIDS BLOCK OF gCFTR ALSO BLOCKS gENaC
We have further explored the role of \( I_{\rm{Cl}^{-}}\) through CFTR by blocking gENaC while maintaining the lumen-to-cell Cl− gradient intact. We adopted this strategy in order to avoid a potential effect of luminal Cl− removal on gENaC [26]. We used DIDS to block gCFTR because we have previously shown that cytosolic DIDS most effectively blocked gCFTR in this tissue as compared to other known CFTR blockers [26]. We found that blocking cAMP/ATP-activated gCFTR with DIDS also inhibited gENaC, even though cAMP and ATP were present in the cytoplasmic bath. DIDS by itself does not seem to have an effect on the residual ENaC either in normal ducts or in ΔF508 CF ducts (Figs. 3 and 4).

Blocking gCFTR with DIDS inhibits gENaC. In this experiment, we measured gENaC before and after blocking gCFTR with DIDS. Notice that gCFTR was activated in the presence of 0.1 mM cAMP + 5 mM ATP, as indicated by the amiloride-sensitive Na+ diffusion potentials and conductance (increase in conductance is indicated by a decrease in the amplitude of transepithelial current pulse-induced voltage deflections). However, gENaC was completely abolished after blocking gCFTR with 10−3 M DIDS in the cytoplasmic bath. DIDS by itself had no effect on spontaneous gENaC before CFTR activation (Fig. 3). These results indicate that Cl− conductance through CFTR is essential for activation of gENaC.
LACK OF EFFECT OF PKA PHOSPHORYLATION CONDITIONS ON gENaC
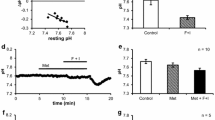
We have examined whether activation of gENaC following phosphorylation-activation of CFTR also requires physiological concentration (5 mM) of ATP. We activated PKA by application of 0.1 mM cAMP in the presence of 100 μM (instead of 5 mM) ATP. This low concentration of ATP was sufficient to support phosphorylation, but not enough to stimulate gCFTR [21, 22]. Under these conditions, neither gCFTR nor gENaC were stimulated in this suboptimal concentration of ATP to support gCFTR (Fig. 5).

The effect of PKA phosphorylation conditions on ENaC. To determine whether PKA phosphorylation conditions alone can activate ENaC in the absence of gCFTR activation, we applied 0.1 mM cAMP + 100 μM ATP to the cytoplasmic bath after washing out amiloride from the lumen. Under these conditions, the ATP concentration is sufficient to support PKA phosphorylation but not gCFTR activation (because stimulating CFTR requires physiological concentration of ATP, ca. 5 mM). (A) Representative electrical trace showing lack of effect of phosphorylation conditions on ENaC. Notice that cAMP and ATP had no effect either on the lumen-to-cell Na+ diffusion potential or apical Na+ conductance, even though the perfusion conditions remained the same as in Fig. 1, except for the low ATP concentration and lack of CFTR-gCl. In contrast, washing out amiloride resulted in a small but significant increase in lumen-negative Na+ diffusion potential as well as apical membrane amiloride-sensitive conductance, as indicated by a decrease in the amplitude of transepithelial constant current pulse-induced voltage deflections. (B) Summary of the data acquired from experiments similar as shown in (A).
LACK OF EFFECT OF ALTERNATIVE PHOSPHORYLATION CONDITIONS ON gENaC
We previously showed that both cGMP-dependent kinase (PKG, with cGMP) and apical G-proteins (with GTP-γ-S) independently stimulate gCFTR, causing simultaneous activation of gENaC [22, 24]. We activated PKG and G-protein-coupled kinase [22, 24] by application of 0.1 mM cGMP and GTP-γ-S, respectively, in the presence of 100 μM ATP. This low concentration of ATP was sufficient to support phosphorylation but not enough to stimulate gCFTR [21]. Under these conditions, neither gCFTR nor gENaC were stimulated by either of these agonists (Figs. 6 and 7).

Lack of effect of alternative phosphorylation conditions on ENaC. These experiments were designed to test the effect of activating endogenous cGMP-dependent kinase (PKG) (A) and apical G-protein kinase activated by GTP-γ-S (B) in the presence of 100 μM ATP on ENaC. Notice that these two kinases (other than PKA) also failed to stimulate ENaC, as indicated by the lack of effect of cGMP and GTP-γ-S on Na+ diffusion potentials and conductance after washing out luminal amiloride. Also notice that only luminal but not cytoplasmic amiloride blocked ENaC.

Lack of effect of CFTR agonists on ENaC of homozygous ΔF508 CF duct. This experiment was designed to investigate the effect of CFTR agonists cAMP, cGMP and glutamate in the presence of 5 mM ATP on ENaC of CF ducts that lack CFTR in the apical plasma membrane. Notice that none of these agonists could stimulate ENaC, as indicated by lack of effect on Na+ diffusion potentials (generated by 150 mM Na+ in the lumen and 150 mM K+ in the cytoplasmic bath) and conductance. Also notice that the ENaC in the sweat duct is sensitive to changes in cytosolic pH, as indicated by complete inhibition of Na+ diffusion potentials and conductance by lowering the pH.
LACK OF ACUTE EFFECT OF 5 mM ATP ALONE ON gENaC
To test whether physiological concentration of ATP alone can stimulate gENaC, we applied 5 mM ATP to the cytoplasmic bath without cAMP. We found that gENaC could not be activated by ATP alone in the presence of 140 mM KGlu in the cytoplasmic bath and 150 mM NaCl in the lumen (Fig. 8).

Lack of effect of ATP alone on gENaC. 5 mM ATP has no effect on gENaC in the absence of cAMP. Washing out amiloride indicated the presence of residual amiloride-sensitive Na+ conductance in ducts after complete inhibition of CFTR Cl− conductance in the absence of CFTR phosphorylation.
NO EFFECT OF CYTOSOLIC GLUTAMATE METABOLITES ON ENaC IN THE ABSENCE OF gCFTR
Cytoplasmic application of glutamate metabolites did not have any effect on gENaC in the absence of Cl− in the lumen and cytoplasmic bath (Fig. 9). Glutamate metabolites also did not have any effect on gENaC in the absence of CFTR in the apical plasma membrane of homozygous ΔF508 CF ducts (See Fig.7).

Lack of effect of glutamate on gENaC in the absence Cl−. Neither glutamate nor α-ketoglutarate stimulated Na+ diffusion potentials and conductance when the Cl− is completely removed from lumen and cell by substituting with the impermeant anion gluconate. The effect of amiloride indicates the presence of a large gENaC in this duct.
ENaC ACTIVATION IS SENSITIVE TO THE DIRECTION OF Cl− CURRENT
We investigated whether the direction of Cl− current affects ENaC function. The data indicate that while CFTR Cl− conductance was mostly unaffected by reversing the Cl− gradient from either lumen to cell or vice versa, the gENaC was virtually abolished when the Cl− gradient was either reversed in the secretory direction (cell to lumen) (Fig. 10A, n = 11, p < 0.001) or abolished by perfusing the lumen and bath with Ringer’s solution containing equimolar [Cl−] (Fig. 10B, n = 11, p < 0.001).

Effect of reversed Cl− gradient on gENaC. (A) This experiment was performed to test the effect of reversing the Cl− gradient from lumen to cell (150 mM NaCl in the lumen vs 140 mM KGlu in the cell, Cl− gradient in the direction of absorption) to cell to lumen (150 mM NaGlu in the lumen vs 140 mM KCl in the cell, Cl− gradient in the direction of secretion) in the presence of cAMP and ATP. Notice that following the reversal of Cl− gradient the Cl− diffusion potential changed from ∼+50 mV (lumen positive) to ∼−50 mV (lumen negative). Also notice that a significant amiloride-sensitive gENaC was present when the Cl− gradient was in the absorptive direction that was completely absent following the reversal of Cl− gradient towards secretion. (B) In this experiment, gENaC was compared in the same ducts under two different conditions, that is, in the presence of lumen-to-cell Cl− gradient (150 mM NaCl and 140 mM KGlu in the lumen and cytosol, respectively) and in the absence of Cl− gradient (150 mM NaCl and 140 mM KCl in the lumen and cell, respectively). Notice that the lack of electrical driving force prevents gENaC activation even though gCFTR was activated by cAMP + ATP and Cl− was present.
Discussion
The main function of human reabsorptive sweat duct is to absorb salt (NaCl) from the primary sweat secreted by the sweat gland secretory coil. CFTR and ENaC are the only ion channels detected in the apical membrane of duct. Salt absorption requires simultaneous activation of both these ion channels because genetic abnormality in either one of these channels in patients with CF or pseudohypoaldosteronism prevents absorption of Na+ and Cl− [20, 22, 25]. Therefore, we might expect that the activities of these ion channels are well coordinated during transepithelial salt transport. However, the physiological mechanisms involved in coordinating this process are unclear.
SIMULTANEOUS ACTIVATION OF CFTR AND ENaC REGARDLESS OF PHOSPHORYLATION AND ATP
We have recently shown [22] and illustrate here that phosphorylation and ATP simultaneously activate CFTR and gENaC in this purely absorptive epithelium (Fig. 1). We interpreted these data to mean that Cl− channel function of phosphorylation-activated CFTR is required for activation of ENaC [22]. Since cAMP and ATP phosphorylate and activate CFTR Cl− channel function, we could not distinguish between direct (phosphorylation-dependent) and indirect (CFTR Cl−-function dependent) effects on gENaC. Previous reports that phosphorylation and ATP not only stimulate CFTR [31], but also directly activate ENaC in a heterologous expression system [5, 11, 33, 34], raises a question of whether simultaneous activation of ENaC simply depends on CFTR function or whether ENaC is phosphorylated along with CFTR. We recently found that CFTR can be activated by glutamate in a process that is independent of ATP and phosphorylation [25]. We exploited this new finding to investigate whether phosphorylation and ATP are required for CFTR-dependent ENaC activity. Figure 2 shows that both gCFTR and gENaC were simultaneously activated following application of either glutamate or α-ketoglutarate in the complete absence of cAMP and ATP. These results suggest that neither phosphorylation nor ATP is required for ENaC activation. To show further that it is CFTR function, not phosphorylation that determines ENaC activity, we investigated the effects of Cl− channel function activated by different mechanisms on ENaC activity.
ENaC ACTIVATION REQUIRES CFTR Cl− CHANNEL FUNCTION
Early reports suggested that PKA phosphorylation might activate ENaC [5, 11, 33, 34]. PKA phosphorylation of purified ENaC channels in lipid bilayers apparently altered the biophysical properties of the channel, including cation selectivity [12, 13, 30]. The carboxy termini of β and γ ENaC were shown to be phosphorylated by PKA in epithelial cell lines stably cotransfected with ENaC subunits [32]. Similarly, ENaC expressed alone in fibroblasts was activated by PKA by increasing the open probability (Po) and mean open time of ENaC channel. ENaC co-expressed with CFTR exhibited reduced Po and mean open time [34]. ENaC expressed in MDCK epithelial cells was stimulated by cAMP, whereas coexpression of human CFTR with rENaC (rat ENaC) generated smaller sodium currents that were inhibited by cAMP [33]. However, the physiological relevance of these observations in heterologous model systems remains unclear.
No Direct Effect of Phosphorylation on gENaC
In view of the aforementioned observations, we sought to determine the role of phosphorylation in regulating ENaC in native epithelial cells in the complete absence of CFTR Cl− channel function, achieved either by: 1) removing Cl− from the medium (Fig. 3); 2) Pharmacologically blocking gCFTR with inhibitor DIDS (Fig. 4) while stimulating with cAMP + 5 mM ATP in the cytoplasmic bath; 3) activating endogenous kinases in the presence of concentrations of ATP (100 μM vs. 5 mM ATP), too low to activate CFTR, but sufficient to support phosphorylation [21] (Figs. 5 and 6); 4) examining homozygous ΔF508 CF ducts that lack CFTR in the plasma membrane (Fig. 7). No ENaC activity was observed with any of these experimental maneuvers to remove CFTR function. These results indicate that the ENaC activation by cAMP, cGMP or G-proteins as previously reported [22] is not due to direct effects on ENaC, but rather due to activation of CFTR Cl− channel function in this tissue.
No Direct Effect of ATP on gENaC
Early reports suggested that ATP alone activated ENaC in the presence of actin in planar lipid bilayers [11, 29]. Studies on ex vivo systems also suggested that the NBD1 (nucleotide binding domain-1) of CFTR might interact with the C-terminus of α-rENaC [29] and that PKA phosphorylation activation of CFTR actually inhibited ENaC [33, 34]. In light of these early observations and taken together with the fact that NBD is well known to interact with ATP, we investigated the role of ATP alone (in the absence of cAMP) in regulating gENaC [33, 34]. We found that 5 mM ATP in the cytoplasmic bath by itself had no effect on gENaC (Fig. 8) in the absence of cAMP. Since 5 mM ATP + cAMP also did not stimulate ENaC in the absence of CFTR Cl− current (150 mM NaGlu in the lumen and 140 mM KGlu in the cytoplasm) in normal ducts or in the absence of CFTR in the apical plasma membrane of homozygous ΔF508 CF ducts as previously mentioned, we can rule out direct regulation of ENaC either by ATP or by phosphorylation in this tissue. However, since phosphorylation and ATP were indispensable components for stimulating CFTR Cl− conductance in these studies, it is not clear whether in addition to Cl− channel function, phosphorylation of either CFTR and/or ENaC is also required for this interaction between these two ion channels.
No Direct Activation of ENaC by Glutamate
We further investigated whether the activation of gENaC in the presence of glutamate is also dependent on CFTR Cl− channel function or whether the activation is independent of CFTR. Application of either glutamate or α-ketoglutarate had no effect on ENaC in the absence of Cl− in the lumen and cytoplasmic bath, as shown in Fig. 7. Further, we found that there was no effect of glutamate on gENaC in the apical membrane of a homozygous ΔF508CF duct that lacks CFTR in the plasma membrane, indicating that CFTR Cl− channel function itself is the determinant of gENaC activity (Figs. 7 and 9).
HOW TO RECONCILE THE RESULTS WITH APPARENT CONTRADICTIONS IN REGARD TO ENaC REGULATION?
Simultaneous activation of CFTR and ENaC in sweat duct is also surprising in light of the observations that activation of CFTR seems to cause reciprocal inhibition of ENaC in both heterologous expression systems [11, 15, 16, 33, 34] as well as in epithelial cells endogenously expressing these channels, including airways and colonic epithelium [3, 4, 6, 14, 17]. This leads to the question of whether structural differences in the ENaC channel could explain some of these apparent discrepancies in the properties of regulation.
Sweat Duct ENaC Is Functionally Similar to Those in Other CF-affected Epithelia
The evidence suggests that the ENaC channels in the sweat duct are similar to those found in other epithelial cells, such as airways and kidney tubules, as indicated by the following shared properties: that this ENaC channel is also 1) comprised of the three sub-units (mRNA for α, β and γ sub-units in sweat duct [21]; 2) severely affected in type-1 pseudohypoaldosteronism (PHA-1), as indicated by complete loss of electrogenic Na+ absorption [27]; 3) sensitive to cytosolic pH (activated by basic pH) [27]; 4) subject to aldosterone regulation [28]; and 5) inhibited by luminal and not cytosolic amiloride because the inhibitor-binding site is located on the extracellular loop of α-ENaC [7] (Fig. 6). In light of these observations one wonders whether the differences in the direction of Cl− current, such as in the case of absorption vs secretion, could determine the nature of interaction between CFTR and ENaC.
Direction of Cl− Current Influences Interaction between CFTR and ENaC
While the CFTR Cl− channel function plays a critical role in activating ENaC in salt-absorbing sweat duct, in which the direction of Cl− current is from lumen to cell, it is unclear what effect CFTR channel function might have on ENaC in epithelial cells with secretory function (e.g., airway epithelial cells), in which the Cl− current is in the opposite direction, that is, from cell to lumen [22, 29]. Previous experiments were conducted under conditions of a strong NaCl gradient (150 mM NaCl in the lumen and 140 KGlu in the cell), causing a significant increase in the electrical gradient following CFTR activation either by phosphorylation or by glutamate, as shown in Figs. 1 and 2. In fact, early studies indicated that changes in the electrical gradient apparently activate Na+ conductance [18, 19]. We further investigated the effect of reversing electrical driving force for Na+ on gENaC. This was achieved by changing the direction of Cl− current from absorption lumen to cell) to secretion (cell to lumen) by perfusing with 140 mM KCl and 150 mM NaGlu in the cytoplasmic bath and lumen, respectively. Under these conditions, stimulation of CFTR with cAMP + ATP almost reversed the electrical gradient to about −50 mV, thereby reducing the driving force by ∼ 100 mV (from ∼ + 50 mV to ∼−50 mV). Figure 10 shows that gENaC is virtually abolished, as indicated by the fact that amiloride application had no detectable effect on either Na+ diffusion potentials or conductance.
It was previously reported that the changes in cytosolic Cl− might directly inhibit the ENaC channel activity mediated by Cl−-sensitive heterotrimeric G-proteins [8]. However, the fact that activation of G-proteins with GTP-γ-S had no effect on gENaC in the absence of CFTR Cl− channel function (because of low ATP, which can only support phosphorylation) (Fig. 6) seems to indicate that inhibition of ENaC is not mediated by the G-proteins. Furthermore, inhibition of gENaC by reversed Cl− gradient (140 mM KCl and 150 mM NaGlu in the cytosol and lumen, respectively) cannot be due to inhibition of PKA phosphorylation activity because PKA itself is unaffected by changes in cytosolic Cl− concentration, as indicated by the fact that irrespective of the direction of Cl− gradient cAMP can activate gCFTR as efficiently (Fig. 10). These results taken together with the observations that gENaC is virtually abolished in the absence of electrical driving force (i.e., in the absence of Cl− current, Figs. 3, 7, 9, and 10) is consistent with significant electrical coupling between the activities of ENaC and CFTR, as shown in heterologous systems [18, 19]. However, this data alone cannot exclude the potential role of changing Cl− concentration and protein-protein interactions in this coordination process between CFTR and ENaC, as previously suggested (2, 8, 9, 11, 15, 16).
Tissue-specific Phosphorylation Activation of ENaC
The lack of effect of phosphorylation conditions on endogenous ENaC in this native tissue also seems to conflict with the early reports of direct phosphorylation activation of ENaC in ex vivo systems [11–13, 30, 33, 34], respiratory epithelial cells [4, 5] and human lymphocytes [5]. These observations suggest that despite several functional similarities, as previously mentioned, subtle differences in the stoichiometry of the channel (with respect to α, β and γ sub-units) between tissues may influence the channel properties [1, 30] or variable expression of a key regulatory protein or proteins linking CFTR or ENaC to the cytoskeleton or to each other in different tissues could explain this discrepancy with respect to tissue-specific phosphorylation and CFTR-dependent regulation of ENaC [10, 29].
In conclusion, we have shown that phosphorylation and ATP has no direct role in the activation of ENaC in this native tissue. CFTR Cl− channel function is critically required for ENaC activation regardless of phosphorylation and ATP. The direction of Cl− current seems to influence the mechanism of interaction between CFTR and ENaC in that ENaC is activated when Cl− is being absorbed, whereas ENaC is almost completely inhibited when Cl− is being secreted. Such functionally dependent CFTR regulation of ENaC may, at least partially, explain tissue-specific differences in the mechanism of interaction between these two epithelial ion channels.
References
Awayda M.S., Tousson A., Benos D.J. 1997. Regulation of a cloned epithelial Na+ channel by its beta- and gamma-subunits. Am. J. Physiol 273:C1889–1899
Bachhuber T., Konig J., Voelcker T., Murle B., Schreiber R., Kunzelmann K. 2005. Cl- Interference with the epithelial Na+ channel ENaC. J. Biol. Chem. 280:31587–31594
Boucher R.C., Cotton C.U., Gatzy J.T., Knowles M.R., Yankaskas J.R. 1988. Evidence for reduced Cl- and increased Na+ permeability in cystic fibrosis human primary cell cultures. J. Physiol. (Paris) 405:77–103
Boucher R.C., Stutts M.J., Knowles M.R., Cantley L., Gatzy J.T. 1986. Na+ transport in cystic fibrosis respiratory epithelia: abnormal basal rate and response to adenylate cyclase activation. J. Clin. Invest. 78:1245–1252
Bubien J.K., Watson B., Khan M.A, Langloh A.L., Fuller C.M., Berdiev B., Tousson A., Benos D.J. 2001. Expression and regulation of normal and polymorphic epithelial sodium channel by human lymphocytes. J. Biol. Chem. 276:8557–8566
Cotton C.U., Stutts M.J., Knowles M.R., Gatzy J.T., Boucher R.C. 1987. Abnormal apical cell membrane in cystic fibrosis respiratory epithelium. An in vitro electrophysiologic analysis. J. Clin. Invest. 79:80–85
de La Ros.a D.A., Canessa C.M., Fyfe G.K., Zhang P. 2000. Structure and regulation of amiloride sensitive sodium channels. Annu. Rev. Physiol. 62:573–594
Dinudom A., Komwatana P., Young J.A., Cook D.I. 1995. Control of the amiloride -sensitive Na+ current in mouse salivary ducts by intracellular anions is mediated by a G protein. J. Physiol. 487:549–555
Dinudom A., Young J.A., Cook D.L. 1993. Na+ and Cl- conductances are controlled by cytosolic Cl- concentration in the intralobular duct cells of mouse mandibular glands. J. Membrane Biol. 135:289–295
Ishikawa T., Marunaka Y., Rotin D. 1998. Electrophysiological characterization of the rat epithelial Na+ channel (rENaC) expressed in MDCK cells. Effects of Na+ and Ca2+. J. Gen. Physiol. 111:825–846
Ismailov II, Berdiev BK, Shlyonsky VG, Fuller CM, Prat AG, Jovov B, Cantiello HF, Ausiello DA, Benos DJ. 1997 Role of actin in regulation of epithelial sodium channels by CFTR. Am J Physiol 272:C1077–1086
Ismailov II, Berdiev BK, Benos DJ. 1995. Biochemical status of renal epithelial Na+ channels determines apparent channel conductance, ion selectivity, and amiloride sensitivity. Biophys J 69:1789–1800
Ismailov I.I., McDuffie J.H., Benos D.J. 1994. Protein kinase A phosphorylation and G-protein regulation of purified renal Na+ channels in planar bilayer membranes. J. Biol. Chem. 269:10235–10241
Iwase N., Sasaki T., Shimura S., Yamamoto M., Suzuki S., Shirato K. 1997. ATP-induced Cl- secretion with suppressed Na+ absorption in rabbit tracheal epithelium. Respir. Physiol. 107:173–180
Ji H.L., Chalfant M.L., Jovov B., Lockchart J.P., Parker S.B., Fuller C.M., Stanton B.A., Benos D.J. 2000. The cytosolic termini of the beta- and gamma-ENaC subunits are involved in the functional interactions between cystic fibrosis transmembrane conductance regulator and epithelial sodium channel. J. Biol. Chem. 275:27947–27956
Kunzelmann K., Kiser G.L., Schreiber R., Riordan J.R. 1997. Inhibition of epithelial Na+ currents by intracellular domains of the cystic fibrosis transmembrane conductance regulator. FEES Lett. 400:341–344
Letz B., Korbmacher C. 1997. cAMP stimulates CFTR-like Cl- channels and inhibits amiloride-sensitive Na+ channels in mouse CCD cells. Am. J. Physiol. 212:C657–666
Nagel G., Barbry P., Chabot H., Brochiero E., Hartung K., Grygorczyk R. 2005. CFTR fails to inhibit the epithelial sodium channel ENaC expressed in Xenopus laevis oocytes. J. Physiol. 564:671–682
Nagel G., Szellas T., Riordan J.R., Friedrich T., Hartung K. 2001. Non-specific activation of the epithelial sodium channel by the CFTR chloride channel. EMBO Rep. 2:249–254
Quinton P.M. 1999. Physiological basis of cystic fibrosis: a historical perspective. Physiol. Rev. 79:S3–S22
Quinton P.M., Reddy M.M. 1992. Control of CFTR chloride conductance by ATP levels through non-hydrolytic binding. Nature 360:79–81
Reddy M.M., Light M.J., Quinton P.M. 1999. Activation of the epithelial Na+ channel (ENaC) requires CFTR Cl- channel function. Nature 402:301–304
Reddy M.M., Quinton P.M. 1992. cAMP activation of CF-affected Cl-conductance in both cell membranes of an absorptive epithelium. J. Membrane Biol 130:49–62
Reddy M.M., Quinton P.M. 2001. cAMP-independent phosphorylation activation of CFTR by G proteins in native human sweat duct. Am. J. Physiol. 280:C604–613
Reddy M.M., Quinton P.M. 2003. Control of dynamic CFTR selectivity by glutamate and ATP in epithelial cells. Nature 423:756–760
Reddy M.M., Quinton P.M. 2002. Effect of anion transport blockers on CFTR in the human sweat duct. J. Membrane Biol. 189:15–25
Reddy M.M., Wang X.F., Gottschalk M,. Jones K., Quinton P.M. 2005. Normal CFTR Activity and Reversed Skin Potentials in Pseudohypoaldosteronism. J. Membrane Biol 203:151–159
Rossier B.C., Pradervand S., Schild L., Hummler E. 2002. Epithelial sodium channel and the control of sodium balance: interaction between genetic and environmental factors. Annu. Rev. Physiol. 64:877–897
Schwiebert E.M., Benos D.J., Egan M.E., Stutts M.J., Guggino W.B. 1999. CFTR is a conductance regulator as well as a chloride channel. Physiol. Rev. 79:S145–S166
Senyk O., Ismailov L, Bradford A.L., Baker R.R., Matalon S., Benos D.J. 1995. Reconstitution of immunopurified alveolar type II cell Na+ channel protein into planar lipid bilayers. Am. J. Physiol. 268:C1148–C1156
Sheppard D.N., Welsh MJ. 1999. Structure and function of the CFTR chloride channel. Physiol. Rev. 79:S23–S45
Shimkets R.A., Lifton R., Canessa C.M. 1998. In vivo phosphorylation of the epithelial sodium channel. Proc. Natl. Acad. Sci. USA 95:3301–3305
Stutts M.J., Canessa C.M., Olsen J.C., Hamrick M., Cohn J.A., Rossier B.C., Boucher R.C. 1995. CFTR as a cAMP-dependent regulator of sodium channels. Science 269:847–850
Stutts M.J., Rossier B.C., Boucher R.C. 1997. Cystic fibrosis transmembrane conductance regulator inverts protein kinase A-mediated regulation of epithelial sodium channel single channel kinetics. J. Biol. Chem. 272:14037–14040
Acknowledgment
We thank Kirk Taylor and Sucharitha Reddy for technical assistance and Mrs. Joanne Albrecht for clerical assistance. This work was funded by grants from the USPHS-NIH-DE 014352, NIH-DK 51899, Nancy Omsted Trust, and Cystic Fibrosis Res. Inc.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s00232-006-1003-4.
Rights and permissions
About this article
Cite this article
Reddy, M., Quinton, P. ENaC Activity Requires CFTR Channel Function Independently of Phosphorylation in Sweat Duct. J Membrane Biol 207, 23–33 (2005). https://doi.org/10.1007/s00232-005-0798-8
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00232-005-0798-8