
Abstract
In rabbit corneal epithelial cells (RCEC), we determined whether capacitative calcium entry (CCE) mediates the mitogenic response to epidermal growth factor, EGF. [Ca2+]i was measured with single-cell fluorescence imaging of fura2-loaded RCEC. EGF (5 ng/ml) maximally increased [Ca2+]i 4.4-fold. Following intracellular store (ICS) calcium depletion in calcium-free medium with 10 µM cyclopiazonic acid (CPA) (endoplasmic reticulum calcium ATPase inhibitor), calcium addback elicited plasma membrane Ca2+ influx as a result of activation of plasma membrane store operated channel (SOC) activity. Based on Mn2+ quench measurements of fura2 fluorescence, 5 ng/ml EGF enhanced such influx 2.3-fold, whereas with Rp-cAMPS (protein kinase A inhibitor) plus EGF it increased by 5.3-fold. In contrast, SOC activation was blocked with 100 µM 2-aminoethyldiphenylborate (2-APB, store-operated channel inhibitor). During exposure to either 50 µM UO126 (MEK-1/2 inhibitor) or 10 µM forskolin (adenylate cyclase activator), 5 ng/ml EGF failed to affect [Ca2+]i. RT-PCR detected gene expression of: 1) transient receptor potential (TRP) protein isoforms 1, 3, 4, 6 and 7; 2) IP3R isoforms 1–3. Immunocytochemistry, in conjunction with confocal and immunogold electron microscopy, detected plasma membrane localization of TRP4 expression. Inhibition of CCE with 2-APB and/or CPA, eliminated the 2.5-fold increase in intracellular [3H]-thymidine incorporation induced by EGF. Taken together, CCE in RCEC mediates the mitogenic response to EGF. EGF induces CCE through its stimulation of Erk1/2 activity, whereas PKA stimulation suppresses these effects of EGF. TRP4 may be a component of plasma membrane SOC activity, which is stimulated by ICS calcium depletion.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Corneal epithelial renewal is a continuous process that is essential for the maintenance of corneal transparency and normal vision. In addition, it assures the maintenance of the epithelial barrier function, which prevents noxious agents from damaging corneal integrity. This process is needed to replace terminally differentiated cells that eventually lose their ability to elicit net ion transport towards the tears and prevent the cornea from becoming swollen and translucent. Numerous cytokines have been identified that modulate the events involved in epithelial regeneration. They regulate it by either stimulating or inhibiting the growth, differentiation and demise of the cells in this layer. One of them is EGF, which in vivo and in vitro is a highly efficacious and potent stimulator of both corneal epithelial cell proliferation and migration. This realization has clinical relevance in the search for more effective strategies to hasten wound closure following injury to the corneal epithelial surface (Lu, Reinach & Kao, 2001).
There is emerging evidence showing that EGF receptor regulation of epithelial cell proliferation and migration is mediated by a number of different, interacting signaling pathways. This cascade includes components eliciting increases in the activity of: 1) the ERK limb of the mitogen-activated protein kinase (MAPK) superfamily and PKA, [EGF activation of PKA has a negative feedback effect on EGF-induced activation of the ERK limb of the MAPK cascade at the level of Raf-1. Such suppression ultimately dampens the mitogenic response to EGF (Kang et al., 2000)]; 2) protein kinase C (PKC), phosphoinositide 3-kinase, PI3-kinase (Zhang et al., 1999). 3) phospholipases Cγ and D [Phospholipase Cγ stimulation results in the generation of diacylglycerol and inositol trisphosphate, IP3 (Zhang & Akhtar, 1998)]. Diacylglycerol in turn stimulates specific PKC isoforms, whereas IP3 mobilizes calcium release from intracellular stores (ICS). One ICS calcium release pathway identified in corneal epithelial cells is a ryanodine-sensitive efflux route (Socci et al., 1993). However, the physiological agonist for this pathway is unclear. Stimulation of calcium-sensitive PKC isoforms with either hepatocyte growth factor or keratinocyte growth factor induces increased wound healing as a result of stimulation of corneal epithelial cell proliferation and migration (Chandrasekher, Kakazu & Bazan, 2001). Direct stimulation of PKC has a similar effect as these cytokines on this response (Hirakata, Gupta & Proia, 1993). However, there is no information in RCEC regarding the role of calcium signaling in eliciting EGF-induced control of proliferation.
Calcium mobilization in response to adrenergic and muscarinic receptor stimulation has been described in corneal epithelial cells (Reinach et al., 1992; Socci et al., 1993; Socci et al., 1996). In addition, two different endothelin receptor isoforms (ETA and ETB) elicit calcium transients as the result of IP3-mediated calcium release from ICS. Another component contributing to these rises involves increases in plasma membrane Ca2+ influx resulting from stimulation of receptor-operated channels by different endothelin isoforms (Tao et al., 1997). There is evidence in some other tissues that IP3 receptor control of calcium efflux is a consequence of the expression of different IP3 receptor isoforms (i.e., 1–3) (Thrower, Hagar & Ehrlich, 2001). The physiological significance of these three isoforms to calcium signaling is unknown.
In corneal epithelial cells as well as numerous other tissues, receptor-activated calcium mobilization in some cases can be accounted for in terms of capacitative calcium entry (CCE) (Takagi et al., 1994; Berridge, 1995; Tao et al., 1997; Wu et al., 1997; Putney & Ribeiro, 2000). According to this paradigm, calcium release from IP3-sensitive ICS is followed by increases in plasma membrane calcium influx, which is mediated by a feedback mechanism involving either conformational coupling or release of a calcium influx factor. Activation of this mechanism has been shown in mouse mammary epithelial cells to be required for the mitogenic response to EGF (Ichikawa et al., 2000). However, in a human salivary cell line EGF-induced calcium depletion of ICS does not result in the activation of CCE (Zhang et al., 2002). Recent studies suggest that transient receptor potential (TRP) protein homologs first identified in Drosophila mutant eyes are a component(s) of different types of ionic channels, including SOC in a variety of many inexcitable and excitable cells (Meir, 2002). A signature of SOC activity is plasma membrane localization of TRP4 expression (Freichel et al., 2001). There is no information regarding the roles of TRPs in mediating calcium homeostasis in corneal epithelial cells.
We report here on the relationship between the mitogenic response to EGF and its activation of CCE in RCEC. Furthermore, the roles are considered of EGF-induced stimulation of the ERK limb of the MAPK superfamily and PKA in mediating CCE. Finally, the involvement is addressed of TRP gene and TRP4 protein expression in CCE.
Materials and Methods
CELL CULTURE
Primary RCEC and SV40-immortalized rabbit corneal epithelial cells (tRCEC) were cultured in Dulbecco's modified Eagle medium/F12, which contained 6% FBS, 5 ng/ml EGF, 1 µg/ml insulin and 40 µg/ml gentamicin (Araki-Sasaki et al., 1993). The cells were kept subconfluent and grown for 1 to 2 days in a mixture containing 5% CO2, 95% ambient air at 37°C. They were serum-starved for 24 h prior to experimentation and 0.5% bovine serum albumin (BSA) was added to maintain osmotic balance. In addition, during this period the cells were no longer exposed to EGF.
FLUORESCENCE CELL IMAGING
The cells were grown on circular coverslips and loaded at room temperature for 45 min in serum-free medium with the calcium-sensitive fluorescent dye (i.e., 2 µM fura2-AM). The cells were then washed with NaCl Ringers containing (in millimolar): 137.5 NaCl, 5 KCl, 1 CaCl2, 1 KH2PO4, 1 MgCl2, 10 glucose and 10 HEPES (pH 7.4). A Ca-free solution was supplemented with 0.5 mM EGTA. A coverslip formed the base of a perfusion chamber that was placed on the stage of an inverted microscope (Nikon Diaphot 200) and the cells were continuously superfused at 34°C. Control and experimental solutions were administered through a two-way valve and their turnover time (<5 s) was kept at a constant rate. The cells were alternately illuminated at 340 and 380 nm and their emission was monitored at 510 nm with a Roper Scientific CCD camera. The emissions were sampled every 5 sec. The field of interest contained roughly 6 to 10 cells and a mean running ratio was calculated for each region. [Ca2+]i was calculated as described. Mn2+ quench rates of fluorescence resulting from excitation at 360 nm were measured as described to characterize plasma membrane calcium influx (Wu et al., 1997). Six to eight cells were evaluated per experiment. The N values provided indicate the number of experiments per data point. Values are shown as means ± SEM. Statistical significance was determined by Student's unpaired t-test.
CELL PROLIFERATION
[3H]-thymidine incorporation was used as an index of proliferation following 24 h serum starvation in medium supplemented with 0.5% BSA. Cells were incubated at 37°C for 1 h with 1 µCi/ml [3H]-thymidine (3.3 to 4.8 TBq/mmol) and washed three times with ice-cold 5% TCA. Cell lysis was obtained with 0.2 N NaOH/2% SDS. The radioactivity was monitored in a liquid scintillation counter and the data were normalized to cellular protein content determined by a modified Lowry assay.
IMMUNOCYTOCHEMICAL LOCALIZATION
Cells were subcultured on a Lab-Tek chamber slide system (Nunc, Naperville, IL) for two days, allowing them to reach confluence. Cell layers were washed twice with HEPES-buffered Ringer solution containing (in millimolar): 142.3 NaCl, 4.2 KCl, 0.8 CaCl2, 2 KH2PO4, 0.2 MgCl2, 5.5 glucose and 10 HEPES (pH 7.4), fixed 30 min in 4% paraformaldehyde (PFA) PBS solution on ice, washed three times with this solution; permeabilized with 0.1% Triton in PBS for 20 min and then washed with PBS. Normal goat serum (0.1%) was used at room temperature to blot the cells for 30 min. The cell monolayers were exposed to anti-TRPC4 at a dilution of 1:200 (Alomone Labs, Jerusalem, Israel) plus 1% BSA at room temperature for at least 60 min. After three washes with HEPES Ringer solution, the cell monolayers were incubated in Cy™ 3-conjugated AffiniPure goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) at a dilution of 1:800 for 30 min at room temperature. This was done with a Zeiss 410 confocal microscope (Zeiss, Thornwood, NY) with a 60× oil objective lens (N.A. 1.4). Serial images were taken through a Z-axis at intervals of 0.5 µm and stacked to achieve the final image. Images were processed using LSM PC software (Zeiss, Thornwood, NY) and Photoshop 5.5 (Adobe Systems, San Jose, CA).
REVERSE TRANSCRIPTION POLYMERASE CHAIN REACTION (RT-PCR)
Total RNA was extracted from both cultured and fresh rabbit corneal epithelium using TRIzol Reagent (Gibco BRL), according to the manufacturer's instructions. To generate the first-strand cDNA, the total RNA extract (0.5–5 mg) was reverse-transcribed (total incubation mixture, 20 µl) at 42°C for 50 min in first-strand buffer (50 mM Tris, 75 mM KC1, 3.0 mM MgCl2, pH 8.4), containing 10 mM dithiothreitol, 0.5 mM of each dNTP, Oligo (dT) primer (3 mg/µl, 1.5 µl) (Gibco BRL) and superscript II reverse transcriptase (40 U/µl, Gibco BRL). First-strand cDNA was used in PCR amplification reactions (total incubation mixture, 25 µl) in a reaction buffer containing 20 mM Tris (pH 8.4), 50 mM KCl, 2.0 mM MgCl2, TaKaRa Ex Taq™ polymerase (2.5 units, TaKaRa Shuzo Co., Otsu, Japan) and 0.2 mM of each dNTP, with the specific primer set for each TRP protein homologue and IP3R isoform described in Table 1. Final concentration of the primers was 0.2 µM. Using mRNA as the template isolated from tRCEC and RCEC, RT-PCR experiments were performed twice with each different primer pair set to probe for the gene expression of each of the 7 different TRP homologs (TRP1, 2, 3, 4, 5, 6 and 7) and IP3R isoforms (IP3R 1–3).
PCR amplifications were carried out in a thermocycler under the following conditions: denaturation at 94°C for 3 minutes for one cycle, 34 cycles of denaturation at 94°C for 45 seconds, annealing at 61°C for 45 seconds, extension at 72°C for 1 minute, and a final extension for one cycle at 72°C for 15 minutes. The PCR products were loaded onto 1% agarose gels, electrophoresed and stained with 0.5 mg/ml ethidium bromide.
SUBCLONING AND SEQUENCING
All PCR products were purified using a 1% low melting point agarose gel. Freshly purified products were mixed for 5 min with PCR® 4TOPO® vector (Invitrogen; San Diego, CA). The TOPO® cloning reaction was added into a vial of One Shot® cells for plasmid transformation. The transformed E. coli bacteria were plated on an agar culture medium containing ampicillin (50 µg/ml) and incubated at 37°C overnight. Inserts from selected clones were digested with EcoR1 (GIBCO BRL) and their size was confirmed using 1% agarose minigel electrophoresis. The vectors with predicted inserts were isolated using a plasmid miniprep kit (Qiagen; Chatsworth, CA). Sequencing was performed using the ABI Prism® BigDye™ Terminator Cycle Sequencing Ready Reaction mix (PE Applied Biosystems) according to the manufacturer's instructions. Sequencing electrophoresis was run on the ABI Prism 3700 DNA analyzer and was assembled and compared using Vector NTI ver5.5 software (InforMax; North Bethesda, MD).
MEDIA AND REAGENTS
Dulbecco's modified Eagle medium/F12; fetal bovine serum and phosphate-buffered salines were from GIBCO (Grand Island, NY). Fura2-AM was from Molecular Probes (Eugene, OR). RG13022, U73122, PD98059, U0126, forskolin; adenosine 3′,5′-cyclic monophosphate, Rp-isomer triethylammonium salt (Rp-cAMPS), 8-(4-chlorophenylthio) adenosine 3′,5′-cyclic monophosphate (CPT-cAMP), were from Sigma RBI (Natick, MA). 2-APB was from Calbiochem (La Jolla, CA). Epidermal growth factor and insulin were from Upstate Biotechnology (Lake Placid, NY). Gentamicin, cyclopiazonic acid and all other agents were from Sigma (St. Louis, MO). All primers were from Invitrogen Corporation (Carlsbad, California).
Results
The concentration-dependent effects of EGF were compared in RCEC and tRCEC on [Ca2+]i over the same range as employed to assess its effects on proliferation, activation of Erk1/2 and the Na:K:2Cl cotransporter (NKCC) (Kang et al., 2000; Tao et al., 1995; Yang et al., 2001). Figure 1 shows that between 1 and 5 ng/ml the maximal response increased from 2.5-fold above its control value to 4.4-fold. The inset shows in RCEC that 5 ng/ml EGF elicited a maximal increase of about 5-fold after 2 min. It was identical to that observed in tRCEC (not shown). On the other hand, over the range from 5 to 60 ng/ml, this response decreased to reach a value that was similar to the one obtained with 1 ng/ml EGF. The correspondence between the effects of EGF in RCEC and tRCEC on [Ca2+]i, coupled with those on proliferation and those previous described on Erk1/2 and NKCC activation, validated the use of tRCEC as a physiologically relevant model to characterize EGF-induced calcium signaling in RCEC (Kang et al., 2000; Kang et al., 2001).

EGF induces dose dependent increases in [Ca2+]i in RCEC and tRCEC. The maximum transient rises in [Ca2+]i were measured following exposure to the indicated EGF concentrations in NaCl Ringer solution containing 1 mM Ca2+. There were eight and ten replicates for RCEC and tRCEC, respectively. Six to eight cells were monitored in each experiment. Inset shows a typical [Ca2+]i transient obtained with 5 ng/ml EGF. Note that the maximal rise was obtained with 5 ng/ml EGF, which is the same concentration previously described to maximally stimulate growth, NKCC activity and Erk1/2 activation. The data are shown as their mean ± SEM.
A relatively specific tyrosine kinase receptor inhibitor was used to block EGF receptor activation, 50 µM RG13022 (Reddy, Keshamouni & Chen, 1999). Following a 10-min exposure to it, [Ca2+]i was unchanged. Subsequently, EGF failed to elicit a calcium transient (data not shown). As EGF elevates IP3 levels in tRCEC through increases in phospholipase Cγ activity, the effect was determined of inhibition of phospholipase Cγ activity with 5 µM U73122 for 20 min on EGF-induced rises in [Ca2+]i (Islam & Akhtar, 2000). During this preincubation period, [Ca2+]i remained unchanged. EGF (5 ng/ml) then failed to induce a transient, suggesting that activation of phospholipase Cγ activity is a prerequisite for EGF-induced rises in [Ca2+]i.
Figure 2 compares the effects of EGF on [Ca2+]i in the presence and absence of extracellular calcium. Calcium removal did not change the [Ca2+]i transient height, but its peak was reached sooner than in the presence of extracellular calcium. Furthermore, with extracellular calcium, the plateau phase was maintained at higher concentrations for a longer period of time. These differences suggest that both intracellular calcium release and increases in plasma membrane influx contribute to EGF-induced calcium signaling.

Comparison in tRCEC of EGF-induced [Ca2+]i transients in Ca2+ containing and Ca2+ free solution. Time-dependent rises in [Ca2+]i are shown resulting from exposure to 5 ng/ml EGF in Ca2+-containing (1 mM) and Ca2+-free solution buffered with 0.5 mM EGTA. Without calcium in the medium, [Ca2+]i fell more rapidly to reach lower plateau values than in the presence of 1 mM calcium in the bathing solution. This difference reflects mobilization of calcium from both extracellular and intracellular sources. The number of experiments in each case is shown in parentheses.
To probe for whether SOC activity is needed for EGF to induce CCE, its effect was determined on calcium addback (cf Fig. 3) (Rivera et al., 1995). We first determined whether inhibition of ICS calcium pump activity with 10 µM CPA affected the calcium addback response. In the absence of CPA, medium supplementation of a Ca-free solution with 1 mM calcium had a minimal effect on [Ca2+]i. Exposure to CPA caused calcium to transiently rise within about 5 min by nearly 3-fold followed by a rapid decline to a stable value lower than the initial one. Subsequently, calcium addback to the bathing solution resulted in [Ca2+]i rising to a stable value about 3-fold higher than in calcium-free solution that lasted for at least 12 min. There is evidence that this rise is in part accounted for by the activation of non-voltage-gated permeation pathways (Rich & Rae, 1995).

Relationship in tRCEC between store depletion by CPA and Ca2+ entry. CPA (10 µM) was applied at the downward-directed arrow in Ca-free Ringer medium containing 0.5 mM EGTA. Following attainment of a nadir, the Ca-free Ringer was isosmotically substituted with Ca-rich (1 mM) Ringer medium containing CPA (10 µM). (N = 5)
To determine whether EGF affects ionic influx through these pathways, the effect of 0.5 mM Mn2+ was measured on fura2 fluorescence in the absence and presence of 5 ng/ml EGF. Such measurements are reflective of plasma membrane calcium influx because Mn2+ acts as a surrogate for calcium. Fura2 fluorescence was excited at 360 nm and the cells were exposed to Mn2+ following depletion of ICS calcium with 10 µM CPA in calcium-free medium containing 0.5 mM EGTA. Average quench curves are shown in Fig. 4 A and reveal that EGF increased this quench rate 2.3-fold, whereas with EGF plus 10 µM Rp-cAMPS it increased 5.9-fold. Furthermore, 100 µM 2-APB completely blocked the increase in Mn2+ influx induced by EGF, suggesting that a component of the EGF-induced CCE results from EGF stimulation of SOC activity.

(A) Manganese quench of fura2 fluorescence in tRCEC after exposure to EGF (5 ng/ml). Following depletion of ICS calcium with 10 µM CPA in Ca-free medium containing 0.5 mM EGTA and fluorescence stabilization, 0.5 mM Mn2+was added to the control and EGF-containing bathing medium. Also shown are the effects of preincubation for 20 min with either 100 µM 2-APB or 10 µM Rp-cAMPS on EGF-induced increases in the rate of Mn2+ quench. Mn2+ induced a quench in the isobestic point indicated by a linear fit. The slopes were: control (−0.00042 ± 0.0003 units/5 s); 2-APB plus EGF (−0.00043 ± 0.0001 units/5 s); EGF (−0.00096 ± 0.0006 units/5 s); Rp-cAMPS plus EGF (−0.002 ± 0.001 units/5 s) (N = 6 for each condition). (B) 2-APB inhibits Ca2+ addback. tRCEC were incubated initially without exogenous calcium. CPA time of addition is indicated by the downward-directed arrow. 2-APB (100 µM), DMSO [0.1% (v/v), final concentration] was present thoroughout the entire period from the first upward-directed arrow. CaCl2 (1.0 mM) was added at the time of the second downward-directed arrow (N = 5).
There is suggestive evidence in some studies that 2-APB is an inhibitor of plasma membrane SOC activity (Gregory, Rychkov & Barritt, 2001). As the rise in [Ca2+]i following addback is at least in part due to increases in SOC activity, we used the protocol of Gregory et al. to determine whether 100 µM 2-APB has such an effect in tRCEC. Before and after depleting the ICS of their calcium content with 20 µM CPA, the cells were continuously exposed to 100 µM 2-APB. As is evident in Fig. 4 B, 2-APB blocked a rise in [Ca2+]i following calcium addback. Therefore, 2-APB appears to act as an inhibitor of SOC activity in these cells.
A component of EGF-linked signaling in tRCEC is activation of the Erk1/2 limb of the MAPK superfamily (Kang et al., 2000). As the cause and effect relationship between EGF-induced MAPK and CCE activation was unknown, we determined whether inhibition of Erk1/2 activation affected the transients caused by this mitogen. This was done by preincubating the cells for 25 min with one of two selective inhibitors of MEK-1/2 and Erk1/2 activation, 50 µM UO126 or 15 µM PD98059, respectively. Neither of these agents had any effect on [Ca2+]i. In both cases, 5 ng/ml EGF had no subsequent effect on [Ca2+]i suggesting that Erk1/2 activation precedes EGF-induced rises in [Ca2+]i (data not shown).
EGF-induced activation of Erk1/2 in tRCEC is associated with a negative feedback effect that involves PKA-mediated inhibition of Raf-1 activation (Kang et al., 2000). This inhibitory limb suppresses EGF-induced stimulation of Erk1/2 because Raf-1 is situated upstream of Erk1/2. Given this negative feedback mechanism, we hypothesized that activation of PKA by 10 µM forskolin inhibits EGF-induced stimulation of Erk1/2 activity and CCE. Forskolin caused [Ca2+]i to rise about 1.8-fold from 46 ± 4 nM to a stable value of 83 ± 4 nM after 5 min (n = 6 different coverslips). Subsequent to this rise, which remained stable for the next 20 min, EGF had no effect on [Ca2+]i. To validate that the forskolin-induced rise in [Ca2+]i was a result of adenylate cyclase stimulation, the effects were determined on [Ca2+]i of either 100 µM CPT-cAMP alone or following inhibition of PKA with 10 µM Rp-cAMPS. CPT-cAMP had nearly the same effect on [Ca2+]i as forskolin, whereas preincubation with Rp-cAMPS obviated the rise in [Ca2+]i induced by CPT-cAMP (data not shown). These effects suggest that increases in PKA activity suppress EGF-induced CCE and are consistent with our finding that inhibition of EGF-induced activation of Erk1/2 activity blocks EGF-induced rises in [Ca2+]i.
TRP proteins are components of different channels involved in the regulation of intracellular calcium and their activation is needed for calcium signaling. The number of its homologs expressed in different cell types is variable and it is suggested that combinations of its numerous homologs with other proteins are needed to elicit activity of a specific channel type (Meir, 2002; Elliott, 2001). To identify their involvement in calcium signaling in tRCEC, we profiled the gene expression pattern of TRP1-7 and probed for TRP4 protein expression. TRP1, 3, 4, 6 and 7 mRNA transcripts were detected in these cells, as seen in Fig. 5 A. Sequencing of the products revealed in all cases more than 95% homology with the published human sequence.

(A) TRP mRNA expression in RCEC. RT-PCR was performed using TRP gene-specific primers shown in Table 1. The products were resolved on a 1% agarose gel and visualized with 0.5 mg/ml ethidium bromide. Based on the primer designs, the anticipated products were obtained for TRPs 1, 3, 4, 6 and 7. Their identity was validated with sequence analysis. In all cases, the products had more than 95% homology with published human sequences. The cDNA marker ladder is shown on the left. (B) Heterogeneous IP3 receptor isoform gene expression. RT-PCR was performed with specific primer pairs for IP3R1-3. Anticipated cDNA products were obtained for all three IP3R isoforms. Their identity was validated with sequence analysis. In all cases, each of the products had more than 95% homology with their corresponding published human sequence. The cDNA marker ladder is shown on the left.
There is also diversity in the number of different IP3 receptor genes expressed in cells wherein calcium signaling was described (Elliott, 2001). Three different IP3R isoforms have been identified. The physiological significance of this heterogeneity remains unclear other than suggestions regarding their possible role in the shaping of calcium transients (Thrower et al., 2001; Elliott, 2001). To obtain baseline information regarding such expression, we also probed for IP3R gene expression heterogeneity. RT-PCR experiments were performed twice with cDNA prepared from tRCEC and RCEC mRNA and the template and primer sets (cf Table 1) specific for each of the three IP3R isoforms (Patel, Joseph & Thomas, 1999). The gene transcripts were detected for all three of the described IP3R isoforms, as shown in Fig. 5 B. Sequencing of the products revealed in all cases more than 95% homology with the published human sequence.
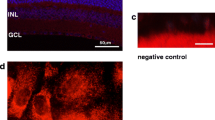
The physiological significance of heterogeneous TRP gene expression was examined by probing for localization of TRP protein expression with immunocytochemistry in conjunction with fluorescence confocal microscopy and immunogold electron microscopy. Figure 6 A shows a representative micrograph of cells stained with a primary and secondary antibody pair specific for TRP4. There is defined delimited staining around the plasma membrane perimeter for this isoform. This localization is consistent with other studies, in which TRP4 protein expression is associated with plasma membrane calcium influx pathways, some of which are SOC (Freichel et al., 2001). Figure 6 B shows the staining obtained following omission of the primary antibody. As can be seen, there is mostly nonspecific staining that appears to be essentially nuclear and there is little staining near the plasma membrane boundaries. To further validate that there is extensive plasma membrane TRP4 localization, we performed in parallel immunogold-labeling electron microscopy. Figure 7 shows a typical electron micrograph, in which there is specific TRP4 labeling, some of which is localized to near or within the plasma membrane domain. In addition, some of the other labeling suggests that this represents TRP4 expression in domains from which it is exported to the plasma membrane.

Immunofluorescent staining of TRP4 Protein in tRCEC. (A) Immunofluorescent labeling using TRPC4 antibody at a dilution of 1:200 viewed with confocal microscopy. (B) Control tRCEC stained in the absence of primary antibody. Omission of the primary antibody reveals nonspecific background staining partially of nuclear origin due to membrane permeabilization with Triton X-100, whereas the plasma membrane is not clearly demarcated due to absence of staining.

Immunogold EM localization of TRP4 protein expression. Electron micrograph shows cells with microplicae projecting into the tear-side compartment labeled as AP. Desmosomes between neighboring apposed cells are labeled DES. Arrowheads indicate TRP4 expression in plasma membrane region as well as intracellular sites.
To determine whether the mitogenic response to EGF is dependent on EGF stimulation of SOC activity resulting from IP3 formation, the effects were determined of 5 µM CPA, 100 µM 2-APB and 10 µM U73122 (phospholipase C inhibitor) on the mitogenic response to 5 ng/ml EGF. The results shown in Fig. 8 of measurements of thymidine uptake indicate that following 24 h of serum starvation EGF increased proliferation by nearly 2.5-fold above its control level. Despite the presence of calcium in the medium, either 100 µM 2APB, 5 µM CPA or 10 µM U73122 inhibited by 73%, 71% and 68% the rises in proliferation induced by EGF, respectively. As the individual inhibitory effects of CPA and 2-APB on proliferation were nearly identical to one another, the mitogenic response to EGF is dependent on CCE. On the other hand, they suppressed proliferation to levels that were slightly below the control level. This slightly higher level of inhibition suggests that there may be autocrine-mediated stimulation of proliferation by EGF. This is tenable because gene expression of EGF and other mitogens was detected in these cells. (Wilson, Lloyd & He, 1992). It is unlikely that these compounds have some toxic effect because in their presence, calcium levels declined and stabilized at values similar to those measured prior to the CPA-induced transient. The inhibition by U73122 is consistent with its inhibition of PLC activation and indicates that the EGF-induced calcium transient needed to trigger a rise in proliferation is dependent on IP3-mediated release of Ca2+ from ICS.

Dependence of mitogenic response to EGF on CCE. Cells were initially incubated in serum-free medium for 24 h (see Methods) under the indicated conditions. [3H] thymidine incorporation was monitored as an index of proliferation. Each experiment was performed three times. Data are expressed as mean ± SEM (N = 6 for each condition). Statistical significance relative to the EGF-treated cells is indicated with (***) (p < 0.001).
Discussion
Both RCEC and tRCEC were used to characterize the mechanism of EGF-induced calcium signaling associated with the mitogenic response to this cytokine. The following considerations indicate that the results obtained with tRCEC have physiological relevance: 1) the EGF-induced calcium transients were identical to those obtained with primary cultures; 2) the physiological and phenotypic properties of primary cultures are conserved during the immortalization process (Kang et al., 2001). Furthermore, use of subconfluent cultures of tRCEC is preferable because responses to receptor stimulation are more reproducible among their different cell populations. Under this condition, nearly all of the cells are proliferating rather than undergoing differentiation.
EGF-elicited calcium transients depend on stimulation of its cognate receptor because this mitogen failed to elicit a calcium transient following inhibition of EGF receptor associated tyrosine kinase receptor activity with RG13022. Such transients reflect mobilization of calcium from CPA-sensitive ICS and extracellular sources because in the presence of extracellular calcium the plateau level was higher and it persisted for a longer period of time than in the absence of extracellular calcium. As described in numerous other excitable and inexcitable cells, there is an inverse relationship between the magnitude of calcium influx occurring across the plasma membrane and the ICS calcium filling state. Should this be the case, such influx may be dependent on the activation of SOC as a result of rises in IP3, which elicits release of calcium from ICS (Putney et al., 2000; Berridge, 1995). Accordingly, cytosolic calcium levels are determined by the balance between the activity of the endoplasmic reticulum calcium pump mediating calcium uptake into the ICS and calcium efflux from the ICS through various different types of ICS calcium efflux channels. The effects of CPA on [Ca2+]i and that of calcium addback are consistent with CCE (Fig. 3). In calcium-free solution, [Ca2+]i initially rose because ICS calcium accumulation was blocked due to inhibition by CPA of ICS calcium pumps, which resulted in depletion of its calcium content. The subsequent [Ca2+]i fall in all likelihood reflects activation of plasma membrane ATP-dependent calcium pump and Na/Ca exchange activity (Rich et al., 1995; Reinach, Holmberg & Chiesa, 1991). Two lines of evidence indicate that calcium release occurs through IP3 receptor-controlled calcium channels: 1) inhibition of phospholipase C activity with U73122 prevented the EGF-induced rises in [Ca2+]i; 2) gene transcripts of three different IP3 receptor isoforms were detected (Fig. 5 B). As per the CCE model, EGF increased Mn2+ influx 2.3-fold, which indicates that subsequent to IP3-induced ICS calcium emptying cytosolic calcium repletion was hastened as a result of marked rises in calcium influx across the plasma membrane (Fig. 4 A). Such increases are in agreement with those described in human glomerular mesangial cells, in which it was directly shown that EGF activates SOC (Ma & Sansom, 2001). The finding that the EGF-induced rise in Mn2+ influx was fully suppressed following exposure to 2-APB is consistent with the notion that SOC activity accounts for plasma membrane calcium influx. The feedback mechanism linking falls in ICS calcium content with increases in plasma membrane calcium influx is not yet well understood.
There is earlier evidence in RCEC for a nonselective ionic influx pathway for calcium refilling based on the findings that calcium influx increased following membrane voltage hyperpolarization and that during exposure to SKF96365 (nonselective cation channel blocker) the magnitude of the calcium addback response was markedly suppressed (Tao et al., 1997; Rich et al., 1995). More recent studies in other tissues have shown that the inhibition of [Ca2+]i transients with 2-APB can be attributed to its direct inhibition of SOC activity. Nevertheless, its mechanism of action appears to be environment-specific in that in some studies 2-APB blocks IP3 receptor-mediated calcium release from ICS (Cui & Kanno, 1997; Maruyama et al., 1997; Ascher-Landsberg et al., 1999; Ma et al., 2000; van Rossum et al, 2000).
The results shown in Fig. 4 B are consistent with those of Gregory et al.; namely, that 2-APB can be a selective inhibitor of SOC activity (Gregory et al., 2001). Our findings are consistent with this interpretation because the results shown in Fig. 4 B indicate that the [Ca2+]i transient induced by CPA was comparable to those measured in the absence of 2-APB (Fig. 3). On the other hand, 2-APB entirely blocked the calcium-addback response, which is consistent with the finding that 2-APB is an inhibitor of SOC activity. Had 2-APB instead blocked IP3 receptor-controlled calcium efflux, the [Ca2+]i transient induced by CPA should have been suppressed. This inhibitory effect of 2-APB on calcium addback suggests that a component of the 2.3-fold increase in Mn2+ influx shown in Fig. 4 A is accountable for by increases in SOC channel activity.
TRP protein family members (TRP1-7) are components of plasma membrane nonselective cationic channels that are either specifically or ubiquitously expressed in excitable and non-excitable cells. Different members of the three subfamilies comprising all of the TRP proteins can form heteromers. In heterologous TRP expression system studies, there is emerging evidence that some of the TRP proteins are needed for activation of SOC activity. A product of one of the twenty different TRP genes is TRPC4 and it corresponds to the SOC in endothelial cells, which controls vasorelaxation in blood vessels (Freichel et al., 2001). On the other hand, there is evidence that some of the other six different TRP proteins are components of store-independent pathways (Meir, 2002). We identified and verified TRPC1, 3, 4, 6 and 7 gene expression. The results of immunocytochemistry reveal extensive specific TRPC4 staining in the plasma membrane proximity, suggesting functional SOC activity in this region, which was corroborated with immunogold labeling electron microscopy.
EGF (5 ng/ml) is the optimal dose in all cases for eliciting [Ca2+]i transients, stimulating NKCC, Erk1/2 activity and mitogenesis (Tao et al., 1995; Kang et al., 2000; Yang et al., 2001). Mitogenesis is in turn dependent on [Ca2+]i transients because 2-APB and CPA blocked EGF-induced increases in proliferation (Fig. 8). These transients are dependent on Erk1/2 simulation by EGF because either PD98059 or UO126 fully suppressed them. The dependence of EGF-induced [Ca2+]i transients on Erk1/2 activation is consistent with the finding in platelets that inhibition of Erk1/2 activation reduced ICS Ca2+ depletion and subsequent activation of Ca2+ entry across SOC (Rosado & Sage, 2001).
Another approach to determining the dependence of EGF-induced [Ca2+]i transients on Erk1/2 activation entailed blocking EGF stimulation of Erk1/2 with forskolin. We previously showed that cAMP induced stimulation of PKA suppressed EGF-induced Erk1/2 activation by inhibiting upstream activation of Raf-1 (Kang et al., 2000). In the current study, we at first maximally stimulated PKA activity with forskolin prior to exposing the cells to EGF. Forskolin caused [Ca2+]i to increase to a stable plateau value. This effect of forskolin reflects PKA stimulation because CPT-cAMP had a similar effect and inhibition of PKA activation with Rp-cAMPS prevented CPT-cAMP from raising [Ca2+]i. Furthermore, the fact that Rp-cAMPS further increased EGF-induced Mn2+ influx about 2.5-fold is consistent with the notion that EGF-induced stimulation of PKA blunts Erk1/2-mediated [Ca2+]i rises. The intracellular calcium transients caused by PKA stimulation are in agreement with its effect on [Ca2+]i in bovine corneal epithelial cells (Reinach et al., 1992). As apparent, PKA stimulation suppressed the subsequent EGF-induced [Ca2+]i transient, EGF-induced [Ca2+]i transients are dependent on Erk1/2 activation. It is not yet possible to determine which components in the calcium signaling pathway are dependent on EGF-induced stimulation of Erk1/2 activity for them to elicit a [Ca2+]i transient.
In summary, EGF-induced [Ca2+]i transients in RCEC can be accounted for by CCE. One of its components includes activation of 2-APB-sensitive SOC activity. Such activity is consistent with our identification of TRP4 gene transcripts and localization of its protein expression to the plasma membrane. This signaling cascade is essential for EGF-induced stimulation of proliferation and its activation occurs as a consequence of EGF-induced increases in the ERK limb of the MAPK superfamily. To integrate the results of the current study with those previously described regarding EGF receptor-linked signaling, a working model of our current understanding of such control is shown in Figure 9.

Working model of associations between EGF receptor-linked signaling pathways. EGF-mediated CCE is dependent on: 1) EGF activation of the ERK limb of the MAPK cascade (Depicted site in the CCE paradigm of dependence on Erk1/2 stimulation agrees with data obtained by Rosado, J.A., Sage, S.O. 2001, in platelets.); 2) EGF-induced stimulation of PLC results in IP3 formation. IP3 in turn mediates calcium release from CPA-sensitive ICS. Depletion of ICS calcium elicits, through a feedback signal of unknown origin, opening of plasma membrane SOC. A protein component of this complex is the TRP isoform, TRP4. EGFR stimulation results in PKA activation as the result of increases in cPLA2 activity, which results in the release of arachidonate (Ara) from membrane phospholipid (PL) followed by cyclooxygenase-2 (COX-2)-mediated rises in prostaglandin (PG) formation. PG stimulates its cognate receptor (PR) resulting in stimulation of adenylate cyclase (AC) and PKA activity. PKA activation in turn has a negative feedback effect on EGF-induced stimulation of the MAPK cascade at the level of Raf-1 in the ERK limb of this superfamily. Also shown is an ICS ryanodine-sensitive Ca2+-release pathway (RyR) whose activation in tRCEC is required for mediating a regulatory volume decrease in response to an imposed hypotonic stress.
References
K. Araki-Sasaki Y. Ohashi T. Sasabe K. Hayashi X.Z. Yang Y. Hosaka S. Aizawa H. Handa (1993) ArticleTitleImmortalization of rabbit corneal epithelial cells by recombinant SV40-adenovirus vector. Invest. Ophthalmol. Vis. Sci. 34 2665–2671 Occurrence Handle1:STN:280:ByyA2cnhs1w%3D Occurrence Handle7688358
J. Ascher-Landsberg T. Saunders M. Elovitz M. Phillippe (1999) ArticleTitleThe effects of 2-aminoethoxydiphenyl borate, a novel inositol 1,4,5-trisphosphate receptor modulator on myometrial contractions. Biochem. Biophys. Res. Commun. 264 979–982 Occurrence Handle10.1006/bbrc.1999.1602 Occurrence Handle1:CAS:528:DyaK1MXmvFOmsrw%3D Occurrence Handle10544041
M.J. Berridge (1995) ArticleTitleCapacitative calcium entry. Biochem. J. 312 1–11 Occurrence Handle1:CAS:528:DyaK2MXpsFShu7k%3D Occurrence Handle7492298
G. Chandrasekher A.H. Kakazu H.E. Bazan (2001) ArticleTitleHGF- and KGF-induced activation of PI-3K/p70 s6 kinase pathway in corneal epithelial cells: its relevance in wound healing. Exp. Eye Res. 73 191–202 Occurrence Handle10.1006/exer.2001.1026 Occurrence Handle1:CAS:528:DC%2BD3MXkvFyjsrs%3D Occurrence Handle11446769
Z.J. Cui T. Kanno (1997) ArticleTitlePhotodynamic triggering of calcium oscillation in the isolated rat pancreatic acini. J. Physiol. 504 47–55 Occurrence Handle1:CAS:528:DyaK2sXntVKrsLY%3D Occurrence Handle9350616
A.C. Elliott (2001) ArticleTitleRecent developments in non-excitable cell calcium entry. Cell Calcium 30 73–93 Occurrence Handle1:CAS:528:DC%2BD3MXlsFagu7w%3D Occurrence Handle11440466
M. Freichel S.H. Suh A. Pfeifer U. Schweig C. Trost P. Weissgerber M. Biel S. Philipp D. Freise G. Droogmans F. Hofmann V. Flockerzi B. Nilius (2001) ArticleTitleLack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nat. Cell Biol. 3 121–127 Occurrence Handle10.1038/35055019 Occurrence Handle1:CAS:528:DC%2BD3MXht1KmsL8%3D Occurrence Handle11175743
R.B. Gregory G. Rychkov G.J. Barritt (2001) ArticleTitleEvidence that 2-aminoethyl diphenylborate is a novel inhibitor of store-operated Ca2+ channels in liver cells, and acts through a mechanism which does not involve inositol trisphosphate receptors. Biochem. J. 354 285–290 Occurrence Handle10.1042/0264-6021:3540285 Occurrence Handle1:CAS:528:DC%2BD3MXitFWrs7c%3D Occurrence Handle11171105
A. Hirakata A.G. Gupta A.D. Proia (1993) ArticleTitleEffect of protein kinase C inhibitors and activators on corneal re-epithelialization in the rat. Invest. Ophthalmol. Vis. Sci. 34 216–221 Occurrence Handle1:STN:280:ByyC2cnotVQ%3D Occurrence Handle8425827
J. Ichikawa K. Furuya S. Miyata T. Nakashima T. Kiyohara (2000) ArticleTitleEGF enhances Ca2+ mobilization and capacitative Ca2+ entry in mouse mammary epithelial cells. Cell Biochem. Funct. 18 215–225 Occurrence Handle10.1002/1099-0844(200009)18:3<215::AID-CBF875>3.3.CO;2-J Occurrence Handle1:CAS:528:DC%2BD3cXmtFOqtrw%3D Occurrence Handle10965359
M. Islam R.A. Akhtar (2000) ArticleTitleEpidermal growth factor stimulates phospholipase cgamma1 in cultured rabbit corneal epithelial cells. Exp. Eye Res. 70 261–269 Occurrence Handle10.1006/exer.1999.0783 Occurrence Handle1:CAS:528:DC%2BD3cXhsFylsLY%3D Occurrence Handle10712812
S.S. Kang L. Wang W.W. Kao P.S. Reinach L. Lu (2001) ArticleTitleControl of SV-40 transformed RCE cell proliferation by growth-factor-induced cell cycle progression. Curr. Eye Res. 23 397–405 Occurrence Handle10.1076/ceyr.23.6.397.6965 Occurrence Handle1:STN:280:DC%2BD38zgtlGqsQ%3D%3D Occurrence Handle12045889
S.S. Kang T. Li D. Xu P.S. Reinach L. Lu (2000) ArticleTitleInhibitory effect of PGE2 on EGF-induced MAP kinase activity and rabbit corneal epithelial proliferation. Invest. Ophthalmol. Vis. Sci. 41 2164–2169 Occurrence Handle1:STN:280:DC%2BD3czkvV2ntQ%3D%3D Occurrence Handle10892858
L. Lu P.S. Reinach W.Y. Kao (2001) ArticleTitleCorneal epithelial wound healing. Exp. Bio. Med. 226 653–664 Occurrence Handle1:CAS:528:DC%2BD3MXkvFSrt7Y%3D
R. Ma S.C. Sansom (2001) ArticleTitleEpidermal growth factor activates store-operated calcium channels in human glomerular mesangial cells. J. Am. Soc. Nephrol. 12 47–53 Occurrence Handle1:CAS:528:DC%2BD3MXkvFGqsA%3D%3D Occurrence Handle11134249
H.T. Ma R.L. Patterson D.B. van Rossum L. Birnbaumer K. Mikoshiba D.L. Gill (2000) ArticleTitleRequirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science 287 1647–1651 Occurrence Handle1:CAS:528:DC%2BD3cXhs1Oqtro%3D Occurrence Handle10698739
T. Maruyama Z.J. Cui T. Kanaji K. Mikoshiba T. Kanno (1997) ArticleTitleAttenuation of intracellular Ca2+ and secretory reponses by Ins(1,4,5)P3-induced Ca2+ release modulator, 2APB, in rat pancreatic acinar cells. Biomed. Res. 18 297–302 Occurrence Handle1:CAS:528:DyaK2sXmsVeju7k%3D
Meir, A. 2002. The molecular diversity of TRP channels and related proteins. Modulator. Issue #16 Summer. pp 14–16, Alomone Labs
S. Patel S.K. Joseph A.P. Thomas (1999) ArticleTitleMolecular properties of inositol 1,4,5-trisphosphate receptors. Cell Calcium 25 247–264 Occurrence Handle10.1054/ceca.1999.0021 Occurrence Handle1:CAS:528:DyaK1MXjsVyru74%3D Occurrence Handle10378086
J.W. Putney Jr. C.M. Ribeiro (2000) ArticleTitleSignaling pathways between the plasma membrane and endoplasmic reticulum calcium stores. Cell Mol. Life Sci. 57 1272–1286 Occurrence Handle1:STN:280:DC%2BD3cvnslGnug%3D%3D Occurrence Handle11028918
K.B. Reddy V.G. Keshamouni Y.Q. Chen (1999) ArticleTitleThe level of tyrosine kinase activity regulates the expression of p21/WAF1 in cancer cells. Int. J. Oncol. 15 301–306 Occurrence Handle1:CAS:528:DyaK1MXltVWhtbY%3D Occurrence Handle10402241
P.S. Reinach N. Holmberg R. Chiesa (1991) ArticleTitleIdentification of calmodulin-sensitive Ca2+ -transporting ATPase in the plasma membrane of bovine corneal epithelial cell. Biochim. Biophys. Acta 1068 1–8 Occurrence Handle10.1016/0005-2736(91)90054-C Occurrence Handle1:CAS:528:DyaK3MXmtVWgs70%3D Occurrence Handle1832560
P.S. Reinach R.R. Socci C. Keith M. Scanlon (1992) ArticleTitleAdrenergic receptor-mediated increase of intracellular Ca2+ concentration in isolated bovine corneal epithelial cells. Comp. Biochem. Physiol. Comp. Physiol. 102 709–714 Occurrence Handle10.1016/0300-9629(92)90728-9 Occurrence Handle1:STN:280:By2A1c%2FgsVE%3D Occurrence Handle1355034
A. Rich J.L. Rae (1995) ArticleTitleCalcium entry in rabbit corneal epithelial cells: evidence for a nonvoltage dependent pathway. J. Membrane Biol. 144 177–184 Occurrence Handle1:CAS:528:DyaK2MXkslyrsL8%3D
A.A. Rivera C.R. White L.L. Guest T.S. Elton R.B. Marchase (1995) ArticleTitleHyperglycemia alters cytoplasmic Ca2+ responses to capacitative Ca2+ influx in rat aortic smooth muscle cells. Am. J. Physiol. 269 C1482–1488 Occurrence Handle1:CAS:528:DyaK28XjslKqsA%3D%3D Occurrence Handle8572177
J.A. Rosado S.O. Sage (2001) ArticleTitleRole of the ERK pathway in the activation of store-mediated calcium entry in human platelets. J. Biol. Chem. 276 15659–15665 Occurrence Handle10.1074/jbc.M009218200 Occurrence Handle1:CAS:528:DC%2BD3MXjvFalu7g%3D Occurrence Handle11278479
R. Socci A. Chu P. Reinach L.G. Meszaros (1993) ArticleTitleIn-situ Ca2+-induced Ca2+ release from a ryanodine-sensitive intracellular Ca2+ store in corneal epithelial cells. Comp. Biochem. Physiol. B 106 793–797 Occurrence Handle10.1016/0305-0491(93)90032-Z Occurrence Handle1:STN:280:ByuC3s%2Flt1A%3D Occurrence Handle7507809
R.R. Socci S.D. Tachado R.S. Aronstam P.S. Reinach (1996) ArticleTitleCharacterization of the muscarinic receptor subtypes in the bovine corneal epithelial cells. J. Ocul. Pharmacol. Ther. 12 259–269 Occurrence Handle1:CAS:528:DyaK28XlsVSiur4%3D Occurrence Handle8875332
W. Tao G.I. Liou X. Wu T.O. Abney P.S. Reinach (1995) ArticleTitleETB and epidermal growth factor receptor stimulation of wound closure in bovine corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 36 2614–2622 Occurrence Handle1:STN:280:BymD1MrgsFY%3D Occurrence Handle7499084
W. Tao X. Wu G.I. Liou T.O. Abney P.S. Reinach (1997) ArticleTitleEndothelin receptor mediated Ca2+ signaling and isoform expression in bovine corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 38 130–141 Occurrence Handle1:STN:280:ByiC287jvVU%3D Occurrence Handle9008638
H. Takagi P.S. Reinach S. Tachado N. Yoshimura (1994) ArticleTitleEndothelin-mediated cell signaling and proliferation in cultured rabbit corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 35 134–142 Occurrence Handle1:STN:280:ByuC3s7ntlw%3D Occurrence Handle8300340
E.C. Thrower R.E. Hagar B.E. Ehrlich (2001) ArticleTitleRegulation of Ins (1,4,5) P3 receptor isoforms by endogenous modulators. Trends Pharmacol. Sci. 22 580–586 Occurrence Handle10.1016/S0165-6147(00)01809-5 Occurrence Handle1:CAS:528:DC%2BD3MXotFSmsbc%3D Occurrence Handle11698102
D.B. van Rossum R.L. Patterson H.T. Ma D.L. Gill (2000) ArticleTitleCa2+ entry mediated by store depletion, S-nitrosylation, and TRP3 channels. Comparison of coupling and function. J. Biol. Chem. 275 28562–28568 Occurrence Handle10.1074/jbc.M003147200 Occurrence Handle1:CAS:528:DC%2BD3cXms1Cqsrs%3D Occurrence Handle10878007
S.E. Wilson S.A. Lloyd Y.G. He (1992) ArticleTitleEGF, basic FGF, and TGF beta-1 messenger RNA production in rabbit corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 33 1987–1995 Occurrence Handle1:STN:280:By2B2Mbhs1M%3D Occurrence Handle1582803
X. Wu H. Yang P. Iserovich J. Fischbarg P.S. Reinach (1997) ArticleTitleRegulatory volume decrease by SV40-transformed rabbit corneal epithelial cells requires ryanodine-sensitive Ca2+-induced Ca2+ release. J. Membrane Biol. 158 127–136 Occurrence Handle10.1007/s002329900250 Occurrence Handle1:CAS:528:DyaK2sXlt1Sitbk%3D
H. Yang Z. Wang Y. Miyamoto P.S. Reinach (2001) ArticleTitleCell signaling pathways mediating epidermal growth factor stimulation of Na:K:2Cl cotransport activity in rabbit corneal epithelial cells. J. Membrane Biol. 183 93–101 Occurrence Handle10.1007/s00232-001-0057-6 Occurrence Handle1:CAS:528:DC%2BD3MXntFWiu7w%3D
B.X. Zhang X. Ma C.K. Yeh M.D. Lifschitz M.X. Zhu M.S. Katz (2002) ArticleTitleEpidermal growth factor-induced depletion of the intracellular Ca2+ store fails to activate capacitative Ca2+ entry in a human salivary cell line. J. Biol. Chem. 277 48165–48171 Occurrence Handle10.1074/jbc.M208077200 Occurrence Handle1:CAS:528:DC%2BD38Xptlegsrk%3D Occurrence Handle12368284
Y. Zhang G.I. Liou A.K. Gulati R.A. Akhtar (1999) ArticleTitleExpression of phosphatidylinositol 3-kinase during EGF-stimulated wound repair in rabbit corneal epithelium. Invest. Ophthalmol. Vis. Sci. 40 2819–2826 Occurrence Handle1:STN:280:DC%2BD3c%2FhsFygtg%3D%3D Occurrence Handle10549641
Y. Zhang R.A. Akhtar (1998) ArticleTitleEpidermal growth factor stimulates phospholipase D independent of phospholipase C, protein kinase C or phosphatidylinositol-3 kinase activation in immortalized rabbit corneal epithelial cells. Curr. Eye Res. 17 294–300 Occurrence Handle10.1076/ceyr.17.3.294.5223 Occurrence Handle1:STN:280:DyaK1c3gsFahsg%3D%3D Occurrence Handle9543638
Acknowledgements
The authors deeply appreciate the outstanding support of Wei Li who documented TRP localization with confocal microscopy. This work was supported by NIH grant EY04795 (PR).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yang, H., Sun, X., Wang, Z. et al. EGF Stimulates Growth by Enhancing Capacitative Calcium Entry in Corneal Epithelial Cells . J. Membrane Biol. 194, 47–58 (2003). https://doi.org/10.1007/s00232-003-2025-9
Received:
Issue Date:
DOI: https://doi.org/10.1007/s00232-003-2025-9