
Abstract
Purpose
We aimed to investigate the regulatory approval of drugs for cancers by the US Food and Drug Administration based on the cancer type (major vs. minor), including the use of expedited development programs and duration from Investigational New Drug application (IND) to marketing approval.
Methods
From publicly available records and through a Freedom of Information Act request, we gathered data to evaluate regulatory characteristics and pivotal study design for 115 anticancer drug approvals between 2012 and 2017 and the data were analyzed based on cancer incidence (major vs. minor cancers) and how expedited programs, orphan drug designation, and pivotal study design contribute to expedited approval was studied.
Results
Drugs targeting minor cancers more frequently (67%; P = 0.0155) utilized breakthrough therapy designation and/or accelerated approval, both of which significantly contributed to expedited drug approval (median time from IND to approval, 6.4 years; P = 0.0008, 6.2 years; P < 0.0001). Drug approvals for pivotal study design without a comparator arm took significantly less time from IND to approval (median time from IND to approval, 6.2 years; P < 0.0001).
Conclusions
Drugs targeting minor cancers have frequently utilized the expedited development programs; thus, efficiently shortening time to approval. As many of such drugs are approved based on non-comparative pivotal studies, meticulous evaluation and follow-up should be performed for such drugs after their approval.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The United States (US) Food and Drug Administration (FDA) has implemented several programs to facilitate and expedite the development and review of new drugs to address the unmet medical need in treating serious or life-threatening conditions. These programs include fast track designation (FT), breakthrough therapy designation (BT), accelerated approval (AA), and priority review designation (PR) [1]. The FDA recognizes that specific aspects of drug development that are feasible for common diseases may not be feasible for rare diseases and development challenges are often greater with the increasing rarity of diseases. Therefore, various incentives are granted for drugs that meet specific requirements to reduce the development burden. Since cancer is a life-threatening disease, especially when diagnosed at a late stage, it is speculated that many anticancer drugs can utilize expedited development programs for their development. A previous report revealed that 95% of newly approved anticancer drugs between 2012 and 2017 in the US have utilized one of the expedited development programs and obtained marketing approval [2].
The Orphan Drug Act (ODA) provides a special status, upon request from a sponsor, to a drug or biologic for the treatment of a rare disease or condition that affects < 200,000 persons in the US [3]. A previous report found that 56% of newly approved anticancer drugs between 2004 and 2010 were designated as orphan drugs. We can therefore assume that the orphan drug designation program has been widely utilized for the development of anticancer drugs [4].
Given the considerable time and cost required to develop pharmaceutical products, drugs with greater marketability may be prioritized for development. Therefore, the development of drugs for rare cancers (< 6 cases per 100,000 individuals) may be deprioritized compared with drugs for non-rare cancers. It remains unknown, however, whether drugs for minor cancers have benefited from the expedited development programs in development and marketing approvals.
We aimed to investigate the characteristics for the regulatory approval of drugs for cancers based on cancer type (major vs. minor), including the use of expedited development programs and the duration from Investigational New Drug application (IND) (submission to the FDA for approval to initiate human clinical studies in the US) to marketing approval.
Methods
Sample identification
We identified drugs approved for efficacy of cancer indications including both New Drug Application (NDA)/Biological License Application (BLA) and supplemental NDA (sNDA)/supplemental BLA (sBLA) approved by the FDA between January 1, 2012, and December 31, 2017, using the FDA’s Drugs@FDA database and Approved Cellular and Gene Therapy Products from the website of the FDA (https://www.fda.gov/Drugs/default.htm). We excluded drugs whose approval was for diseases that were not cancers, supportive or palliative care, a new treatment line, or pediatric approval in an existing indication, and approvals not for efficacy.
Data collection
We extracted the dates for IND from the Federal Register (FR; https://www.federalregister.gov/). For drugs whose IND dates were not available from FR, the dates were searched using documents such as medical review, correspondence review, pharmacology review, chemistry review, risk review, or others from the FDA’s website (Drugs@FDA and Approved Cellular and Gene Therapy Products). We used the date when the IND became effective as IND date; if such date was unavailable, IND submission or FDA receipt date, whichever was later, was used. When multiple IND dates with different IND numbers were available, we used the earliest date as IND date. For drugs for which only month and year, or only year was available, we treated the first day of the month or year as the IND date. As we could not identify the IND dates for 2 drugs (inotuzumab ozogamicin and copanlisib), they were excluded from the present study.
Using the database, we also collected information on the type of application (NDA/BLA/sNDA/sBLA) and its FDA receipt dates, indication, cancer type, and key clinical studies to evaluate efficacy (pivotal study), which are shown on the approved labels. We defined the receipt date by the FDA as the NDA/BLA/sNDA/sBLA date; if this date was unavailable, we used the submission date. If there were multiple dates for such dates, the earliest date was adopted. For each of the pivotal studies, study design information (comparative or non-comparative) was also collected.
We used publicly available lists and annual new drugs summary reports published by the FDA’s Center for Drug Evaluation and Research for orphan drug designation and the expedited development program (BT, AA, and/or PR) for all drugs. For FT, we obtained such information from the FDA through a Freedom of Information Act request, because no list was available after June 1, 2010, on the FDA’s website.
Incidence of cancer
We used SEER*Stat analytic software version 8.3.5 (http://seer.cancer.gov/resources/) in client-server mode to obtain cancer rates for each cancer type [5]. In the database, we selected 2000–2014 patient data with age-adjusted to the 2000 US standard population to obtain incidence rates [6]. For specific tumor type or biomarker characteristics, the distribution was obtained from published articles to estimate their incidences. These exceptions included basal cell carcinoma [7] (estimated without using SEER*Stat data), EGFR mutation–positive NSCLC [8], EGFR T790M mutation–positive NSCLC [8, 9], ALK-positive NSCLC [10], programmed cell death-ligand 1–positive NSCLC [11], ROS1-positive NSCLC [12], melanoma with BRAF mutation [13], ovarian cancer with BRCA mutation [14], microsatellite instability-high (MSI-H) or mismatch repair deficient solid tumors/colorectal cancer [15], acute myeloid leukemia (AML) with FLT3 mutation positive [16], and chronic lymphocytic leukemia (CLL) with 17p deletion [17]. Some cancers with specific populations or biomarker profiles with an incidence < 6 per 100,000 individuals prior to the selection of populations or biomarkers (e.g., pediatric patients with high-risk neuroblastoma, AML with an isocitrate dehydrogenase 2 (IDH2) mutation) were categorized as minor cancers (as defined below), without identifying their specific incidences.
For the definition of minor cancer in this study, we utilized the definition for rare cancer (those with an incidence of < 6 cases per 100,000) by International Rare Cancers Initiative (http://www.irci.info/). To investigate the regulatory characteristics and time from IND to approval by cancer incidence, cancer indicated for each approved drug was categorized by incidence as major (≥ 6 cases per 100,000 individuals per year) or minor (< 6/100,000).
Statistical methodology
To calculate time from IND to approval, the IND date was subtracted from the US approval date; review time by the FDA was calculated by subtracting the NDA/BLA/sNDA/sBLA date from the approval date.
Time from IND to approval in the US by cancer incidence (major vs. minor) was described using box-and-whisker plots featuring 25th percentile, median, and 75th percentile values. We compared time from IND to approval and review time in the US by cancer incidence (major vs. minor) between groups (use of expedited development programs (FT, BT, AA, PR, and combination of these), orphan drug designation status (yes or no), and pivotal trial design (comparative vs. non-comparative)) using the Mann–Whitney U test. All P values were based on a 2-sided hypothesis, and P values < 0.05 were considered statistically significant. For analytical calculations, we used StatsDirect software version 3.1.18 (StatsDirect Ltd., Cambridge, UK).
Results
Anticancer drugs investigated
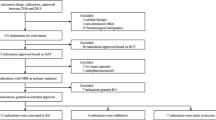
As shown in Fig. 1, we identified 115 approvals (NDA/BLA, 73; sNDA/sBLA, 42) for analysis, after excluding approvals for diseases that were not anticancer indications, new treatment lines within the existing indications, pediatric indications for which adult indications exist; approvals not granted for efficacy; approval under the Federal Food, Drug, and Cosmetic Act (FD&C Act) section 505(B)(2); and approvals for which IND was not identified. Three drugs (vincristin sulfate liposome injection, irinotecan liposome injection, and liposome-encapsulated combination of daunorubicin and cytarabine) approved under 505(B)(2) were included in the present study as they have been listed as recommended therapies for acute lymphoblastic leukemia [18], adenocarcinoma of the pancreas [19], and acute myeloid leukemia [20], respectively, in the National Comprehensive Cancer Network (NCCN) guidelines. Based on the drug approvals analyzed, 54 (NDA/BLA, 33; sNDA/sBLA, 21) and 61 approvals (NDA/BLA, 40; sNDA/sBLA, 21) were for major (22 types) and minor (42 types) cancers, respectively (Tables 1 and 2).

Anticancer drugs approved by the US FDA between January 1, 2012, and December 31, 2017
Expedited development programs and pivotal study design
Orphan drug designation was significantly more often employed for drugs targeting minor cancers than major cancers. As shown in Table 1, the proportion of approvals using AA, BT and/or AA, and BT and/or AA and/or PR was significantly greater in drugs for minor cancers compared with those for major cancers (97% vs. 78%; P = 0.003). Specifically, AA was significantly more often used for approvals of drugs targeting minor cancers than major cancers (48% vs. 24%; P = 0.0117).
Based on pivotal study design, significantly more pivotal studies were comparative studies for drugs treating major cancers compared with those treating minor cancers; more than half of these pivotal studies were non-comparative (proportion of non-comparative study, 24% vs. 56%; P = 0.0006).
Time from IND to approval
Figure 2 shows the time from IND to approval for anticancer drugs approved in the US between January 1, 2012, and December 31, 2017. The median time from IND to approval for the 115 identified approvals was 8.3 years. Median time from IND to approval was numerically shorter for drugs targeting minor cancers (n = 61, 7.8 years) when compared with those for major cancers (n = 54, 8.9 years); however, no significant difference in time from IND to approval was found. There was no difference in review time between major and minor cancers, and the median time was nearly identical (median, 181 vs. 182 days).

Time from IND to approval in the US by cancer incidence. The upper and lower boundaries of the central box indicate the first and third quartiles, respectively, with median marked with diamond. Whiskers indicate maximum (upper) and minimum (lower) values, unless an outlier is present, in which case whiskers indicate the lower or upper quartiles plus (upper) and minus (lower) 1.5 times the interquartile range (IQR); data points outside those boundaries are plotted as white circles (mild outlier, 1.5–3 IQR) or black circles (extreme outlier, > 3 IQR). Differences in time from IND to approval were evaluated using the Mann–Whitney U test
We analyzed time from IND to approval by the expedited development programs: FT, BT, AA, PR, BT and/or AA, and BT and/or AA and/or PR (Table 3). Time from IND to approval was significantly shorter when BT (median, 6.4 vs. 9.6 years; P = 0.0008), AA (median, 6.2 vs. 9.6 years; P < 0.0001), BT and/or AA (median, 6.7 vs. 10.2 years; P < 0.0001), and FT and/or BT and/or AA and/or PR (median, 8.0 vs. 11.4 years; P = 0.0368) were employed. These categories except FT and/or BT and/or AA and/or PR had a significantly shorter time to approval when the analyses were limited to new molecular entities/original BLAs (data not shown). Time to approval between orphan drugs and non-orphan drugs did not statistically differ.
We also analyzed time from IND to approval by pivotal study design (comparative vs. non-comparative study). The time was significantly shorter when a non-comparative study was used as the pivotal study for drug approval (median, 10.0 vs. 6.2 years; P < 0.0001).
Pivotal study design for AA
As AA was significantly more often used for drugs targeting minor cancers than major cancers, and independently significantly shortened the time from IND to approval, we further analyzed pivotal study design for drugs utilizing AA. Among 42 drug approvals that used AA, 37 approvals (88%) were granted based on non-comparative studies. In all such non-comparative pivotal studies, objective response rate (proportion of patients who had tumor burden reduction) was used as a primary endpoint.
Discussion
We found that time from IND to approval did not differ between drugs for major and minor cancers approved between 2012 and 2017; however, median time was numerically shorter for drugs targeting minor cancers. To our knowledge, this is the first study to investigate time from IND to approval of anticancer drugs by cancer incidence. We have also revealed that no difference exists in review time between minor and major cancer drugs. Therefore, the development time of drugs for minor cancers is not extended compared with that for major cancers, at least for approved drugs. It might be more time-consuming and expensive to conduct clinical studies for minor than major cancers. Fewer patients might be enrolled per investigational site and relatively more sites are required in clinical studies for minor cancers when compared with those for major cancers.
Expedited development programs (FT, BT, AA, and PR) were introduced by the FDA to expedite development and review of drugs to address unmet medical needs in the treatment of serious or life-threatening conditions. Cancer is a life-threatening disease; thus, in many cases, anticancer drugs are expected to be subjected to these programs. Previously, it was reported that 95% of newly FDA-approved drugs (NDA/BLA) utilized at least one of the expedited programs (FT, BT, AA, or PR) [2]. In the present study, we showed that 93% of anticancer drug approvals (NDA/BLA/sNDA/sBLA) from 2012 to 2017 utilized any or combination of FT, BT, AA, or PR. Furthermore, 97% of drug approvals for minor cancers utilized any or combination of BT, AA, or PR; therefore, expedited development programs are widely utilized in anticancer drug development, especially for drugs targeting minor cancers. According to a previous report, BT-designated cancer drugs were associated with faster time to approval (NDA/BLA) in the US; median time to first FDA approval was 5.2 years for BT-designated drugs versus 7.1 years for non-BT-designated drugs (difference, 1.9 years; P = 0.01) [2]. In our analysis, BT as well as AA and combined BT and/or AA significantly shortened time to approval (difference of median, 3.2, 3.5, and 3.5 years, respectively). We found that the development of anticancer drugs targeting minor cancers significantly more frequently (67%; P = 0.0155) utilized the expedited development programs that could potentially shorten the time from IND to approval (BT or AA); approximately 44% of drugs targeting major cancers utilized BT or AA. Furthermore, the pivotal study design for drugs targeting minor cancers more frequently (56%; P = 0.0006) used a non-comparative study design which also has the potential to shorten time to approval. This may contribute to reducing hurdles in developing drugs for minor cancers and lead to shortened development time. Specifically, a feature of AA is granting approval based on an effect on a surrogate endpoint or an intermediate clinical endpoint that is reasonably likely to predict a drug’s clinical benefit [1]. As shown in our study, 88% of approvals using AA were based on non-comparative studies with the endpoint of objective response rate, and this finding reflects the feature of AA. According to a previous report [4], pivotal trials for orphan drugs approved from 2004 to 2010 for cancer were more likely to use nonrandomized, non-blinded trial designs and surrogate end points to assess efficacy. In the present study, 85% of drugs targeting minor cancers received orphan drug designation, whereas 30% were for major cancers. The higher proportion of orphan designated drugs in minor cancer drugs may be responsible for the outcome of the previous report.
Expediting drug approvals means that approvals are given based on limited information, especially for safety information, compared with standard approvals. There may be a safety concern for drugs approved that used small studies; this has been identified and investigated in previous studies. It was reported that BT-designated drugs were less likely to act via a novel mechanism of action (36% vs. 39%; P = 1.00) [2]. Rates of deaths (6% vs. 4%; P = 0.99) and serious adverse events (38% vs. 36%; P = 0.93) were also similar in BT-designated and non-BT-designated drugs. Another report found that more treated patients had serious adverse events in trials of orphan drugs vs trials of non-orphan drugs (48% vs 36%; odds ratio, 1.72; 95% confidence interval, 1.02–2.92; P = 0.04) [4]. As patient population and the mechanism of action of investigational drugs differ for each clinical study, we cannot draw a definitive conclusion by comparing clinical studies of different populations and drugs. Therefore, a careful approach should be adopted in the development and review of drugs approved from small studies under the expedited development programs.
This study has some limitations. First, we excluded drugs approved for a different treatment line within the same indication or for pediatric use with existing adult indications; therefore, time to approval for such cases was not evaluated. Second, we tabulated approval numbers by cancer type based on information provided in “Indications and usage” sections. Therefore, some approvals were counted twice, even if both approvals were based on the same clinical study; our results may have differed if such cases were counted as one. Finally, for some cancer types such as basal cell carcinoma, MSI-H, or mismatch repair deficient solid tumor or colorectal cancer, the proportion of patients subjected to treatment with anticancer drugs was small, although they were categorized as major cancer based on their incidences.
In conclusion, we found that time from IND to approval for drugs targeting minor cancers did not significantly differ from those targeting major cancers. The median time was however numerically shorter, although a higher development hurdle may exist for drugs targeting minor than major cancers. These minor cancer drugs more frequently utilized the expedited development programs and pivotal studies without comparator arm, which may shorten their development time. The expedited development programs have been widely used for the development of anticancer drugs, especially those targeting minor cancers and have aided in the expedited approval of such drugs. However, given the limited information available from the evaluated clinical studies for some approvals, meticulous evaluation and follow-up are required to ensure that minor cancer drugs are continuously developed for patients with such conditions, without a serious safety and/or efficacy concerns following their approval.
References
US Food and Drug Administration. Guidance for industry. Expedited programs for serious conditions - drugs and biologics. https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm358301.pdf. Accessed 13 October 2018
Hwang TJ, Franklin JM, Chen CT, Lauffenburger JC, Gyawali B, Kesselheim AS, Darrow JJ (2018) Efficacy, safety, and regulatory approval of Food and Drug Administration–designated breakthrough and nonbreakthrough cancer medicines. J Clin Oncol 36(18):1805–1812
Pub. L. 97–414 (1983) codified as amended at 21 U.S.C. §§ 360aa - 360ee
Kesselheim AS, Myers JA, Avorn J (2011) Characteristics of clinical trials to support approval of orphan vs nonorphan drugs for cancer. JAMA 305(22):2320–2326
Surveillance Research Program, National Cancer Institute, SEER*Stat software version 8.3.5 (http://seer.cancer.gov/resources/)
Surveillance, Epidemiology, and End Results (SEER) Program (http://www.seer.cancer.gov) SEER*Stat Database. Incidence - SEER 18 Regs Research Data + Hurricane Katrina Impacted Louisiana Cases, Nov 2016 Sub (2000–2014) <Katrina/Rita Population Adjustment> − Linked To County Attributes - Total U.S., 1969–2015 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, released April 2017, based on the November 2016 submission
Goldenberg G, Karagiannis T, Palmer JB, Lotya J, O’Neill C, Kisa R, Herrera V, Siegel DM (2016) Incidence and prevalence of basal cell carcinoma (BCC) and locally advanced BCC (LABCC) in a large commercially insured population in the United States: a retrospective cohort study. J Am Acad Dermatol 75(5):957–966
Sequist LV, Joshi VA, Jänne PA et al (2007) Response to treatment and survival of patients with non-small cell lung cancer undergoing somatic EGFR mutation testing. Oncologist 12(1):90–98
Mok TS, Wu Y-L, Ahn M-J, Garassino MC, Kim HR, Ramalingam SS, Shepherd FA, He Y, Akamatsu H, Theelen WS, Lee CK, Sebastian M, Templeton A, Mann H, Marotti M, Ghiorghiu S, Papadimitrakopoulou VA, AURA3 Investigators (2017) Osimertinib or platinum–pemetrexed in EGFR T790M–positive lung cancer. N Engl J Med 376(7):629–640
Fan L, Feng Y, Wan H, Shi G, Niu W (2014) Clinicopathological and demographical characteristics of non-small cell lung cancer patients with ALK rearrangements: a systematic review and meta-analysis. PLoS One 9(6):e100866
Herbst RS, Baas P, Kim DW, Felip E, Pérez-Gracia JL, Han JY, Molina J, Kim JH, Arvis CD, Ahn MJ, Majem M, Fidler MJ, de Castro G Jr, Garrido M, Lubiniecki GM, Shentu Y, Im E, Dolled-Filhart M, Garon EB (2016) Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387(10027):1540–1550
Bergethon K, Shaw AT, Ou SH et al (2012) ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 30(8):863–870
Liu W, Kelly JW, Trivett M, Murray WK, Dowling JP, Wolfe R, Mason G, Magee J, Angel C, Dobrovic A, McArthur GA (2007) Distinct clinical and pathological features are associated with the BRAF(T1799A(V600E)) mutation in primary melanoma. J Invest Dermatol 127(4):900–905
Risch HA, McLaughlin JR, Cole DE et al (2001) Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet 68(3):700–710
Cortes-Ciriano I, Lee S, Park WY, Kim TM, Park PJ (2017) A molecular portrait of microsatellite instability across multiple cancers. Nat Commun 8:15180. https://doi.org/10.1038/ncomms15180
Levis M (2013) FLT3 mutations in acute myeloid leukemia: what is the best approach in 2013? Hematology Am Soc Hematol Educ Program 2013:220–226
Zenz T, Benner A, Döhner H, Stilgenbauer S (2008) Chronic lymphocytic leukemia and treatment resistance in cancer: the role of the p53 pathway. Cell Cycle 7(24):3810–3814
National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: acute lymphoblastic leukemia, version 1.2018. [Online]. Available: http://www.nccn.org. Accessed 30 September 2018
National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: pancreatic adenocarcinoma, version 2.2018. Available: http://www.nccn.org. Accessed 14 October 2018
National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: acute myeloid leukemia, version 2.2018. http://www.nccn.org. Accessed 30 September 2018
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Kenji Yamashita is an employee of MSD K.K. (a subsidiary of Merck & Co., Inc., Kenilworth, N.J., USA). Masayuki Kaneko declares that he has no conflict of interest. Mamoru Narukawa declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals.
Informed consent
For this study, a formal consent is not required.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yamashita, K., Kaneko, M. & Narukawa, M. Regulatory characteristics and pivotal study design of US Food and Drug Administration approval of drugs for major vs. minor cancer. Eur J Clin Pharmacol 75, 1193–1200 (2019). https://doi.org/10.1007/s00228-019-02695-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-019-02695-0