
Abstract
Purpose
This is the first study that connects pharmacokinetics of tolperisone with genetic polymorphism of the enzymes involved in its metabolism in human. We aimed to identify the influence of polymorphism of two main enzymes (CYP2D6 and CYP2C19) on pharmacokinetic profile of parent drug.
Methods
In a single-dose study, 28 healthy Caucasian male volunteers received an oral dose of 150 mg of tolperisone. The subjects were genotyped with respect to CYP2D6 and CYP2C19 enzymes. Plasma was sampled for up to 12 h post dose, followed by quantification of tolperisone by a fully validated HPLC-tandem mass spectrometry (MS/MS) method. The pharmacokinetic parameters were estimated using a non-compartmental method and compared statistically at level p < 0.05 across the genotyped groups.
Results
High variability (exceeded 100 %) of main bioavailability parameters (AUCt, AUCinf, Cmax) was observed in the whole group of subjects. An essential difference in the pharmacokinetics of tolperisone of quick metabolizers whose genotype expressed wild homozygote CYP2D6 *1/*1 with respect to heterozygous *1/*4 and *1/*5 subjects was demonstrated. The mean AUCinf was 2.1- and 3.4-fold higher in *1/*4 and *1/*5, respectively, than in *1/*1 subjects. In case of Cmax, the differences were greater and reached maximally 3.8 times (mean values 54.00, 98.85, and 205.20 ng/mL for CYP2D6 *1/*1, *1/*4, and *1/*5, respectively). Values of the parameters for the one subject that expressed *4/*4 genotype were even 8.5 times higher than in subjects with extensive or intermediate phenotype. Although CYP2C19 *1/*2 subjects had higher AUCt, AUCinf, and Cmax values than *1/*1, no statistically significant differences were observed. Oral clearance (CL/F) significantly decreased by 65.7 % in heterozygous *1/*2 relative to homozygous *1/*1 extensive metabolizers.
Conclusion
In this study, we first demonstrated the effect of CYP2D6 polymorphism on pharmacokinetics of tolperisone in Caucasian subjects. The contribution of CYP2C19 enzyme seems to be less important.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genetic picture of cytochrome P450’s hemoproteins which catalyze the biotransformation of various xenobiotics as well as metabolic state of individuals can vary largely even in the same population. These differences can influence significantly the bioavailability of many drugs and consequently induce essential pharmacokinetic, pharmacodynamic differences and efficacy, and toxicity between individuals [1–4]. Discovery and evaluation of pharmacogenetic differences in the drug response and tolerability are an essential concept of personalized medicine [5].
Data about the biotransformation of tolperisone, a centrally acting muscle relaxant [6], in human are limited. It is known that it is rapidly absorbed after oral administration, undergoes an extensive first pass effect, and only 20 % of an administered dose appears unchanged in the blood. The human organism oxygenates the methyl group of the aromatic ring to a hydroxymethyl group producing the main tolperisone metabolite [7]. The hydroxymethyl metabolite formation revealed to be the main P450-mediated metabolic pathway. CYP2D6 was identified as the key enzyme in the metabolism; however, CYP2C19, CYP2B6, and CYP1A2 are also involved to some extent [6].
The in vitro study [6] indirectly evidenced the substantial involvement of microsomal carbonyl reductase in the metabolism of tolperisone suggesting that the parent compound undergoes also the P450-independent microsomal biotransformation. No further supportive studies evaluating reductase role in tolperisone metabolism have been identified; therefore, investigators of the presented study worked on the assumption that tolperisone mainly undergoes CYP-dependent metabolism.
A few papers are available on the metabolic path of tolperisone [6–9], but to the best of our knowledge, no study has addressed the pharmacokinetic variability of the compound connected with genetic polymorphism of the enzymes involved in tolperisone metabolism in human so far.
The goal of the present study was to identify the influence of genetic polymorphism of two main biotransformation enzymes on pharmacokinetic profile of tolperisone following a single oral dose administration of 150 mg in healthy subjects. Our interest was focused on debrisoquine/sparteine monooxygenase CYP2D6 and S-mephenytoin hydroxylase CYP2C19.
The CYP2D6 was chosen as it was identified as the key enzyme in tolperisone metabolism, and it metabolizes a wide range of common drugs including antidepressants, antipsychotics, beta-blockers, and antiarrhythmics, which can generate drug-drug interaction. Approximately 5 % of Europeans lack CYP2D6 activity. Ninety-five percent of European poor metabolizers (PM) can be identified by screening the *3, *4, *5, and *6 alleles [10]. The remaining 5 % of PM are likely to be homozygous or heterozygous for a range of different inactive, relatively rare alleles.
The CYP2C19, enzyme of second importance in tolperisone biotransformation, revealed 60 % of CYP2D6 activity in both parent compound loss and main metabolite formation in vitro, but the role of the enzyme at “physiological” level may be less essential [6]. Frequency of the enzymatic deficiency associated with poor metabolism of (S)-mephenytoin is approximately 3 % [10, 11], and in Caucasian, it is connected mainly (80 %) with CYP2C19*2 allele; CYP2C19*3 is extremely rare in the population [11]. Significant ethnic differences can be observed in the frequencies of genotyped variants of *17 allele contributing to extensive, intermediate, or poor phenotypes [12].
Methods
Study design and study group
It was a single-dose study at which Mydocalm Forte® (Gedeon Richter) 150 mg film-coated tablets were administered in fasting condition with at least 250 mL of water.
The study protocol as well as other applicable documents had been approved by the local independent Institutional Review Board (IEC of District Council of Physicians, Baśniowa 3, Warsaw; resolution no. KB/596/07), and the trial was registered in EudraCT (no. 2007-003676-18). Each volunteer had been properly informed and signed an informed consent form.
The study was performed in a group of 28 healthy Caucasian males aged 27.3 ± 7.7 years, with BMI 23.3 ± 1.7 kg/m2. The volunteers have no significant diseases in medical history and no abnormalities in medical, biochemical, and hematological profiles and were not addicted to drugs and alcohol. All participants fulfilled standard inclusion/exclusion criteria for healthy subject studies. The criteria did not pertain to a genotype/phenotype assessment in advance; therefore, the genotyping was performed after the screening, for all 28 volunteers included in the study. The aim of the study was to find out how genotypes of cytochrome P450 enzymes often presented in a population and essential for tolperisone metabolism affect its pharmacokinetics in healthy volunteers. Therefore, the study was carried out on a random group, selected from a population of healthy men, expecting that the individual genotypic groups will differ in sample size and may not reflect the full spectrum of genotypes observed in the population.
The phenotype characteristics of the study group with respect to CYP2D6 and CYP2C19 enzymes are presented in Table 1. All volunteers had normal enzyme activity (EM) of CYP2C19 and represented the wild-type allele or were phenotypically extensive. With respect to CYP2D6, the group was more differential with poor, intermediate, and extensive phenotypes.
Genotyping
Real-time PCR with fluorescent detection (TaqMan® Drug Metabolism Genotyping Assays; Applied Biosystems, USA) was used. The test allowed detection of the following variants of the gene CYP2D6: allele *1 (wild type determining the normal enzyme activity), the alleles *3 and *4 conditioning lack of enzyme activity, and additional test evaluated the number of copies of the gene CYP2D6, i.e., the occurrence of additional alleles *1 (CYP2D6 *1 × N—multiplication) and CYP2D6 deletion of the whole gene (allele CYP2D6 *5). The analysis of CYP2C19 differentiation was based on wild allele *1 with normal activity and two most frequent alleles (*2 and *3) responsible for the lack of enzyme activity. Based on the results, all subjects have been divided into a few phenotypic groups, most often mentioned in the literature.
Sampling schedule
The sampling design took into account an expected rapid absorption of tolperisone and ensured a precise determination of pharmacokinetic parameters, including Cmax. Blood samples were collected at predefined protocol time points just before dosing and at 10, 20, 30, and 45 min and 1.0, 1.25, 1.50, 2.0, 2.50, 3.0, 4.0, 5.0, 6.0, 8.0, 10, and 12 h after drug administration.
Analytical method
Tolperisone plasma concentrations were determined using a fully validated liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) method.
For the sample preparation, 0.5 mL of human plasma were mixed with the internal standard (indapamide), shaked with organic phase n-heksan: tert-butyl methyl ether mixture (1/5, V/V), evaporated at 30 °C under a stream of nitrogen and reconstructed in methanol:water (1/1, V/V) solution. Chromatography was performed on Acquity UPLC BEH C8 1.7 μm 2.1 × 50 mm (Waters) column using a mobile phase of 0.05 % formic acid in methanol and 0.05 % formic acid in water (45/55, V/V). Quantification was performed using ES+ ionization (m/z 246.29→m/z 98.01 for tolperisone and m/z 366.07→m/z 132.08 for indapamide). The limit of quantification was 0.50 ng/mL; the calibration curve covers concentrations at range 0.50–200.00 ng/mL. The method meets all requirements referred to selectivity, precision, and accuracy.
Data analysis
Pharmacokinetic parameters were calculated using standard non-compartmental methods.
Area under the plasma concentration-time curve from point 0 to the last determined concentration (AUCt) was calculated by the linear trapezoidal method. AUCinf is a sum of AUCt and AUCrest (ratio of the last estimated concentration and elimination rate constant). The clearance (CL/F) was calculated from the ratio of oral dose and AUCinf.
Maximum concentration (Cmax) and the time to reach Cmax (Tmax) were determined by inspection of the plasma concentration-time curves. The elimination rate constant (k) was determined by a linear regression of the terminal portion of the log-concentration-time curve, and the apparent elimination half-life (t1/2) was calculated based on formula 0.693/k. Mean residence time (MRT) was calculated as ratio of area under the first-order moment curve (AUMC) and area under the zero-order moment curve (AUCinf).
As tolperisone absorption is fast, AUCpartial to the time 0.75 h when median Tmax occurs was calculated additionally.
Statistics
The group with the poor metabolizer of CYP2D6 had been excluded from the statistical evaluation due to the limited sample size.
Homogeneity of variance and distribution of continuous data were checked by Levene test and Kolmogorow-Smirnow test, respectively, and parametric or nonparametric tests were applied consequently when appropriate. The pharmacokinetic parameters were compared across the three or two genotyped groups within CYP2D6 and CYP2C19 groups, respectively, by one-way Anova and either nonparametric Kruskal-Wallis or Mann-Whitney test. A post hoc Duncan test or Dunn test was used to assess the presence of statistical differences between the genotyped groups when a statistically significant association was described by Anova or Kruskal-Wallis, respectively. For all analyses, a statistical significance was assumed when p < 0.05.
The parametric tests, as more powerful in their nature than their nonparametric counterparts, were followed by power calculation for detection of differences between the compared two groups [13]. The statistical evaluation was performed using software Statistica v.10.
Results
The calculated pharmacokinetic parameters for the whole study group revealed significant differences (Table 2, Supplementary Figures 1 and 2).
An essential difference in CYP2D6 *1/*1, *1/*4, *1/*5, and *4/*4 group is observed in mean plasma concentration-time curves of tolperisone as well as pharmacokinetic (PK) parameters (Fig. 1, Table 3).

Mean plasma concentration-time curves of tolperisone in CYP2D6 *1/*1, *1/*4, *1/*5, and *4/*4 groups (N = 28)
It has been demonstrated an essential difference in the tolperisone pharmacokinetics of quick metabolizers whose genotype expressed wild homozygote with respect to heterozygous subjects. CYP2D6 *1/*4 and *1/*5 subjects had higher (statistically significant in case of *1/*5) AUCt, AUCinf, and AUCpartial values than *1/*1 subjects. The same tendency was also observed for Cmax. However, the time-dependent parameters, i.e., Tmax, t1/2, and MRT, did not differ within the analyzed groups. The clearance that is directly related to the metabolic activity was the highest for homozygous subjects and perceptibly lower for heterozygotes; however, the differences within single pairs had not been proved with a statistical significance.
The AUCt, AUCinf, and Cmax values for the one subject that expressed *4/*4 genotype were essentially higher while the CL/F was essentially lower than the parameters in groups of subjects with extensive or intermediate phenotype.
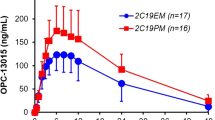
Although CYP2C19 *1/*2 subjects had higher AUCt, AUCinf, AUCpartial, and Cmax values than *1/*1, no statistically significant differences were observed in the subjects with these CYP2C19 genotypes (Table 4). The significant differences in metabolic activity in homo- and heterozygous extensive metabolizers of CYP2C19 are statistically proved by clearance. A huge variability of the tested parameters and plasma concentrations was confirmed (Table 4, Fig. 2)

Plasma concentration-time curves of tolperisone in CYP2C19 1/*1 and *1/*2 groups (mean ± SD) (N = 27)
Large differences in the power values for respective pharmacokinetic parameters were found within the compared genotypes. The maximum power in CYP2D6 analysis was calculated for AUCpartial comparing genotype *1/*1 and *1/*5 (80.22 %). In other cases, the power was below 80 %, e.g., for a key bioavailability parameter (AUCt), the power was 48.89, 74.30, and 30.68 % for the comparison of the genotypes *1/*1 vs. *1/*4, *1/*1 vs. *1/*5, and *1/*4 vs. *1/*5, respectively. Among CYP2C19, statistical analysis indicated the maximum power for AUCt comparison (54.98 %) and the smallest one for AUCpartial (15.48 %).
Discussion
In the presented study, high variability of tolperisone pharmacokinetic parameters was observed. The coefficients of variation of main bioavailability parameters (AUCt, AUCinf and Cmax) and parameter related to the metabolic activity (CL/F) were close to 100 % in the whole-tested group of Caucasian subjects. The high variability of PK parameters was indicated in some previous publications [14–16], but those studies were performed on Asian subjects. CV was close to 70 % in case of AUCinf and Cmax and about 30 % in case of Tmax, kel, and t1/2. Similar results were obtained from our study—the diversity is greater for AUCinf and Cmax (CV 106.59 and 94.67 %, respectively) than for the parameters associated with time (CV 54.09 % for Tmax, 46.05 % for t1/2). The disproportion within variability can result from a rapid absorption and distribution after oral administration immediate release formulation of tolperisone.
In the publication of Bae et al. [15], authors suggested that it is the genetic polymorphism of metabolizing enzymes that can be responsible for large variability in plasma concentration of tolperisone. Our observation reveals very variable pharmacokinetics on Caucasian subjects that is a result of phenotype variability of an evaluated population. The presented study proved an important variability demonstrated on the bioavailability parameters within genotype CYP2D6.
The metabolism of tolperisone in CYP2D6 *4/*4 subject was remarkably different from those of the other three groups. The homozygotes *1/*1 with normal metabolic activity of the enzyme differ with statistical confidence from heterozygotes *1/*5 with reduced activity. Mean AUCt, AUCinf, AUCpartial, and Cmax were essentially higher for IMs *1/*5 metabolizers compared with EMs group (increase by 243, 238, 278, and 280 %, respectively). The same tendency is observed in comparison with CYP2D6 *1/*1 and *1/*4; however, it could not be statistically proved. The clearance was 5.9- and 1.7-fold lower for heterozygotes *1/*5 and *1/*4, respectively, in relation to EMs *1/*1. The results indicated that CYP2D6 plays an important role in the pharmacokinetics of tolperisone. This observation is consistent with Dalmadi et al.’s [6] conclusion about the significant involvement of the enzyme CYP2D6 in the tolperisone metabolism.
The study group was not differentiated phenotypically widely with respect to CYP2C19, but it was decided to include this cytochrome to the analysis because its influence on other active substances’ pharmacokinetics in the homo- and heterozygous rapid metabolizers had been demonstrated [17–20].
In the homo EMs CYP2C19 group, mean AUCt reached 98.01 ng*h/mL compared with 191.46 ng*h/mL in hetero EMs group (increase by 95 %; similar data for AUCinf—increase by 92 %), and also Cmax was 70.89 and 115.44 ng/mL (increase by 63 %) between homo EMs and hetero EMs, respectively. These differences were not statistically significant. Nevertheless, it should be emphasized that clearance that is directly related to the metabolic activity was 2.9 times lower for hetero EMs and was found to be statistically significant.
The frequency of the PM phenotype of CYP2C19 and slow metabolizers in the Caucasian population is only at level 1–6 % [11]. The limitations of this study were that none of the individuals participated in the tolperisone kinetic study were PM ((*2–10, *12)/(*2–10, *12)) as well as no *17 variants were demonstrated, so no data were available, and comparisons could not have been done between the EMs and the others.
The findings obtained in this study indicate that CYP2C19-related pharmacogenetics is one of the causes of the interindividual variability in the plasma concentration of tolperisone in healthy men subjects. The data dispersion (Supplementary Figures 1 and 2) suggests that the CYP2C19 *1/*2 may increase the PK variability in the group of volunteers who are simultaneously homozygote *1/*1 with respect to CYP2D6. Due to the small number of respondents in each subgroup, this thesis cannot be statistically proved. Taking into account the allele frequencies for the white population, we can predict that metabolizers with genotype *1/*17 (EM) might have been at the level of about 27 % [12] thus partly contributing to the growth of the observed PK variability.
The pharmacokinetic differences observed in genetically variable groups may be associated with differences in the pharmacological effect and therefore in the effectiveness of the drug product. In order to achieve a satisfied therapeutic effect, one should pay attention not only to the appropriate dose selection for the particular disease, symptoms, and patient condition but also to the individual enzymatic profile related to biotransformation of a therapeutic compound. Results of the study confirm the suggestion in previous publications [15, 21] that the oral tolperisone hydrochloride therapy might need to be individualized due to a genetically variable metabolism.
Further studies performed on a larger population and taking into account a broader range of alleles within the key cytochrome P450 enzymes will be needed for a complete understanding of the impact of the phenotypic differentiation of CYPs on tolperisone biotransformation. It would be fruitful to determine, beside parent drug, the metabolites and include them into the future assessment.
References
Nagata K, Yamazoe Y (2002) Genetic polymorphism on human cytochrome P450 involved in drug metabolism. Drug Metabol Pharmacokinet 17(3):167–189
Zanger UM, Schwab M (2013) Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 138:103–141. doi:10.1016/j.pharmthera.2012.12.007
Callegari E, Kalgutkar AS, Leung L, Obach RS, Plowchalk DR, Tse S (2013) Drug metabolites as cytochrome p450 inhibitors: a retrospective analysis and proposed algorithm for evaluation of the pharmacokinetic interaction potential of metabolites in drug discovery and development. Drug Metab Dispos 41:2047–2055. doi:10.1124/dmd.113.052241
Daly AK (2012) Genetic polymorphisms affecting drug metabolism: recent advances and clinical aspects. Adv Pharmacol 63:137–167. doi:10.1016/B978-0-12-398339-8.00004-5
Howland RH (2012) Where are we today with personalized medicine? J Psychosoc Nurs Ment Health Serv 50(11):11–13. doi:10.3928/02793695-20121005-01
Dalmadi B, Leibinger J, Szeberenyi S, Borbas T, Farkas S, Szombathelyi Z, Tihanyi K (2003) Identification of metabolic pathways involved in the biotransformation of tolperisone by human microsomal enzymes. Drug Metab Dispos 31(5):631–636
Balint J, Hell Z, Markovits I, Parkanyi L, Fogassy E (2000) Synthesis and resolution of a tolperisone metabolite. Tetrahedron-Asymmetry 11:1323–1329
Balint J, Markovits I, Egri G, Tuza Z, Parkanyi L, Fogassy E (2001) Synthesis, resolution and absolute configuration of a tolperisone metabolite. Tetrahedron-Asymmetry 12:719–724
Balint J, Egri G, Markovits I, Czugler M, Marthi K, Demeter A, Temesvari-Takacs K, Fogassy E (2002) Synthesis of some tolperisone metabolites in racemic and optically active form. Tetrahedron-Asymmetry 12:3417–3422
Daly AK (2003) Pharmacogenetics of the major polymorphic metabolizing enzymes. Fundam Clin Pharmacol 17:27–41
Desta Z, Zhao X, Shin J-G, Flockhart DA (2002) Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin Pharmacokinet 41(12):913–958
Strom CM, Goos D, Crossley B, Zhang K, Buller-Burkle A, Jarvis M, Quan F, Peng M, Sun W (2012) Testing for variants in CYP2C19: population frequencies and testing experience in a clinical laboratory. Genet Med 14(1):95–100. doi:10.1038/gim.0b013e3182329870
Cohen J (1988) Statistical power analysis for the behavioral sciences, LEA Publishers
Bae JW, Park YS, Sohn UD, Myung CS, Ryu BK, Jang CG, Lee SY (2006) HPLC determination of tolperisone in human plasma. Arch Pharm Res 29(4):339–342
Bae JW, Kim MJ, Park YS, Myung CS, Jang CG, Lee SY (2007) Considerable interindividual variation in the pharmacokinetics of tolperisone HCl. Int J Clin Pharmacol Ther 45(2):110–113
Choi CI, Park JI, Lee HI, Lee YJ, Jang CG, Bae JW, Lee SY (2012) Determination of tolperisone in human plasma by liquid chromatography/tandem mass spectrometry for clinical application. J Chromatogr B Analyt Technol Biomed Life Sci 12(911):59–63. doi:10.1016/j.jchromb.2012.10.027
Ruan CJ, Liu DY, Jiang J, Hu P (2012) Effect of the CYP2C19 genotype on the pharmacokinetics of icotinib in healthy male volunteers. Eur J Clin Pharmacol 68(12):1677–1680. doi:10.1007/s00228-012-1288-4
Gawrońska-Szklarz B, Adamiak-Giera U, Wyska E, Kurzawski M, Gornik W, Kaldonska M, Drozdzik M (2012) CYP2C19 polymorphism affects single-dose pharmacokinetics of oral pantoprazole in healthy volunteers. Eur J Clin Pharmacol 68(9):1267–1274. doi:10.1007/s00228-012-1252-3
Bohanec Grabar P, Grabnar I, Rozman B, Logar D, Tomsic M, Suput D, Trdan T, Peterlin Masic L, Mrhar A, Dolzan V (2009) Investigation of the influence of CYP1A2 and CYP2C19 genetic polymorphism on 2-cyano-3-hydroxy-N-[4-(trifluoromethyl)phenyl]-2-butenamide (A77 1726) pharmacokinetics in leflunomide-treated patients with rheumatoid arthritis. Drug Metab Dispos 37(10):2061–2068. doi:10.1124/dmd.109.027482
Mamiya K, Hadama A, Yukawa E, Ieiri I, Otsubo K, Ninomiya H, Tashiro N, Higuchi S (2000) CYP2C19 polymorphism effect on phenobarbitone. Pharmacokinetics in Japanese patients with epilepsy: analysis by population pharmacokinetics. Eur J Clin Pharmacol 55(11–12):821–825
Quasthoff S, Mockel C, Zieglgansberger W, Schreibmayer W (2008) Tolperisone—a typical representative of a class of centrally acting muscle relaxants with less sedative side effects. CNS Neurosci Ther 14:107–119. doi:10.1111/j.1527-3458.2008.00044.x
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 34 kb)
Rights and permissions
About this article

Cite this article
Pawlowska, M., Bogiel, M., Duda, J. et al. Influence of CYP2D6 and CYP2C19 genetic polymorphism on the pharmacokinetics of tolperisone in healthy volunteers. Eur J Clin Pharmacol 71, 699–705 (2015). https://doi.org/10.1007/s00228-015-1856-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-015-1856-5