
Abstract
Purpose
As an inhibitor of HMG-CoA reductase that catalyses the first step of cholesterol synthesis, pitavastatin undergoes little hepatic metabolism; however, it is a substrate of uptake and efflux transporters. Since pitavastatin is potentially co-administered with agents that affect transporter activities, the pharmacokinetics of pitavastatin was investigated on the effects of a single-dose rifampin in healthy volunteers.
Methods
Twelve Chinese healthy male volunteers took 4 mg pitavastatin orally with 150 ml water or with a single dose of 600 mg rifampin on separate occasions and the plasma concentrations of pitavastatin were measured over 48 h by HPLC-MS/MS.
Results
A single dose of rifampin significantly increased the mean area under the plasma concentration-time curve(AUC)(0-48h) and Cmax of pitavastatin by 573.5 %(95%CI, 373.3–773.7 %, p < 0.001) and 819.2 %(95 % CI, 515.4–1123.0 %, p < 0.001) respectively, while significantly decreased the t1/2 and CL/F of pitavastatin by 38.8 % (95 % CI, 18.2–59.4 %, p < 0.001) and 81.4 % (95 % CI, 75.0–87.7 %, p < 0.001) respectively.
Conclusions
Co-administration of pitavastatin with a single dose of rifampin resulted in a significant increase in plasma levels of pitavastatin in Chinese healthy subjects.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pitavastatin is newly developed as a reductase (3-hydroxy-3-methylglutaryl-coenzyme) inhibitor and potent for the treatment of hyperlipidemia [1], Most of the bio-available fraction of an oral dose is excreted unchanged in the bile because of the minimal metabolism of pitavastatin, which is reabsorbed by the small intestine ready for enterohepatic recirculation [2]. The pharmacokinetics and LDL-C-lowering efficacy were significantly improved at low doses due to its characteristic structure, with heptenoate as the basic structure coupled with a core quinoline ring and side chains with fluorophenyl and cyclopropyl moieties inside [2]. Most statins are metabolized in part by one or more hepatic cytochrome P450 enzymes, leading to an increased potential for drug interactions and problems with certain foods, such as grapefruit juice [3]. Compared with other statins, pitavastatin could be diverted away from its metabolism by cytochrome P450(CYP)3A4, while only amall amount is insignificantly metabolized by CYP2C9 due to the cyclopropyl group in the molecular structure [2]. Although pitavastatin undergoes little hepatic metabolism, it is a substrate of uptake and efflux transporters. The hepatic uptake of pitavastatin is mediated by carriers, especially OATP1B1, which is encoded by the SLCO1B1 gene. Because the liver is a target organ of pitavastatin, OATP1B1 is responsible for both the pharmacodynamic action and clearance of pitavastatin [4].
Rifampin is a potent inhibitor of OATP1B1 [5]; it was reported to inhibit OATP1B1 and restrain the uptake of olmesartan, a substrate for OATP1B1 in oocytes [6]. OATP1B1 plays an important role in the transport of rifampin, the SLCO1B1*15 haplotype was found to associated with susceptibility to cholestatic or mixed injury in patients treated with rifampin [7]. Recent reports also have demonstrated clinically relevant interactions of rifampin with numerous other drugs, such as warfarin [8], cyclosporine [9], ketoconazole [6, 10], digoxin [11], human immunodeficiency virus-related protease inhibitors, zidovudine, delavirdine mesylatee [12], tacrolimus [13], ondansetron hydrochloride and so on [14, 15]. The underlying mechanisms may be the influence of rifampin on the drug metabolism enzymes and the transporter, such as CYP3A4/CYP2C9 enzymes or OATP1B1 transporter [2], which were shared by other drugs simultaneously.
Since pitavastatin and rifampin were all transported by OATP1B1, there may be a significant drug–drug interactions (DDIs) when two drugs are used at the same time, the consequences of DDIs would be very different due to the combinations the two drugs; single-dose and multiple doses of rifampin would result in different influences on the pharmacokinetics of pitavastatin due to the completely different mechanisms. The current research was mainly carried on the effect of a single-dose rifampin on the pharmacokinetics of pitavastatin in healthy volunteers, and provided the first direct evidence for DDIs between pitavastatin and a single dose of rifampin.
Materials and methods
Materials and reagents
Pitavastatin calcium standard reference (Lot: 0502–1, purity: 99.5 %) and pitavastatin calcium tablets (specification: 2 mg/tablet, batch number: 04pr09028, Kowa Company Ltd, Japan) were provided by Zhejiang Haizheng Parmaceutical Co., LTD. Rifampin capsule (specification: 0.15 g/capsule, batch number: H20102905, Shanghai Yan’an Pharmaceutical Factory), the internal standard rosuvastatin reference (Lot:147098-20-2, purity:99.5 %) was purchased from Shanghai Secco Chemical Technology Co., LTD. HPLC-grade ammonium formate, formic acid, acetonitrile and methanol were purchased from Chemical Reagent Factory of Hunan (Changsha, Hunan, China). Ultrapure water was produced by water purification system (Aquapro Co.., LTD, Shanghai, China).
Subjects
Twelve unrelated healthy adult men (mean ± SD: age 24.1 ± 1.9 years; mass index 25.6 ± 4.3 kg/m2) were recruited for the study. Ethical approval for the study protocol was given by the Xiangya Ethics Committee of Central South University, Changsha, Hunan, China. This clinical trial was registered with the Chinese Clinical Trial Registry (No. ChiCTR-TRC-10001277). All informed consents were signed before the experiment. Participants were healthy with no clinically relevant conditions identified from medical history, physical examinations, electrocardiogram and routine laboratory tests (blood chemistry, hematology and urine analysis). All subjects refrained from the use of any prescription or nonprescription medication 2 weeks before and throughout the study. They also abstained from taking grapefruit juice, apples, onions, red wines, herbal dietary supplements, caffeine-containing beverages including coffee, green tea, cola and chocolate or any other medications 2 weeks before the study and during the study period. The volunteers were served standard meals and were monitored during the experimental period for the development of any possible adverse effects.
Study design
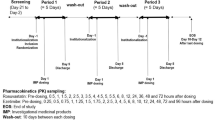
The study had a randomized crossover design with two periods that were separated by a 2-week washout period. During the study periods, the volunteers received administrations of either 4 mg pitavastatin calcium, or 4 mg pitavastatin calcium in combination with 600 mg rifampin capsule swallowed by 150 ml water, then a series venous blood samples of 5 ml were collected into EDTA-containing tubes at 0, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, 24 and 48 h. After the washout period, the experiment was carried out by crossover. The plasma samples were separated by centrifugation and immediately stored in polypropylene tubes at −40 °C until analysis.
HPLC-MS/MS method for pitavastatin
Concentrations of pitavastatin in plasma samples were determined using validated liquid chromatography with tandem mass spectrometric detector (HPLC-MS/MS) method. Samples were rapidly thawed under ambient temperature. A volume of 200 μl of the plasmas samples were precipitated by adding 400 μl acetonitrile contained internal standard solution (100 ng/ml rosuvastatin), and vortex mixed for 10 min, after centrifugation for 10 min at 7,280 g, 200-μl upper phases were removed to sample bottles, and 20-μl volume of aliquots were injected into the analytical column. HPLC-MS/MS analysis was performed by liquid chromatography-mass spectrometry with the Finnigan LCQ Deca XP plus (Thermo Finnigan, San Jose, CA). A Waters Cosmosil Packed Coulmn C18-MS-II(150 mm × 2.0 mm, 5 μm) and a mobile phase(acetonitrile/10 mM ammonium formate and 0.1 % formic acid = 60/40) at a flow rate of 0.3 ml/min were applied. The ion transitions monitored were as follows: m/z 422.1 to 290.4 for pitavastatin, and m/z 482.1 to 446.2 for rosuvastatin. These transitions represent the product ions of the [M + H]+ ions. The lower limit of quantification(LOQ) for pitavastatin was 0.5 ng/mL. The calibration curves were linear over the range 0.5 to 1,000 ng/ml. The intra- and inter-day coefficients of variation were less than 15 %. The standard deviation for quality control samples(1, 25, 800 ng/ml pitavastatin) in analysis batches were all less than 15 %, and the results of method validation have proven to be specific, precise and repetitive.
Pharmacokinetics analysis
Pitavastatin was analyzed by noncompartmental pharmacokinetic method using DAS software (Drug and Statistics, Version 2.0, Chinese Pharmacological Society, Beijing, China). The peak plasma concentration (C max) and time to peak plasma concentration (T max) were directly obtained from the observed concentration-time data. The measured concentrations of samples after Cmax below the LOQ were as unavailable (NA), and the samples before C max below the LOQ were as zero. The area under the plasma concentration-time curve (AUC) from time zero to last measured concentration above LOQ(AUC(0−t)) was calculated according to the linear trapezoidal rule. The terminal elimination rate constant (k) was estimated by linear regression of the terminal portion of the In(concentration)-time curve, and the elimination half-life (t 1/2) was calculated as 0.693/k accordingly, the estimate of oral clearance(CL/F) was calculated as CL/F =Dose (4 mg)/AUC(0–t) (ng · h/ml).
Statistical analysis
Statistical analysis was performed by the SPSS software for Windows (version 11.5, SPSS, Chicago, IL). Pharmacokinetic parameters of AUC(0–t), AUC(0-infinity), t 1/2, C max, T max and CL/F for pitavastatin with and without co-treatment of rifampin were analyzed by paired-samples t test, p < 0.05 was considered statistically significant.
Results
The main pharmacokinetic parameters of pitavastatin with and without co-treatment of rifampin are summarized in Table 1. Mean plasma concentration profiles for pitavastatin with and without co-treatment of rifampin are shown in Fig. 1; it can be seen that subject has higher plasma exposure to pitavastatin after concomitant administration with rifampin. Co-treatment of rifampin significantly increased the C max of pitavastatin by 819.2 % (95 % C I, 515.4–1123.0 %, p < 0.001) and the AUC(0–48 h) by 573.5 % (95 % CI, 373.3–773.7 %, p < 0.001). Co-treating with rifampin, the CL/F of pitavastatin was significantly decreased by 81.4 %(95 % CI, 75.0–87.7 %, p < 0.001) compared with non co-treatment of rifampin. Meanwhile, rifampin decreased the t 1/2 of pitavastatin by 38.8 % (95 % CI, 18.2–59.4 %, p < 0.001) with no significant influence on the T max of pitavastatin in comparison to non co-treatment of rifampin phase. Intra-subject changes in the AUC(0–48 h), Cmax, CL/F and t 1/2 of pitavastatin are depicted in Fig. 2.

Plasma concentration(conc)-time profiles of pitavastatin in 12 healthy subjects after a single oral dose of 4 mg pitavastatin with and without co-treatment of 600 mg rifampin. Inset depicts the same data on a semilogarithmic scale. Open circles indicate without co-treatment of rifampin phase; solid circles indicate co-treatment of rifampin phase, Each value is the mean value ± SD

Comparison of the AUC(0–48), Cmax, T1/2 and CL/F in 12 healthy subjects after a single oral dose of 4 mg pitavastatin with and without co-treatment of 600 mg rifampin. **p < 0.01
Discussion
Rifampin has numerous well documented, clinically relevant drug interactions associated with its use [15]. Rifampicin usually generated drug interactions associated with its delivery method; continuous dosing, and single dose rifampicin with other drugs often has very different results in DDIs, because rifampin could both induce hepatic enzymes and inhibits uptake transporters [16, 17]. Dosing a drug that is a dual substrate of enzymes and uptake transporters may have different pharmacokinetics when co-administrated with rifampicin, so continuous dosing of rifampicin often need to increase the dose of other drugs [10, 11, 18–22], while a single dose of rifampicin often result in a higher blood concentration of other drugs [6, 12, 16, 23]. the underlying mechanism may due to the balance between the induction on the hepatic enzymes and inhibition on uptake transporters by rifampicin [13]. Rifampin co-administered with pitavastatin at multiple doses or continuous using usually have dual functions on drug metabolic enzymes induction and transporter inhibition, if the induction on the hepatic enzymes was more than the inhibition on uptake transporters, rifampin would accelerate the metabolism of pitavastatin, otherwise the metabolic process will be suppressed. When rifampin co-administered with pitavastatin at a single dose, the inhibition on uptake transporters was often more than the induction on the hepatic enzymes, the metabolic process of pitavastatin would be inhibited, so different delivery method of rifampin would produce a very different effect on the pharmacokinetics of pitavastatin.
According to the reports, pitavastatin undergoes little hepatic metabolism, but it is a substrate for uptake and efflux transporters, particularly OATP1B1(gene SLCO1B1) [2, 3]. Factors that affect the activity of OATP1B1, such as SLCO1B1*15 genetic variation, can affect the metabolism of pitavastatin [3, 4, 24–27], also different substrates of OATP1B1 can affect the pharmacokinetics of pitavastatin via the competition mechanism [3]. Rifampin was reported to cause a lot of drug interactions with other statins such as simvastatin due to the competitive influence on the OATP1B1 [15], so the DDI between pitavastatin and rifampin was likely to occur.
In the present study, we found a significant increase in the AUC(0–t) and AUC(0–infinity) for pitavastatin when the drug was taken with rifampin, which may due to an effect of rifampin on the drug transporter expressed in the intestine. Because the liver is a target organ of pitavastatin, OATP1B1 is responsible for both the pharmacological effects and clearance of pitavastatin [4]. The CL/F for pitavastatin was significantly decreased when co-treated with rifampin that reflected the effect of rifampin on the drug transporter in liver especial for OATP1B1. The study also show a significant increase in C max for pitavastatin with co-treatment of rifampin and this result suggest that rifampin change the disposition of pitavastatin by altering its absorption and elimination through its effect on both intestinal uptake and efflux transporters. Rifampin significantly decreased the blood elimination half-life of pitavastatin, and this result suggest that rifampin may induce the activity of enzymes such as CYP3A4 or CYP2C9 that accelerate the metabolism of pitavastatin in the elimination phase. The present study may reveal that rifampin has multiple effects on drug enzymes and transporters simultaneously, and for the absorption phase of pitavastatin, the effect of rifampin on drug transporters play a principal position and mask the induction on the activities of drug metabolism enzymes, while their positions were inversed in the elimination phase of pitavastatin, which needs further study.
Although the probability of co-administration of rifampin and pitavastatin was low in clinic, rifampicin represents a class of agents that could act on the OATP1B1, and thus DDIs could happen when co-administrating these drugs with pitavastatin. Subtherapeutic microdoses of rifampicin producing great drug interactions with atorvastatin was reported in a previous study, which indicates that hepatic uptake via OATPs makes the dominant contribution to the hepatic elimination, so pitavastatin co-administrated with rifampicin should be avoided at the same time. If the co-administration was inevitable, the first dose of pitavastatin should be reduced. If the co-administration was a long-term, adjusting the dosage of pitavastatin was required according to the blood drug concentration monitoring. Certainly, in similar with other research reports, the static mechanistic models was another way to predict the dosing time-dependent pharmacokinetic interactions of pitavastatin with rifampicin [28, 29], which needed further study to provide evidence. All in all, the current research firstly provided the direct evidence for DDI between pitavastatin and rifampin when two drugs were used at the same time, due to the clinical relevance of a large number of DDIs, and whenever pitavastatin and these agents are prescribed, it is prudent to check for DDIs carefully, as optimal management is required for safe and efficacious therapy.
References
Chung JY, Cho JY, Yu KS, Kim JR, Oh DS, Jung HR, Lim KS, Moon KH, Shin SG, Jang IJ (2005) Effect of OATP1B1 (SLCO1B1) variant alleles on the pharmacokinetics of pitavastatin in healthy volunteers. Clin Pharmacol Ther 78:342–350
Saito Y (2011) Pitavastatin: An overview. Atheroscler Suppl 12:271–276
Hu M, Mak VW, Yin OQ, Chu TT, Tomlinson B (2012) Effects of grapefruit juice and SLCO1B1 388A > G polymorphism on the pharmacokinetics of pitavastatin. Drug Metab Pharmacokinet
Choi CI, Lee YJ, Lee HI, Kim BH, Kim MJ, Jang CG, Bae JW, Lee SY (2012) Effects of the SLCO1B1*15 allele on the pharmacokinetics of pitavastatin. Xenobiotica 42:496–501
Anderson MS, Cote J, Liu Y, Stypinski D, Auger P, Hohnstein A, Rasmussen S, Johnson-Levonas AO, Gutstein DE (2013) Effects of rifampin, a potent inducer of drug-metabolizing enzymes and an inhibitor of OATP1B1/3 transport, on the single dose pharmacokinetics of Anacetrapib. J Clin Pharmacol
Choi MK, Jin QR, Choi YL, Ahn SH, Bae MA, Song IS (2011) Inhibitory effects of ketoconazole and rifampin on OAT1 and OATP1B1 transport activities: Considerations on drug-drug interactions. Biopharm Drug Dispos 32:175–184
Li LM, Chen L, Deng GH, Tan WT, Dan YJ, Wang RQ, Chen WS (2012) SLCO1B1 *15 haplotype is associated with rifampin-induced liver injury. Mol Med Rep 6:75–82
Frymoyer A, Shugarts S, Browne M, Wu AH, Frassetto L, Benet LZ (2010) Effect of single-dose rifampin on the pharmacokinetics of warfarin in healthy volunteers. Clin Pharmacol Ther 88:540–547
Bruderer S, Aanismaa P, Homery MC, Hausler S, Landskroner K, Sidharta PN, Treiber A, Dingemanse J (2012) Effect of cyclosporine and rifampin on the pharmacokinetics of macitentan, a tissue-targeting dual endothelin receptor antagonist. AAPS J 14:68–78
Stroh M, Palcza J, McCrea J, Marsilio S, Breidinger S, Panebianco D, Johnson-Levonas A, Kraft WK, Orford K, Murphy G, Agrawal N, Trucksis M, Wagner JA, Iwamoto M (2012) The effect of multiple doses of rifampin and ketoconazole on the single-dose pharmacokinetics of ridaforolimus. Cancer Chemother Pharmacol 69:1247–1253
Greiner B, Eichelbaum M, Fritz P, Kreichgauer HP, von Richter O, Zundler J, Kroemer HK (1999) The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest 104:147–153
Kirby BJ, Collier AC, Kharasch ED, Whittington D, Thummel KE, Unadkat JD (2012) Complex drug interactions of the HIV protease inhibitors 3: Effect of simultaneous or staggered dosing of digoxin and ritonavir, nelfinavir, rifampin, or bupropion. Drug Metab Dispos 40:610–616
Abdel Halim M, Al-Otaibi T, Gheith O, El-Kholy O, Abdel Tawab K, Said T, Nair P, Nampoory MR (2010) Toxic tacrolimus blood levels with rifampin administration in a renal transplant recipient. Ann Transplant 15:57–60
Finch CK, Chrisman CR, Baciewicz AM, Self TH (2002) Rifampin and rifabutin drug interactions: An update. Arch Intern Med 162:985–992
Baciewicz AM, Chrisman CR, Finch CK, Self TH (2013) Update on rifampin, rifabutin, and rifapentine drug interactions. Curr Med Res Opin 29:1–12
Lam JL, Shugarts SB, Okochi H, Benet LZ (2006) Elucidating the effect of final-day dosing of rifampin in induction studies on hepatic drug disposition and metabolism. J Pharmacol Exp Ther 319:864–870
Paine MF, Wagner DA, Hoffmaster KA, Watkins PB (2002) Cytochrome P450 3A4 and P-glycoprotein mediate the interaction between an oral erythromycin breath test and rifampin. Clin Pharmacol Ther 72:524–535
Giessmann T, Modess C, Hecker U, Zschiesche M, Dazert P, Kunert-Keil C, Warzok R, Engel G, Weitschies W, Cascorbi I, Kroemer HK, Siegmund W (2004) CYP2D6 genotype and induction of intestinal drug transporters by rifampin predict presystemic clearance of carvedilol in healthy subjects. Clin Pharmacol Ther 75:213–222
Gurley BJ, Swain A, Williams DK, Barone G, Battu SK (2008) Gauging the clinical significance of P-glycoprotein-mediated herb-drug interactions: comparative effects of St. John’s wort, Echinacea, clarithromycin, and rifampin on digoxin pharmacokinetics. Mol Nutr Food Res 52:772–779
Hamman MA, Bruce MA, Haehner-Daniels BD, Hall SD (2001) The effect of rifampin administration on the disposition of fexofenadine. Clin Pharmacol Ther 69:114–121
Kim KA, Park PW, Liu KH, Kim KB, Lee HJ, Shin JG, Park JY (2008) Effect of rifampin, an inducer of CYP3A and P-glycoprotein, on the pharmacokinetics of risperidone. J Clin Pharmacol 48:66–72
Naesens M, Kuypers DR, Streit F, Armstrong VW, Oellerich M, Verbeke K, Vanrenterghem Y (2006) Rifampin induces alterations in mycophenolic acid glucuronidation and elimination: implications for drug exposure in renal allograft recipients. Clin Pharmacol Ther 80:509–521
Oswald S, Giessmann T, Luetjohann D, Wegner D, Rosskopf D, Weitschies W, Siegmund W (2006) Disposition and sterol-lowering effect of ezetimibe are influenced by single-dose coadministration of rifampin, an inhibitor of multidrug transport proteins. Clin Pharmacol Ther 80:477–485
Ieiri I, Suwannakul S, Maeda K, Uchimaru H, Hashimoto K, Kimura M, Fujino H, Hirano M, Kusuhara H, Irie S, Higuchi S, Sugiyama Y (2007) SLCO1B1 (OATP1B1, an uptake transporter) and ABCG2 (BCRP, an efflux transporter) variant alleles and pharmacokinetics of pitavastatin in healthy volunteers. Clin Pharmacol Ther 82:541–547
Deng JW, Song IS, Shin HJ, Yeo CW, Cho DY, Shon JH, Shin JG (2008) The effect of SLCO1B1*15 on the disposition of pravastatin and pitavastatin is substrate dependent: The contribution of transporting activity changes by SLCO1B1*15. Pharmacogenet Genomics 18:424–433
Oh ES, Kim CO, Cho SK, Park MS, Chung JY (2012) Impact of ABCC2, ABCG2 and SLCO1B1 polymorphisms on the pharmacokinetics of pitavastatin in humans. Drug Metab Pharmacokinet
Zhou Q, Chen QX, Ruan ZR, Yuan H, Xu HM, Zeng S (2013) CYP2C9*3(1075A > C), ABCB1 and SLCO1B1 genetic polymorphisms and gender are determinants of inter-subject variability in pitavastatin pharmacokinetics. Pharmazie 68:187–194
Varma MV, Lin J, Bi YA, Rotter CJ, Fahmi OA, Lam JL, El-Kattan AF, Goosen TC, Lai Y (2013) Quantitative prediction of repaglinide-rifampicin complex drug interactions using dynamic and static mechanistic models: delineating differential CYP3A4 induction and OATP1B1 inhibition potential of rifampicin. Drug Metab Dispos 41:966–974
Maeda K, Ikeda Y, Fujita T, Yoshida K, Azuma Y, Haruyama Y, Yamane N, Kumagai Y, Sugiyama Y (2011) Identification of the rate-determining process in the hepatic clearance of atorvastatin in a clinical cassette microdosing study. Clin Pharmacol Ther 90:575–581
Acknowledgements
Thanks to the grants from China Postdoctoral Science Foundation(2013 M531817), Young Teachers Boost Plans of Chinese Central College (grant #201012200047), Hunan Provincial Natural Science Foundation of China (Key Project, 10JJ2008) and 863 Project(No. 2012AA02A518) for financial support.
Declaration of interest
The authors report no conflicts of interest.
Funding source
This work was supported by research grants from China Postdoctoral Science Foundation(2013 M531817), Young Teachers Boost Plans of Chinese Central College (grant #201012200047), Hunan Provincial Natural Science Foundation of China (Key Project,10JJ2008) and 863 Project(No. 2012AA02A518).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chen, Y., Zhang, W., Huang, Wh. et al. Effect of a single-dose rifampin on the pharmacokinetics of pitavastatin in healthy volunteers. Eur J Clin Pharmacol 69, 1933–1938 (2013). https://doi.org/10.1007/s00228-013-1554-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-013-1554-0