
Abstract
Purpose
Interactions between ticagrelor and atorvastatin or simvastatin were investigated in two-way crossover studies.
Methods
Both studies were open-label for statin; the atorvastatin study was placebo-controlled for ticagrelor. For atorvastatin, volunteers (n = 24) received ticagrelor (loading dose 270 mg; 90 mg twice daily, 7 days) or placebo, plus atorvastatin calcium (80 mg; day 5). For simvastatin, volunteers (n = 24) received simvastatin 80 mg, or ticagrelor (loading dose 270 mg; 180 mg twice daily, 7 days) plus simvastatin (80 mg; day 5). In each study, volunteers received the alternate treatment after washout (≥7 days).
Results
Ticagrelor increased mean atorvastatin maximum plasma concentration (Cmax) and area under the plasma concentration-time curve from zero to infinity (AUC) by 23 % and 36 %, respectively. Simvastatin Cmax and AUC were increased by 81 % and 56 % with ticagrelor. Ticagrelor also increased Cmax and AUC of analysed atorvastatin metabolites by 13–55 % and 32–67 %, respectively, and simvastatin acid by 64 % and 52 %, respectively. Co-administration of ticagrelor with each statin was well tolerated.
Conclusions
Exposure to ticagrelor and its active metabolite, AR-C124910XX, was generally unchanged by a single dose of either statin, except for a minor increase in ticagrelor Cmax in the presence of simvastatin. Effects of ticagrelor on atorvastatin pharmacokinetics were modest and unlikely clinically relevant, while with simvastatin, changes were slightly larger, and simvastatin doses >40 mg with ticagrelor should be avoided.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ticagrelor is a reversibly binding adenosine diphosphate P2Y12 receptor antagonist that, unlike the thienopyridine antiplatelet agent, clopidogrel, does not require metabolic activation to exert its antiplatelet effect [1–4]. The clinical benefits of ticagrelor over standard treatment with clopidogrel have recently been demonstrated in the phase 3 PLATelet inhibition and Outcomes (PLATO) trial, where treatment with ticagrelor vs. clopidogrel significantly reduced the rate of the composite of myocardial infarction, stroke, and death from vascular causes [5]. Furthermore, ticagrelor was associated with a similar rate to clopidogrel of overall major bleeding, but was associated with a higher rate of major bleeding not related to coronary artery bypass grafting; dyspnoea and ventricular pauses also occurred more frequently in patients treated with ticagrelor [5]. Based on these findings, ticagrelor is now approved in more than 70 countries, including the EU [6] and the USA [7], for the reduction of thrombotic events in patients with acute coronary syndromes (ACS). The clinical regimen for ticagrelor is a single 180 mg loading dose, then 90 mg twice daily thereafter [6, 7].
Ticagrelor is rapidly absorbed and exhibits linear and predictable pharmacokinetics over a wide dose range [8–10]. It is metabolised to the major metabolite AR-C124910XX; this metabolite is also active, and present in the plasma at a concentration approximately 30 % that of the parent compound [3, 11, 12]. Cytochrome P450 (CYP) 3A4 and 3A5 are the enzymes predominantly responsible for the metabolism of ticagrelor to AR-C124910XX [13]. In vitro metabolism studies in human liver microsomes have shown that ticagrelor is a substrate and weak inhibitor of CYP 3A [13], suggesting a potential for drug interactions with other CYP 3A substrates. In these studies, complex interactions were observed between ticagrelor and different CYP 3A substrates [13], as would be expected due to well documented substrate-dependent interactions. Ticagrelor partially inhibited testosterone hydroxylation (23–30 % at 5–50 μM ticagrelor) and inhibited midazolam 4-hydroxylation (IC50 8.2 μM), markers of CYP3A activity [13].
Conditions that are often co-morbid with ACS result in patients receiving multiple medications post-ACS [14, 15]. The cholesterol-lowering drugs, statins, are one such class widely used by patients with ACS, because of their ability to significantly reduce cardiovascular risk [16–20]. For example, atorvastatin (10–80 mg once daily) [21] and simvastatin (5–80 mg once daily) [22] are prescribed for the treatment of hypercholesterolaemia. Indeed, in the PLATO trial, of the 18,624 ACS patients, 51 % received atorvastatin and 44 % received simvastatin post-randomisation (AstraZeneca, data on file).
Atorvastatin and simvastatin are both metabolised by CYP3A4 and CYP3A5 [23–25], although CYP3A5 plays a lesser role in the metabolism of atorvastatin [25]. Given the role of the CYP3A enzyme family in the metabolism of both ticagrelor and several statins [13, 23, 24], together with the likelihood of their combination in clinical practice, two studies were carried out early in the clinical development programme to investigate the effects of co-administration of ticagrelor and atorvastatin or simvastatin in healthy volunteers. The primary objectives of the present studies were to examine the pharmacokinetics of each statin when administered in the presence and absence of ticagrelor. The effects of each statin on the pharmacokinetics of ticagrelor, as well as safety and tolerability during co-administration, were also assessed.
Methods
Study populations
Inclusion criteria were: age 18–45 years; weight at least 50 kg; body mass index (BMI) 18–30 kg/m2; normal physical and laboratory examination results; and if female, postmenopausal or surgically sterile. Key exclusion criteria included a history of hypersensitivity or adverse reactions to dicalcium phosphate and lactose excipients; history of myopathy; co-administration of known CYP3A4/5 inhibitors and inducers within 30 days previously; known allergy to statins or history of myositis with statins; previous complications during statin therapy; and use in the previous 2 weeks of aspirin, ibuprofen, or other drugs known to increase the propensity for bleeding.
Since a drug interaction was predicted based on in vitro data [13], the sample sizes were calculated such that any interaction would be fully characterised, i.e. the studies were powered to show a lack of interaction. Sample sizes were based on between-subject coefficients of variation for area under the plasma concentration-time curve from zero to infinity (AUC) and maximum plasma concentration (Cmax) in healthy volunteers receiving 40 mg atorvastatin calcium or simvastatin [26, 27].
For the simvastatin study, assuming an intrasubject correlation of 0.65 for AUC and Cmax, similar variability assumptions and log-normal distribution of data, 16 evaluable volunteers would provide at least 80 % power that 90 % confidence intervals (CIs) for the Cmax ratio would be contained within the range 0.7–1.43, if there was no clinically significant interaction.
For the atorvastatin study, assuming an intrasubject correlation of 0.5, similar variability assumptions and log-normal distribution of data, 20 evaluable volunteers would provide at least 80 % power that 95 % CIs for Cmax ratio would be contained within the range 0.7–1.43, if there was no clinically significant interaction. In this study, two-sided 95 % CIs were used to reflect an adjustment for a planned interim analysis that was performed to ascertain if the trial should be extended (randomising further volunteers) or stopped. As the 95 % CI was not contained within the limits of 0.7–1.43, recruitment of additional volunteers was not required.
All volunteers provided written, informed consent. The final protocols were approved by independent institutional review boards (atorvastatin study: Southern Institutional Review Board, Miami, Florida, USA; simvastatin study: Heartland Institutional Review Board, Lenexa, Kansas, USA). The studies were performed in accordance with the Declaration of Helsinki, and were consistent with International Conference on Harmonisation/Good Clinical Practice guidelines, AstraZeneca’s bioethics policy, and applicable regulatory requirements.
Design and treatment
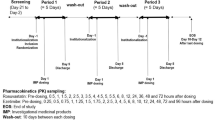
Both studies (D5130C00025 [atorvastatin] and D5130C00024 [simvastatin]) were randomised, two-period, two-way crossover studies (Fig. 1). In each study, volunteers received each of two treatments over two periods, crossing over to the alternate treatment after the washout period. Follow-up visits were scheduled 4–7 days after the end of the last treatment period.

Study designs. T ticagrelor, S simvastatin, A atorvastatin, bid twice daily
In the atorvastatin study, given the side-effect profile of statins and their potential for muscle-related adverse events (AEs), use of ticagrelor was placebo controlled in order to assess the safety of the co-administered combination more objectively. Patients were enrolled between May and June 2005, after the phase 3 study dose (90 mg twice daily) had been selected; this ticagrelor dose is now used clinically [6, 7]. Ticagrelor and placebo treatments were blinded to both investigator and volunteer; however, the study was open label with regard to the statin. The atorvastatin study consisted of two sequential 9-day periods, with a washout period of 7–10 days between treatments. Volunteers remained in the clinical research unit from day −1 until discharge on day 8 of each study period. Patients were randomised (1:1) sequentially to receive initially either ticagrelor or placebo (Fig. 1). After a single loading dose of ticagrelor (270 mg) or placebo, patients received either ticagrelor 90 mg twice daily or placebo twice daily for 7 days. On day 5, patients received a single dose of atorvastatin 80 mg. After the washout period, volunteers were crossed over to receive the other regimen (ticagrelor or placebo). For the atorvastatin study, the blinding remained unbroken for planned analyses until all data evaluability had been established.
The simvastatin study was all open label. Volunteers were randomised (1:1) to receive initially either ticagrelor for 7 days with a single dose of simvastatin on day 5 or just a single simvastatin 80-mg dose on day 5. After a washout period of ≥7 days, volunteers were crossed over to receive the other regimen. The ticagrelor regimen consisted of a single loading dose of ticagrelor (270 mg) followed by ticagrelor 180 mg twice daily for 7 days (Fig. 1). Ticagrelor 180 mg twice daily was used, as this study preceded the phase 2 DISPERSE2 study [2] in which the 90-mg twice-daily dosing regimen was established.
For both studies, all study medications were taken with 240 ml of room-temperature water. All volunteers had standardised meals that were identical for each study period, and no other food was permitted. During the treatment periods, no consumption of alcohol, caffeine-containing products, Seville oranges or grapefruit-containing products was permitted.
Pharmacokinetic sampling
During collection of blood samples, volunteers fasted overnight prior to dosing and until 4 h postdosing; water consumption was restricted.
For the atorvastatin study, blood samples for ticagrelor and AR-C124910XX analyses were collected predose (0) and 0.25, 0.5, 1, 2, 3, 4, 6, 8, 10, 12 h postdose on days 4 and 5. Predose samples were also collected on days 1 and 3. Following atorvastatin dosing on day 5, blood samples for analysis of atorvastatin and its metabolites (atorvastatin lactone, 2-hydroxy atorvastatin and 4-hydroxy atorvastatin) in plasma were collected predose (0) and 0.25, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 18, 24, 36, 48 and 72 h postdose.
For the simvastatin study, blood samples for ticagrelor/AR-C124910XX analyses were collected predose (0) and 0.5, 1, 2, 3, 4, 6, 8, 10, 12 h postdose on days 4 and 5, and additionally at 18, 24, 36, 48 and 72 h postdose following the day 5 dose. Blood samples for analysis of simvastatin and the metabolite simvastatin acid were collected predose (0) and 0.25, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 18, 24, 36, 48 and 72 h postdose.
For all analyses, 2-ml venous blood samples were collected into lithium-heparin tubes and kept on ice. Plasma was prepared within 30 min by centrifugation (1500 g, 10 min, 4 °C), and stored at −20 °C.
Following protein precipitation, reversed phase chromatography and tandem mass spectrometry were used to determine plasma concentrations of atorvastatin and the metabolites atorvastatin lactone, 2-hydroxy atorvastatin and 4-hydroxy atorvastatin (lower limit of quantification [LLOQ] for all compounds 0.25 ng/ml), simvastatin and simvastatin acid (LLOQ 0.25 ng/ml), and ticagrelor and AR-C124910XX (LLOQ 5.0 and 2.5 ng/ml, respectively) [28].
Safety assessments
Safety and tolerability were assessed throughout the studies. As mentioned above, in the atorvastatin study, ticagrelor was placebo-controlled to better characterise the safety profile of co-administered ticagrelor and atorvastatin. Information on serious AEs (SAEs) was collected from the time of informed consent until 14 days after the last dose of study drug (atorvastatin study) or the follow-up visit (simvastatin study). Non-serious AEs were monitored during both studies, from the time of first drug administration to the follow-up visit.
Data analyses
Pharmacokinetic parameters were estimated by standard non-compartmental methods, using WinNonlin Professional (Pharsight Corporation, Mountain View, California, USA).
Steady-state pharmacokinetic parameters estimated for ticagrelor and AR-C124910XX were Cmax, time to Cmax (tmax), area under the plasma concentration-time curve from zero to 12 h (AUC0–12), and metabolite–parent Cmax and AUC0–12 ratios.
Single-dose pharmacokinetic parameters estimated for atorvastatin and simvastatin, and their respective metabolites, were Cmax, tmax, half-life (t½) and AUC. t½ was calculated as 0.693/λz, where λz was the terminal elimination rate constant calculated by log-linear regression of the terminal portion of the concentration-time profile. AUC was calculated using the linear trapezoidal method up to the last measurable concentration and thereafter by extrapolation of the terminal elimination phase to infinity.
Statistical analyses were performed using SAS Version 8.0 (SAS Institute, Cary, North Carolina, USA). Pharmacokinetic data were summarised descriptively by treatment. Following log-transformation, Cmax and AUC data were analysed by analysis of variance fitting terms for period, sequence, and treatment. Volunteer within sequence was included as a random effect in the models. Geometric least squares mean ratio point estimates and CIs (95 % for the atorvastatin study; 90 % for the simvastatin study) for the difference in treatments were constructed using within-volunteer error terms. The no clinically significant effect limits were 0.80–1.25 for 95 % CIs for the effect of ticagrelor on atorvastatin, and for the effect of atorvastatin on ticagrelor and AR-C124910XX pharmacokinetic parameters. The no clinically significant effect limits were 0.80–1.25 for 90 % CIs for the effect of ticagrelor on simvastatin, and vice versa.
Results
Demographics and baseline characteristics
Of the 24 volunteers randomised and treated in the atorvastatin study, 19 (79 %) were male; 18 were Hispanic (75 %), five (21 %) were black, and one (4 %) was Caucasian. Mean (standard deviation [SD]) age was 32.5 (8.8) years and mean (SD) BMI was 24.8 (2.9) kg/m2. Of the 24 volunteers randomised and treated in the simvastatin study, 18 (75 %) were male, 14 (58 %) were Caucasian, and 10 (42 %) were black. Mean (SD) age was 28.8 (8.3) years and mean (SD) BMI was 25.6 (2.5) kg/m2. Three and four volunteers discontinued the atorvastatin and simvastatin studies, respectively. No concomitant medication was taken during the atorvastatin study. During the simvastatin study, six volunteers took concomitant medication preapproved by the investigator, and considered unlikely to have a major effect on the pharmacokinetic data.
Pharmacokinetic findings
Atorvastatin
The atorvastatin median tmax was unchanged during co-administration with ticagrelor (Table 1). However, relative to administration with placebo, Cmax and AUC of atorvastatin were increased in the presence of ticagrelor (Table 1, Fig. 2a): Cmax increased by 23 % and AUC by 36 %, with 95 % CIs outside the no clinically significant effect limits of 0.80–1.25 for each parameter (Table 1). When ticagrelor and atorvastatin were co-administered, considerable interindividual variability in exposure to atorvastatin was observed. However, this variability was no greater than that observed with atorvastatin alone (coefficients of variation for Cmax and AUC were 60.9 % and 59.3 %, respectively).

Mean (± standard deviation) plasma concentrations of (a) atorvastatin, (b) atorvastatin lactone, (c) 2-hydroxy atorvastatin, and (d) 4-hydroxy atorvastatin, over time following administration of a single dose of atorvastatin 80 mg with ticagrelor 90 mg (n = 21) or placebo (n = 21)
Increases in Cmax, AUC and area under the plasma concentration-time curve from zero to time t (AUC0–t) were observed for the metabolites atorvastatin lactone (39 %, 32 %, 33 %, respectively), 2-hydroxy atorvastatin (13 %, 33 %, 35 %, respectively), and 4-hydroxy atorvastatin (Cmax: 55 %; AUC0–t: 67 %), following co-administration with ticagrelor (Table 1, Fig. 2b–d). However, in general, tmax and t½ of the atorvastatin metabolites were unchanged during ticagrelor co-administration (Table 1, Fig. 2b–d).
Simvastatin
Simvastatin tmax and t½ values were similar with or without co-administration of ticagrelor (Table 1). In addition, compared with administration alone, simvastatin Cmax and AUC were increased by 81 % (90 % CI: 1.49–2.21) and 56 % (90 % CI: 1.30–1.87), respectively, during co-administration with ticagrelor (Table 1, Fig. 3a). There was considerable interindividual variability in the magnitudes of the changes in pharmacokinetic parameters when ticagrelor was co-administered with simvastatin, but variability was no greater than that observed with simvastatin alone. Although the mean increases in Cmax and AUC were 81 % and 56 % respectively, in some individual volunteers 2–3-fold increases in the Cmax and AUC of simvastatin were observed (Fig. 4).

Mean (± standard deviation) plasma concentrations of (a) simvastatin and (b) simvastatin acid, over time following administration of a single dose of simvastatin 80 mg alone (n = 20) or with ticagrelor 180 mg (n = 20)

Range of fold-changes in AUC and Cmax of a single dose of simvastatin 80 mg following administration alone or with ticagrelor 180 mg
Relative to administration of simvastatin alone, little change was observed in tmax and t½ for simvastatin acid during ticagrelor co-administration (Table 1, Fig. 3b). Similar to simvastatin, Cmax and AUC for simvastatin acid were increased in the presence of ticagrelor by 64 % (90 % CI: 1.38–1.95) and 52 % (90 % CI: 1.30–1.78), respectively (Table 1, Fig. 3b).
Ticagrelor and AR-C124910XX
Steady-state plasma concentration-time profiles of ticagrelor and AR-C124910XX were similar after treatment with ticagrelor alone and in combination with either atorvastatin or simvastatin (Fig. 5). Relative to administration of ticagrelor alone, no clinically significant changes in the pharmacokinetic parameters of ticagrelor and AR-C124910XX were observed during co-administration with either atorvastatin or simvastatin, except for a 14 % increase in ticagrelor Cmax following co-administration with simvastatin; the geometric mean ratio for ticagrelor Cmax was 1.14 (90 % CI: 1.00–1.30), with the upper confidence limit marginally outside the no clinically significant effect limits (Table 2).

Mean (± standard deviation) plasma concentrations of ticagrelor (a and b) and AR-C124910XX (c and d) over time following administration of ticagrelor alone (90 mg twice daily for a and c; 180 mg twice daily for b and d) or with a single dose of atorvastatin 80 mg (a and c; n = 21) or simvastatin 80 mg (b and d; n = 20)
Safety and tolerability
Study medication was generally well tolerated in both studies. One volunteer discontinued due to atrial fibrillation in the simvastatin study. This event was considered mild and since the volunteer had a history of palpitations with exercise prior to enrolling in the study, it may have predated the study. In the same study, one volunteer experienced an SAE 8 days after the last dose of study medication: chronic mediastinitis considered moderate and not treatment-related.
All other AEs were mild and the majority had resolved before the end of the studies. Two volunteers receiving ticagrelor and atorvastatin had ecchymoses (classified as minor bleeding), which were considered treatment-related. Three volunteers receiving ticagrelor and simvastatin had dyspnoea (two events considered treatment related) and four experienced one minor bleeding event each (epistaxis, cephalhaematoma, haematuria [considered treatment-related], and ecchymosis).
Discussion
Since ticagrelor is a weak inhibitor of CYP 3A in vitro, and several statins that are commonly administered in ACS are also metabolised by CYP 3A, the nature of a possible pharmacokinetic interaction with ticagrelor was investigated early in the clinical development programme. Our findings showed that ticagrelor increased the plasma exposures of simvastatin and atorvastatin, whereas the pharmacokinetic parameters of ticagrelor and AR-C124910XX during co-administration with atorvastatin or simvastatin were generally unchanged relative to the administration of ticagrelor alone, except for a minor increase in ticagrelor Cmax in the presence of simvastatin, considered to be of minimal clinical relevance despite lying outside the no clinically significant effect limits.
Co-administration of ticagrelor with atorvastatin resulted in an increase in the AUC of atorvastatin by a mean of 36 % and Cmax by a mean of 23 %. In addition, the AUC and Cmax of the metabolites of atorvastatin were also increased by 13–67 %. Following co-administration of ticagrelor and simvastatin, the magnitude of the interaction was greater than with atorvastatin, with mean increases in simvastatin AUC and Cmax of 56 % and 81 %, respectively. Increases in the simvastatin acid metabolite were also observed (52–64 %) following co-administration. Given the significance of CYP3A4 in the metabolic pathway of atorvastatin and simvastatin [29, 30], it is considered that the increased exposure to atorvastatin and simvastatin and their respective metabolites during co-administration with ticagrelor is mostly due to inhibition of CYP3A4 by ticagrelor. This is concordant with previously reported data showing that other CYP3A4 inhibitors, such as itraconazole and cyclosporine, increase exposure to atorvastatin and simvastatin when co-administered [31, 32]. A similar effect could also be expected when ticagrelor is co-administered with lovastatin, another substrate for CYP3A [33].
However, the role of organic anion transporting polypeptide (OATP) 1B1 in these interactions cannot be ruled out. Indeed, many statins have been shown to be substrates of OATP1B1, including atorvastatin and simvastatin acid [34]. Itraconazole is also a substrate for OATP1B1, and cyclosporine is an inhibitor [34], which could at least in part explain the increased exposure to atorvastatin and simvastatin when these agents are co-administered. Gemfibrozil is also an inhibitor of OATP1B1 [34]. However, in the absence of data regarding the effect of ticagrelor on OATP1B1 in vitro, the role of OATP1B1 in the interaction between ticagrelor and atorvastatin and simvastatin cannot be further elucidated with the observations in these two studies.
Although the ethnicity of the enrolled volunteers was different in the atorvastatin and simvastatin studies, the variability in the pharmacokinetics of both statins with and without ticagrelor is consistent with other findings. Previously, substantial intra- and interindividual variability were observed in the pharmacokinetics of these agents [35, 36]. Indeed, the pharmacokinetics of atorvastatin have been shown to be affected by several factors, including age, gender, and liver function [37, 38]. Furthermore, since CYP3A5 and P-glycoprotein contribute to atorvastatin and simvastatin metabolism [24, 39], interindividual variability of statin disposition may result from gene polymorphisms in CYP3A5 and ABCB1 (which encodes P-glycoprotein) [39, 40]. These polymorphisms may also help to explain the large interindividual variability in simvastatin exposure. In vitro studies have also shown that ticagrelor is a substrate and inhibitor of the P-glycoprotein transporter (AstraZeneca, data on file), of which atorvastatin and simvastatin are also substrates and inhibitors [30, 31, 39, 41]. Hence, ticagrelor’s effects on transport proteins may also contribute to the increased exposure to atorvastatin and simvastatin in the present study.
The simvastatin study was performed before the phase 2 study DISPERSE2 [2], in which the 90-mg twice-daily and 180-mg twice-daily dosing regimen [6, 7] were assessed and subsequently the 90-mg twice-daily regimen was selected for the phase 3 trial, PLATO. In contrast, the atorvastatin study was performed later in the development of ticagrelor than the simvastatin study, after the 90-mg twice-daily dosing regimen had been established. Thus, the interaction between ticagrelor and simvastatin was examined using ticagrelor 180 mg twice daily, whereas the interaction between ticagrelor and atorvastatin was examined using ticagrelor 90 mg twice daily. Therefore, compared with atorvastatin, the greater increase in exposure to simvastatin reported here may be partly due to the different doses of ticagrelor used in each study. It would be expected that the extent of interaction between ticagrelor and simvastatin would be smaller with ticagrelor 90 mg twice daily, than that observed in the current study with 180 mg twice daily. However, although the mean simvastatin exposure was only modestly increased in the presence of ticagrelor (180 mg twice daily), in some volunteers a greater than two-fold increase in simvastatin AUC was observed. This increase is unlikely to be explained solely by the use of a higher dose of ticagrelor. Atorvastatin is thought to be less reliant than simvastatin on CYP3A4 metabolism [30, 42]. This has previously been suggested as a reason for the relative difference in the impact of CYP3A inhibitors on the pharmacokinetics of atorvastatin vs. other statins [43]. The results of the present study are, therefore, consistent with these observations.
As only a single dose of each statin was administered to volunteers in the present studies, plasma levels of simvastatin or atorvastatin were not at steady state. Therefore, a clinically significant interaction, although unlikely, cannot be fully excluded on the basis of the present data. Aside from this limitation, these findings suggest that given atorvastatin’s favourable safety profile, the level of interaction reported here is considered unlikely to be clinically relevant. However, co-administration of ticagrelor with greater than 40 mg simvastatin could result in exposures exceeding those with 80 mg/day, which is the maximum prescribable dose of simvastatin [44, 45], and which the FDA has recently recommended be restricted. Since at the highest approved simvastatin dose of 80 mg there is an increased risk of muscle injury in patients taking the drug compared to patients taking lower doses of simvastatin [46], patients receiving ticagrelor should avoid simvastatin doses greater than 40 mg.
Conclusion
Co-administration of a single dose of atorvastatin or simvastatin with ticagrelor had little effect on the pharmacokinetic parameters of ticagrelor and AR-C124910XX, except for a minor increase in ticagrelor Cmax in the presence of simvastatin. Co-administration of ticagrelor with a single dose of atorvastatin increased the exposure to atorvastatin and its metabolites (AUC increased by 32–67 %), but only to a magnitude that is unlikely to be clinically relevant, given atorvastatin’s favourable safety profile. However, although co-administration of ticagrelor and a single dose of simvastatin resulted in an overall modest increase in simvastatin exposure (AUC increased by 52–60 %), variability in simvastatin concentrations was considerable, and in some individuals two-fold to three-fold increases in simvastatin exposure were observed. This finding suggests that instructing prescribers to avoid use of simvastatin doses >40 mg daily appears appropriate and sufficient. Indeed, since this study was performed, the FDA has recommended that the 80-mg dose of simvastatin be restricted to patients who have been taking simvastatin 80 mg for 12 months or more without evidence of muscle toxicity, irrespective of whether a patient is receiving ticagrelor [47].
References
Storey RF, Husted S, Harrington RA, Heptinstall S, Wilcox RG, Peters G, Wickens M, Emanuelsson H, Gurbel P, Grande P, Cannon CP (2007) Inhibition of platelet aggregation by AZD6140, a reversible oral P2Y12 receptor antagonist, compared with clopidogrel in patients with acute coronary syndromes. J Am Coll Cardiol 50:1852–1856
Cannon CP, Husted S, Harrington RA, Scirica BM, Emanuelsson H, Peters G, Storey RF, DISPERSE-2 Investigators (2007) Safety, tolerability, and initial efficacy of AZD6140, the first reversible oral ADP receptor antagonist, compared with clopidogrel, in patients with non–ST segment elevation acute coronary syndrome: primary results of the DISPERSE-2 trial. J Am Coll Cardiol 50:1844–1851
Husted S, van Giezen JJJ (2009) Ticagrelor: The first reversibly binding oral P2Y12 receptor antagonist. Cardiovasc Ther 27:259–274
van Giezen JJJ, Berntsson P (2008) AZD6140 displays over 100-fold higher affinity for the P2Y12 receptor vs AZ11702105, a chemical compound indistinguishable from the active metabolite of prasugrel, and is a more potent inhibitor of ADP-induced platelet aggregation. Arterioscler Thromb Vasc Biol 28:e139–e140
Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, Horrow J, Husted S, James S, Katus H, Mahaffey KW, Scirica BM, Skene A, Steg PG, Storey RF, For the Harrington RA for the PLATO Investigators (2009) Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 361:1045–1057
Brilique, summary of product characteristics (2010) http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001241/WC500100494.pdf. Accessed 8 November 2011
BrilintaTM, US full prescribing information (July 2011) http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/022433s000lbl.pdf. Accessed 8 November 2011
Butler K, Teng R (2010) Pharmacokinetics, pharmacodynamics, safety and tolerability of multiple ascending doses of ticagrelor in healthy volunteers. Br J Clin Pharmacol 70:65–77
Teng R, Butler K (2008) AZD6140, The first reversible oral platelet P2Y12 receptor antagonist, has linear pharmacokinetics and provides near complete inhibition of platelet aggregation, with reversibility of effect in healthy subjects. Can J Clin Pharmacol 15:e426
Teng R, Butler K (2010) Pharmacokinetics, pharmacodynamics, tolerability and safety of single ascending doses of ticagrelor, a reversibly binding oral P2Y(12) receptor antagonist, in healthy subjects. Eur J Clin Pharmacol 66:487–496
Husted S, Emanuelsson H, Heptinstall S, Sandset PM, Wickens M, Peters G (2006) Pharmacodynamics, pharmacokinetics, and safety of the oral reversible P2Y12 antagonist AZD6140 with aspirin in patients with atherosclerosis: a double-blind comparison to clopidogrel with aspirin. Eur Heart J 27:1038–1047
Teng R, Oliver S, Hayes MA, Butler K (2010) Absorption, distribution, metabolism, and excretion of ticagrelor in healthy subjects. Drug metabolism and disposition. Drug Metab Dispos 38:1514–1521
Zhou D, Andersson TB, Grimm SW (2011) In vitro evaluation of potential drug-drug interactions with ticagrelor: cytochrome p450 reaction phenotyping, inhibition, induction and differential kinetics. Drug Metab Dispos 39:703–710
Anderson JL, Adams CD, Antman EM, Bridges CR, Califf RM, Casey DE Jr, Chavey WE 2nd, Fesmire FM, Hochman JS, Levin TN, Lincoff AM, Peterson ED, Theroux P, Wenger NK, Wright RS, Smith SC Jr, Jacobs AK, Halperin JL, Hunt SA, Krumholz HM, Kushner FG, Lytle BW, Nishimura R, Ornato JP, Page RL, Riegel B (2007) ACC/AHA 2007 guidelines for the management of patients with unstable angina/non ST-elevation myocardial infarction. Circulation 116:e148–304
Task Force for Diagnosis and Treatment of Non-ST-Segment Elevation Acute Coronary Syndromes of European Society of Cardiology, Bassand JP, Hamm CW, Ardissino D, Boersma E, Budaj A, Fernández-Avilés F, Fox KA, Hasdai D, Ohman EM, Wallentin L, Wijns W (2007) Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. Eur Heart J 28:1598–1660
Scandinavian Simvastatin Survival Study Group (1994) Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian simvastatin survival study (4 S). Lancet 344:1383–1389
LaRosa JC, He J, Vupputuri S (1999) Effect of statins on risk of coronary disease: a meta-analysis of randomized controlled trials. J Am Med Assoc 282:2340–2346
Cannon CP, Braunwauld E, McCabe CH, Rader DJ, Rouleau JL, Belder R, Joyal SV, Hill KA, Pfeffer MA, Skene AM (2004) Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 350:1495–1504
Schwartz GG, Olsson AG, Ezekowitz MD, Ganz P, Oliver MF, Waters D, Zeiher A, Chaitman BR, Leslie S, Stern T, For the Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) Study Investigators (2001) Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study; a randomized controlled trial. J Am Med Assoc 285:1711–1718
Aronow HD, Topol EJ, Roe MT, Houghtaling PL, Wolski KE, Lincoff AM, Harrington RA, Califf RM, Ohman EM, Kleiman NS, Keltai M, Wilcox RG, Vahanian A, Armstrong PW, Lauer MS (2001) Effect of lipid-lowering therapy on early mortality after acute coronary syndromes: an observational study. Lancet 357:1063–1068
LipitorTM, summary of product characteristics (September 2011) http://www.medicines.org.uk/EMC/medicine/1424/SPC/Lipitor+10mg%2c+20mg%2c+40mg%2c+80mg+Tablets/. Accessed 8 November 2011
Zocor®, summary of product characteristics (March 2011). http://www.medicines.org.uk/EMC/medicine/1201/SPC/Zocor+10mg%2c+20mg%2c+40mg++and+80mg+film-coated+tablets/. Accessed 4 October 2011
Lennernäs H (2003) Clinical pharmacokinetics of atorvastatin. Clin Pharmacokinet 42:1141–1160
Prueksaritanont T, Gorham LM, Ma B, Liu L, Yu X, Zhao JJ, Slaughter DE, Arison BH, Vyas KP (1997) In vitro metabolism of simvastatin in humans: identification of metabolizing enzymes and effect of the drug on hepatic P450s. Drug Metab Dispos 25:1191–1199
Park JE, Kim KB, Bae SK, Moon BS, Liu KH, Shin JG (2008) Contribution of cytochrome P450 3A4 and 3A5 to the metabolism of atorvastatin. Xenobiotica 38(9):1240–1251
Lilja JJ, Kivistö KT, Neuvonen PJ (1999) Grapefruit juice increases serum concentrations of atorvastatin and has no effect on pravastatin. Clin Pharmacol Ther 66:118–127
Lilja JJ, Kivisto KT, Neuvonen PJ (1998) Grapefruit juice-simvastatin interaction: effects on serum concentration of simvastatin, simvastatin acid, and HMG-CoA reductase inhibitors. Clin Pharmacol Ther 64:477–483
Sillén H, Cook M, Davis P (2010) Determination of ticagrelor and two metabolites in plasma samples by liquid chromatography and mass spectrometry. J Chromatogr B Analyt Technol Biomed Sci 878:2299–2306
Bhindi R, Ormerod O, Newton J, Banning AP, Testa L (2008) Interaction between statins and clopidogrel: is there anything clinically relevant? Q J Med 101:915–925
Bottorff MB (2006) Statin safety and drug interactions: clinical implications. Am J Cardiol 97(Suppl):27C–31C
Bellosta S, Paoletti R, Corsini A (2004) Safety of statins: focus on clinical pharmacokinetics and drug interactions. Circulation 109(Suppl III):50–57, III
Jacobson TA (2004) Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am J Cardiol 94:1140–1146
Shitara Y, Sugiyama Y (2006) Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme a (HMG-CoA) reductase inhibitors: drug-drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol Ther 112:71–105
FDA Guidance for Industry (2012) Drug interaction studies – study design, data analysis, and implications for dosing and labelling recommendations. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf. Accessed 24 April 2012
Pasanen MK, Neuvonen M, Neuvonen PJ, Niemi M (2006) SLCO1B1 Polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet Genomics 16:873–879
Pasanen MK, Fredrikson H, Neuvonen PJ, Niemi M (2007) Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther 82:726–733
Gibson DM, Bron NJ, Richens A, Hounslow NJ, Sedman AJ, Whitfield LR (1996) Effect of age and gender on pharmacokinetics of atorvastatin in humans. J Clin Pharmacol 36:242–246
Narwal R, Akhlaghi F, Asberg A, Hermann M, Rosenbaum SE (2010) Development of a population pharmacokinetic model for atorvastatin acid and its lactone metabolite. Clin Pharmacokinet 49:693–702
Keskitalo JE, Kurkinen KJ, Neuvoneni PJ, Niemi M (2008) ABCB1 Haplotypes differentially affect the pharmacokinetics of the acid and lactone forms of simvastatin and atorvastatin. Clin Pharmacol Ther 84:457–461
Kim KA, Park PW, Lee OJ, Kang DK, Park JY (2007) Effect of polymorphic CYP3A5 genotype on the single-dose simvastatin pharmacokinetics in healthy subjects. J Clin Pharmacol 47:87–93
Bogman K, Peyer AK, Török M, Künsters E, Drewe J (2001) HMG-CoA reductase inhibitors and P-glycoprotein modulation. Br J Pharmacol 132:1183–1192
Kantola T, Kivisto KT, Neuvonen PJ (1998) Effect of itraconazole on the pharmacokinetics of atorvastatin. Clin Pharmacol Ther 64:58–65
Jacobsen W, Kuhn B, Soldner A, Kirchner G, Sewing KF, Kollman PA, Benet LZ, Christians U (2000) Lactonization is the critical first step in the disposition of the 3-hydroxy-3-methylglutaryl-coa reductase inhibitor atorvastatin. Drug Metab Disp 28:1369–1378
Mauro VF (1993) Clinical pharmacokinetics and practical applications of simvastatin. Clin Pharmacokinet 24:195–202
Merck Manuals online medical library: Simvastatin (2010) http://www.merckmanuals.com/professional/lexicomp/simvastatin.html. Accessed 8 August 2011
Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report (2002) Circulation 106:3143–3421
Merck Sharp & Dohme Corp (2010) Zocor (simvastatin) package insert. Whitehouse Station, NJ, USA. www.merck.com/product/usa/pi_circulars/z/zocor/zocor_pi.pdf. Accessed 8 November 2011
Acknowledgements
The authors would like to thank the principal investigators and the clinical research staff who took part in these studies. They also acknowledge the medical writing support by Patrick Hoggard (medical writer, Gardiner-Caldwell Communications, Macclesfield, UK). Funding to support this service was provided by AstraZeneca.
Author information
Authors and Affiliations
Corresponding author
Additional information
Sources of support
This phase 1 study was funded by AstraZeneca LP. Medical writing assistance was provided by Patrick Hoggard (medical writer, Gardiner-Caldwell Communications, Macclesfield, UK), Macclesfield, UK; financial assistance to support this service was provided by AstraZeneca LP.
Trial details
AZ study numbers: D5130C0025 (ticagrelor/atorvastatin); D5130C00024 (ticagrelor/simvastatin)
Rights and permissions
About this article
Cite this article
Teng, R., Mitchell, P.D. & Butler, K.A. Pharmacokinetic interaction studies of co-administration of ticagrelor and atorvastatin or simvastatin in healthy volunteers. Eur J Clin Pharmacol 69, 477–487 (2013). https://doi.org/10.1007/s00228-012-1369-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-012-1369-4