
Abstract
Purpose
Cholinesterase inhibitors are commonly prescribed to patients with Alzheimer’s disease (AD) to enhance cholinergic neurotransmission. Differential response to these treatments has been observed, and claims have been made that individual genetic variants may influence the pharmacokinetic and pharmacodynamic properties of these drugs. Here we assess the effects of genetic variation at two loci involved in the activity of cholinesterase inhibitors on longitudinal clinical change in AD patients being treated with donepezil, galantamine, and rivastigmine.
Methods
This was an open study in which 171 Italian AD patients treated with donepezil (n = 92), galantamine (n = 33), or rivastigmine (n = 46) were enrolled. Response to treatment was quantified by grading the patient’s cognitive state (Mini-Mental State Examination) and the patient’s ability to perform normal daily activities (Activities of Daily Living, Instrumental Activities of Daily Living) at baseline and after 6 and 12 months of treatment. Genetic variation was comprehensively characterized and analyzed at two loci: CYP2D6, which is involved in donepezil and galantamine metabolism, and BCHE, which codes for an enzyme (butyrylcholinesterase) which is both target and metabolizer of rivastigmine. APOE (coding for apolipoprotein E), which is associated with the risk of AD and inefficacy of specific AD treatments, was genotyped to control for patient stratification. The influence of the CYP2D6 and BCHE genotype on clinical changes after 12 months was evaluated by several tests of association.
Results
After 1 year of treatment, 29, 12, and 12 of the patients receiving donepezil, galantamine, and rivastigmine, respectively, showed a cognitive decrement, while eight patients interrupted the therapy before 12 months of treatment. No significant differences between the three treatments were observed in terms of response and tolerability. Non-responders show a higher proportion of BCHE and CYP2D6 mutated alleles, but genetic variation at the two loci was not a reliable predictor of clinical changes in AD patients treated with cholinesterase inhibitors.
Conclusions
Individualized therapy based on CYP2D6 and BCHE genotypes is unlikely to be beneficial for treating Alzheimer’s disease patients in routine clinical practice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) has become the most common neurodegenerative disease worldwide, representing 60–70 % of all dementia cases. This disease imposes a great burden and costs on caregivers and society alike [1]. In Italy, approximately 1.5% of the general population is estimated to suffer from AD [2]. AD is a progressive neurodegenerative disorder characterized by multiple cognitive deficits, including loss of memory, judgment, comprehension, and deterioration in global functioning accompanied by behavioral and mood disturbances.
Despite intense efforts to identify risk factors, implement preventive measures, and develop effective neuroprotection measures against AD, first-line treatment is still limited to temporary enhancement of cognitive abilities [3, 4]. The most commonly used group of agents consists of the cholinesterase inhibitors, which act by inhibiting the enzymes acetylcholinesterase (AChE) and, in some cases, butyrylcholinesterase (BChE), which are responsible for the breakdown of acetylcholine at the synapse and hence favor an enhancement of cholinergic transmission. In healthy brains, AChE accounts for the majority of cholinesterase activity, whereas BChE plays a minor role. Conversely, over the course of AD, BChE activity progressively increases and AChE partially loses its predominant role [5]. The action of cholinesterase inhibitors results in an accumulation of acetylcholine or prolongs its effects at the synapse. At the present time, the three most commonly prescribed cholinesterase inhibitors are donepezil, galantamine, and rivastigmine [6]. Donepezil and galantamine are reversible or short-acting inhibitors that primarily target AChE. Both are metabolized by two cytochrome P450 hepatic enzymes, CYP2D6 and CYP3A4. Rivatigmine is considered a “pseudo-irreversible” or intermediate-acting cholinesterase inhibitor that inhibits both AChE and BChE with equal potency. It is converted to an inactive metabolite at the site of action by cholinesterases, bypassing hepatic metabolic pathways [6].
Variable therapeutic response to cholinesterase inhibitors has been observed among AD patients, and the same is true for the occurrence of adverse effects and non-compliance. Some authors claim that one cause of differences in the efficacy and tolerability of cholinesterase inhibitors may be the presence of individual genetic variants that influence the pharmacokinetic and pharmacodynamic properties of these drugs [7]. If individuals carrying specific gene variants respond differently to—or tolerate differently—treatment with cholinesterase inhibitors, it would be possible to design individualized therapies for AD patients.
Only a few studies to date have focused on the relationship between individual genotypes and responses to cholinesterase inhibitors in Italian patients [8–11]. For participation in our study, we selected AD patients treated with the three most commonly prescribed cholinesterase inhibitors. We recorded cognitive changes and tolerability of the three drugs over a 12-month period and assessed patient genotypes for CYP2D6 locus, which codes for the most polymorphic drug metabolizing enzyme of donepezil and galantamine, and for BCHE locus, which codes for an enzyme which is both target and metabolizer of rivestigmine. The putative genetic confounder APOE (apolipoprotein E) was also genotyped.
Our first aim was to test if donepezil, galantamine, and rivastigmine differ with respect to their beneficial effects and occurrence of adverse events in the study subjects. Secondly, we wanted to test whether genotyping of CYP2D6 and BCHE loci would be useful in predicting cognitive changes in our AD patients treated with cholinesterase inhibitors.
Patients and methods
Study subjects
For this open study we recruited 171 patients between 55 and 87 years old with a clinical diagnosis of probable AD in accordance with the criteria of the National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association (NINCDS–ADRDA) [12]. These criteria include a mild-to-moderate severity, as defined by the results of a neurological exam, an extensive neuropsychiatric evaluation, and an neuroimaging analysis (computerized tomography or magnetic resonance imaging), and a Mini-Mental State Examination (MMSE) [13] score of 11.0–29 at diagnosis (Table 1). MMSE values were corrected by age and educational level using the score-adjustment coefficients proposed by Magni et al. [14]. After correction, the MMSE scores ranged from 12.0 to 27.7. All patients were recruited at the Alzheimer Center of the Department of Neurology, Sant’Anna Hospital, Ferrara. The study was approved by the Ethics Committee of the University of Ferrara (http://www.ospfe.it/index.phtml?id=1726), and the subjects and the caregivers consented to the study after a full explanation of what was involved. For each patient, age, sex, education, presence of other disease, and use of other drugs were recorded. The CYP drug interaction table [15] was consulted to identify comedications possibly interacting with CYP2D6 or CYP3A4.
Patients were divided in three groups according to the cholinesterase inhibitor treatment: donepezil (Don), galantamine (Gal), rivastigmine (Riv). The treatment regimes were as follows: patients were taking donepezil at a dose of either 5 (70 patients) or 10 mg/day (22 patients), galantamine at a dose of 8 (8 patients), 12 (5 patients), or 16 mg/day (20 patients), and rivastigmine at a dose of <6 (17 patients), 6 (25 patients), or 9 mg/day (4 patients). Treatment efficacy was evaluated by considering the effect of treatment on the cognitive function (MMSE) and on the patient’s ability to perform normal daily activities (Activites of Daily Living, ADL [16], and Instrumental Activities of Daily Living, IADL [17]), with a follow-up every 6 months. The occurrence of adverse events (AEs) was also recorded. Patients were classified as responder (R) and non-responder (NR) based on the corrected MMSE score at 12 months post-initiation of the treatment. Considering that AD symptoms tend to evolve over time, patients that showed no change in MMSE (ΔMMSE: MMSE at 12 months − MMSE at diagnosis) or a small decrement (decrease of the MMSE score ≤1.5 at the end of the first year) after the follow up were considered to be responders.
All patients were genotyped for CYP2D6 and BCHE. We also tested two-thirds of the patients for the presence of APOE allele ε4, which is known to be associated with the late-onset sporadic forms of AD [18].
Genotyping
CYP2D6 (OMIM 124030)
Genomic DNA was isolated from whole blood using the QIAamp DNA Blood Mini kit (QIAGEN, Hilden, Germany). Genotyping of 11 polymorphic positions in the CYP2D6 gene was performed following a protocol based on long PCR and the single nucleotide primer extension reaction (SNaPshot; Applied Biosystems, Foster City, CA) [19]. The same method was used to test for the presence of the whole-gene deletion and of multiple copies of the gene on the same chromosome (commonly referred to as whole-gene duplication). To check for the presence of the 2549delA defining CYP2D6*3, we performed a nested PCR-restriction fragment length polymorphism (RFLP) analysis as previously described [20]. A 200-bp fragment containing a BsaAI restriction site generated by a mismatch primer was amplified and digested with the BsaAI enzyme (New England Biolabs, Ipswich, MA).
BCHE (OMIM 177400)
We amplified a 1,839-bp fragment that included exon 2, using the primer pair pEx2_fwd (5′- GGCCTTTACAGAAGCAGGTT-3′ and pEx2_rev (5′-CACAGGGAGTTGAAATGCAG-3′), and a 549-bp fragment that included exon 4, using the primer pair pEx4_fwd (5′-TTCAGGCAAAGCGAGCTAAT-3′) and pEx4_rev (5′-GAAATTGAACCAGGCCATTG-3′). Of these, 700 bp (from −140 to 560) of exon 2 and the whole 549-bp fragment (including exon 4) were re-sequenced using big dye terminator chemistry (BigDye Terminator v3.1 Cycle Sequencing kit; Applied Biosystems) and PCR primers. Our aim was to test for the presence of two common non-synonymous substitutions (rs1799807, corresponding to A-variant Asp70Gly in exon 2, and rs1803274, corresponding to K-variant Ala539Thr in exon 4) with a known phenotypic effect (OMIM 177400) as well as less common or new mutations.
APOE (OMIM 107741)
To test for the presence of two substitutions, rs429358 and rs7412, that define alleles APOEε2 (334 T/472 T; 112 Cys/158 Cys), APOEε3 (334 T/472 C; 112 Cys/158 Arg), and APOEε4 (334 C/472 C; 112 Arg/158 Arg), we amplified 235 bp of exon 4 in a PCR reaction using the primer pair 3 published in Koch et al. [21]. The DNA fragment was re-sequenced using big dye terminator chemistry (BigDye Terminator v3.1 Cycle Sequencing kit, Applied Biosystems).
Haplotype reconstruction
Maximum-likelihood BCHE and CYP2D6 haplotype frequencies were estimated using an expectation–maximization (EM) algorithm implemented in Arlequin 3.1 [22]. CYP2D6-inferred haplotypes were compared with those available on the web site of the official allele nomenclature (http://www.cypalleles.ki.se/cyp2d6.htm). The CYP2D6*6-defining mutation (1707 del T) was excluded from the inference because it was present on only one chromosome in the whole dataset. Its attribution to haplotype *6 was based only on the official nomenclature.
Definition of genotypic scores
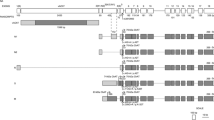
For CYP2D6, the previously published “activity score” system [23] was used to translate genotype information into a phenotypic measure. In this way, specific CYP2D6 variants were associated with metabolic activities based on what is known from in vivo and in vitro studies (http://www.cypalleles.ki.se/cyp2d6.htm). In practice, a value of 1 was given to fully functional haplotypes (*1 and *2), 0 to non-functional haplotypes (*3, *4, *5, and *6), and 0.5 to haplotypes with reduced activity (*9, *10, and *41). Gene duplications received twice the value assigned to the haplotype in single copy regardless of the number of copies of the gene on the same chromosome. The activity score of a genotype was the sum of the values assigned to each haplotype. Thus, the genotype activity score theoretically ranges from 0 (null metabolic activity) to 4 (ultrarapid metabolic activity). This high-resolution CYP2D6 genotyping procedure allowed us to identify seven activity score groups, namely, 0, 0.5, 1, 1.5, 2, 2.5, and 3. However, because only a few individuals fell in the most extreme groups, group 0 and 0.5 (corresponding to null or almost null activity score) at one extreme and group 2.5 and 3 (corresponding to ultrarapid activity) at the other extreme were merged, so that only five groups were considered for the association analyses (Fig. 1).

Distribution of BCHE genotypes (a) and CYP2D6 (b) in different groups (R responders, NR non-responders) of Alzheimer’s patients participating in this study. Don Donepezil, Gal galantamine, Riv rivastigmine. A BCHE variant A (rs1799807), K BCHE variant K (rs1803274), wt wild-type (no mutations). Predicted CYP2D6 enzyme activity is based on genotype scores: 0–0.5 poor, 1 and 1.5 intermediate, 2 normal, 2.5–3 ultrarapid. For the number of subjects in each genotype subgroup, see Fig. 2
BCHE allele K (rs1803274) is known to cause a reduction in the effective number of circulating BChE molecules, and the A variant (rs1799807) actually reduces protein activity [24, 25], but the activity of these two alleles may differ in different parts of the body [26]. For this reason it is impossible to define a simple activity score system for BCHE, and so patients were divided in carriers and non-carriers, respectively, of at least one mutated allele.
Statistical analyses
To detect significant departure from Hardy–Weinberg equilibrium, we used the extended version of Fisher’s exact test implemented in Arlequin 3.01 [22]. Tests of association between genotypes and cognitive changes were performed taking phenotypic outcomes into account in two ways: first, patients were divided in groups of responders and non-responders (binary predicted variable: R, NR after 12 months of treatment). A logistic regression adjusted for age, sex, baseline MMSE, and comedication (only for CYP2D6) was used to estimate odds ratios (ORs) and 95% confidence intervals (CI) for testing for possible association between responders/non-responders to treatment and BCHE genotypes or CYP2D6 genotypic scores. The same analysis was performed for APOE genotypes. This set of analyses was limited to ΔMMSE because the definition of responders and non-responders based on ΔADL and ΔIADL is the same as for the first index. In a second set of analyses we used individual patients ΔMMSE, ΔADL, and ΔIADL as phenotypic outcomes (continuous predicted variable); ΔMMSE, ΔADL, and ΔIADL were checked if normally distributed by performing a Kolmogorov–Smirnov test. ΔMMSE was normally distributed, thus differences in ΔMMSE between CYP2D6 and BCHE genotypes were tested by means of an analysis of covariance (ANCOVA) to control for baseline MMSE and age, whereas a multi-factor analysis of variance (ANOVA) was used to control for sex (both genes) and comedications (only for CYP2D6). Because ΔADL and ΔIADL do not follow a normal distribution, two non parametric tests were used to compare groups of patients with different genotypes. Carriers and noncarriers of at least one BCHE mutation were compared by means of a Mann–Whitney U test. Different groups of CYP2D6 activity scores were compared by means of a Kruskal–Wallis ANOVA. All of the analyses were performed using STATISTICA [27].
Results
Patient characteristics
At diagnosis, the mean corrected MMSE score among the 171 patients was 19.5, with minimum and maximum values of 12.0 and 27.7, respectively. Patient characteristics and cognitive state for each treatment are shown in Table 1. No significant differences across treatments were found with respect to ADL, IADL, and corrected MMSE at diagnosis (Kruskal–Wallis ANOVA, P ADL = 0.11, P IADL = 0.59, P MMSE = 0.95, respectively) and sex (χ 2 test, P = 0.95). The age at diagnosis was found to be significantly different among treatments (Kruskal–Wallis ANOVA, P = 0.003) because the average age of rivastigmine patients was lower (Table 1). Of these 171 patients, eight changed therapy before the end of the first year of treatment because they experienced an AE to galantamine (3 patients, 1 switched to donepezil and 2 to rivastigmine) or rivastigmine (5 patients: 2 switched to donepezil and 3 to galantamine). These eight individuals were excluded from the study. Among patients treated with donepezil, 13 were taking comedications that may influence the activity of the CYP2D6 enzyme: 11 patients were co-treated with citalopram/escitalopram (9 R and 2 NR to donepezil) and 2 with paroxetine (1 R and 1 NR), both CYP2D6 inhibitors. Similarly, three galantamine patients were co-treated with citalopram and two with paroxetine, all of them responders to galantamine. Comedication with CYP2D6 inhibitors was considered a possible confounder in association analyses. None of the subjects included in our study was comedicated with CYP2D6 inducers or with CYP3A4/5 inhibitors or inducers.
Cognitive changes and adverse events
Patients treated with donepezil, galantamine, and rivastigmine showed an average decline in MMSE of 0.3, 0.5, and 0.2, respectively, after 12 months of therapy. The efficacy of the pharmacological treatment was comparable to that observed for the same three drugs by Aguglia et al. [28] after 6 months and for donepezil by Patterson et al. [29] after 3–9 months. However, our study showed a lower cognitive decline compared to other studies with follow-up of 6 months [8, 10] or >1 year [9, 30]. We did not observe significant differences in terms of ΔMMSE between treatments (Kruskal–Wallis ANOVA, P = 0.71).
During the first 12 months of therapy several patients experienced an AE to the treatment. In particular, AEs were reported in seven (7.6%), six (18.2%), and nine (19.5%) patients treated with donepezil, galantamine, and rivastigmine, respectively. The most common side effects of rivastigmine were gastrointestinal complaints (5 cases of epigastralgies and 2 of diarrhea) and, but rarely, irritability (1), vertigo (1), and dizziness (1) were observed. Galantamine caused abdominal pain (2 cases), dizziness (2), nausea (1), diarrhea (1), cardiovascular event (1), rash (1), depression (1), irritability (1), and sweating (1). Donepezil caused fewer side effects than the other two treatments, with epigastralgy (2 cases), nausea (1), allucination (1), fall (1), agitation (1), insomnia (1), and dizziness (1) being the most frequently reported AEs. The number of patients having an AE and the number of AEs do not correspond because the same subject may have experienced more than one side-effect. Even if donepezil showed a higher tolerability, the incidence of AE was not statistically different between the three treatment groups (Fisher’s exact test, P = 0.08). Contrary to what has been found in other studies[6], we did not find any dose-dependent AE for any of the three treatments (Fisher’s exact tests, P > 0.05).
APOE: a possible confounder
To control for possible patient stratification due to the APOE genotype [18], we tested carriers and noncarriers of APOEε4 for association with cognitive changes (Table 2). The increasing number of APOEε4 alleles per genotype did not increase the risk of a poor response to cholinesterase inhibitors after 12 months (logistic regression controlling for age, sex, and baseline MMSE; P > 0.05; OR 0.68, 95% CI 0.34–1.35) or to donepezil, galantamine, and rivastigmine tested separately (P > 0.05; donepezil: OR 0.70, 95% CI 0.27–1.76; galantamine: OR 0.46, 95% CI 0.07–3.34; rivastigmine OR 0.39, 95% CI 0.05–3.02).
Description of BCHE and CYP2D6 genetic variation
Frequencies and distributions of combined BCHE and CYP2D6 genotypes for the total sample and across treatments after 12 months of therapy are summarized in Table 3.
In our re-sequencing analysis of 1,249 bp of BCHE coding regions, no genetic variants other than K (rs1803274) and A (rs1799807) were detected. Of 326 chromosomes, 58 (17.8%) carried the K variant located in exon 4, whereas the rare A variant in exon 2 was found only on six chromosomes (1.8%). The BCHE locus did not show any deviation from Hardy–Weinberg equilibrium. When haplotypes were reconstructed from genotypic information, only three haplotypes could be inferred, namely wtwt, wtK, and AK, indicating the presence of complete linkage disequilibrium between variants K and A.
The great majority of patients enrolled in this study were either heterozygotes at the CYP2D6 locus or homozygous for mutations known to be frequent in populations of European origin, such as those defining CYP2D6*2, *4, or *41 [31, 32] (Table 3). The only exception was the presence of a homozygous individual for the rather rare CYP2D6*3 [32]. To rule out the possibility of an allelic drop-out in the SNaPshot assay, we confirmed our results by a nested PCR-RFLP analysis, testing all the CYP2C6*3 carriers (homo- and heterozygotes) of our dataset, together with a non-*3 individual. Of the 12 CYP2D6 variants (11 SNPs and whole-gene duplication), four significantly deviated from the equilibrium expectation due to a deficit of heterozygosity (P < 0.05). Specifically, three of these variants were in linkage disequilibrium and together define CYP2D6*2 (1661 G > C, 2850 C > T, and 4180 G > C), coding for a fully functional enzyme, while the fourth variant alone defines CYP2D6*3 (2549delA), coding for a null-function enzyme (http://www.cypalleles.ki.se/cyp2d6.htm). None of these deviations remained significant when a Bonferroni correction for multiple tests was performed.
Candidate genes and cognitive changes
Patients defined as NR to donepezil and rivastigmine showed a higher proportion of BCHE genotypes carrying at least one mutation, whereas galantamine patients showed the opposite trend (Fig. 1a). Lower CYP2D6 genotype scores were more frequent among NRs, possibly indicating an association between a decreased 2D6 enzymatic activity and a faster cognitive decline (Fig. 1b). However, BCHE and CYP2D6 trends are not significant when formally tested by means of a logistic regression controlling for age, sex, baseline MMSE, and comedications for CYP2D6 (BCHE: donepezil OR 2.14, 95% CI 0.75–6.11; galantamine OR 0.37, 95% CI 0.06–2.33; rivastigmine OR 3.86 95% CI 0.63–23.6; CYP2D6: donepezil OR 0.99, 95% CI 0.93–1.08; galantamine OR 0.97, 95% CI 0.83–1.13; rivastigmine OR 0.92, 95% CI 0.81–1.04).
In a second group of analyses, individual changes in MMSE, ADL, and IADL after 12 months of therapy were treated as a continuous outcome. Figure 2 shows the distribution of individual ΔMMSE in different BCHE genotype groups (Fig. 2a) and CYP2D6 genotype score groups (Fig. 2b). No significant differences between the genotype groups were evident for MMSE (ANCOVA/multi-factor ANOVA controlling for age, sex, baseline MMSE, and comedications for CYP2D6; P > 0.05), ADL and IADL (Mann–Whitney U test and Kruskal–Wallis ANOVA, P > 0.05), suggesting that neither the BCHE nor the CYP2D6 genotype play a relevant role in explaining the cognitive decline of AD patients after 12 months of treatment with cholinesterase inhibitors.

Individual change in Mini-Mental State Examination scores (ΔMMSE) after 12 months of donepezil (DON), galantamine (GAL) or rivastigmine (RIV) treatment in Alzheimer’s patients with different BCHE (a) and CYP2D6 (b) genotypes. Black symbols Average values. A BCHE variant A (rs1799807), K BCHE variant K (rs1803274), wt wild-type (no mutations). Predicted CYP2D6 enzyme activity based on genotype scores: 0–0.5 poor, 1 and 1.5 intermediate, 2 normal, 2.5–3 ultrarapid
Discussion
In this open study we observed a general trend towards an increased frequency of BCHE and CYP2D6 mutations among AD patients treated with cholinesterase inhibitors showing a faster cognitive decline (NR) (Fig. 1). The tendency observed here for BCHE contrasts with the finding that patients with dementia carrying the BCHE K variant show a slower rate of cognitive decline and improved attention [33]. Although none of the tests performed in our study identified significant associations between the BCHE and CYP2D6 genotype and the phenotypic outcome, this finding may not be irrelevant. The limited availability of DNA samples from well-defined AD patients treated with cholinesterase inhibitors and followed for 1 year led to a relative small sample size in our study. This may have influenced our ability to detect significant associations between genotypes and phenotypic outcomes. However, a lack of association between BCHE genetic variation and donepezil and rivastigmine response was recently observed in a study in which Italian AD patients were characterized for the BCHE K variant and for another common intronic single nucleotide polymorphism (SNP) (rs1355534) [9]. Based on our observations and on those of Scacchi et al. [9], it seems reasonable to conclude that assessing BCHE genetic variation is not useful in terms of predicting cognitive decline in AD patients treated with cholinesterase inhibitors. Conversely, the BCHE genotype was shown to be a predictor of differential response between treatments in AD patients younger than 75 years [30, 34]. The results of a recent study suggest that late response to cholinesterase inhibitors in moderate-to-severe AD may be associated with the BCHE genotype [29], but these conclusions were based on a very limited sample size, the association was evident only for one of three recorded responses, and multiple testing correction was not performed.
Conclusions on the CYP2D6 genotype effect are also controversial. An evaluation of the efficacy of donepezil after 3 months in 42 Italian AD patients showed that poor metabolizers are better responders [8], and a similar result was recently observed in another set of 57 Italian AD patients after 6 months [11]. Conversely, our 92 patients treated with donepezil showed a general tendency towards a higher frequency of faster metabolizers in responders (Fig. 1b, higher scores), and a similar trend was observed for galantamine and rivastigmine. The three studies are difficult to compare for a number of reasons. First, given that AD symptoms tend to evolve over time, different follow-ups are likely to lead to different results. More specifically, the discrepancy between our results and those of Varsaldi et al. [8] is probably the result of the presence of all four groups of CYP2D6 metabolizers among our patients, namely, poor, intermediate, normal, and ultrarapid (scores of 0–0.5, 1–1.5, 2, and 2.5–3, respectively), whereas the patients enrolled in the study of Varsaldi et al. [8] included no poor metabolizers and only two ultrarapid ones. A similar comparison cannot be performed with the data presented by Seripa et al. [11], whose patients were subdivided based on the presence/absence of mutations affecting the enzyme activity. Based on this criterion, the authors found a significant higher frequency of variants conferring a decreased or null enzyme activity in donepezil responders. In other words, the results suggest that being homozygous for CYP2D6 full-function alleles increases the risk of being non-responders to donepezil. This observation does not have a simple biological interpretation, and it is difficult to reconcile with the bimodal distribution of in vivo phenotypes always observed in Europeans, where poor metabolizers (i.e., homozygous for null-function alleles) represent a separate subgroup, while all the other genotypes overlap [35]. In this respect, our choice to translate genotypes to phenotypes by means of the activity score system based on pharmacokinetic data may also be problematic. To control for this, we re-analyzed our CYP2D6 data dividing patients into two subgroups, namely, poor metabolizers and all the rest, and the results did not change (data not shown). A third study on Italian AD patients found a significant association between a CYP2D6 promoter polymorphism and response to donepezil [10]. Unfortunately, rs1080985 is not included in our assay, so a comparison is not possible. The reason we chose not to type this position is that the C > G substitution does not define a specific CYP2D6 variant, and its phenotypic outcome is very controversial [36–38]. However, its unconfirmed effect on the enzyme activity does not exclude a possible phenotypic effect or a role as a genetic marker. In the latter case, it is possible that the causative variant is in linkage disequilibrium with rs1080985 G. It would be worthwhile testing whether this interesting result is reproducible in our patients.
Genetic variation at other genomic regions may have a role in determining differential cognitive declines in AD patients enrolled in this study. The APOEε4 allele is known for its association with risk of AD [18]. One study reported a lack of predicted response to the insulin sensitizing agent rosiglitazone [39] and cholinesterase inhibitors [40–42], although other studies do not support this conclusions [43, 44]. As suggested by our analysis, genetic variation at the apolipoprotein E locus did not affect our results on the role of BCHE and CYP2D6 genotype in determining cognitive changes.
Another possible explanation for the lack of association between CYP2D6 genetic variation and cognitive changes in patients treated with donepezil and galantamine could be the role played by other CYP isoenzymes in their metabolism [6, 45]. In particular, in the presence of CYP2D6 mutations conferring a reduced/absent enzyme activity, the CYP3A4 isoenzyme may have a major role in donepezil and galantamine metabolism.
In conclusion, our results show that, taken on its own, this study of the possible effects of variation at the BCHE and CYP2D6 loci does not seem to help predict cognitive changes in patients treated with donepezil, galantamine, or rivastigmine in Italy. As is the case for many complex traits, genetic and/or nongenetic factors other than BCHE and CYP2D6 contribute substantially to determining the ability of AD patients to respond to treatment. Generalizing this observation is not yet possible because it may well be that in other populations the confounding effects of other loci are less marked. Indeed, the contradictory outcomes of different studies may be due to differences in the general genetic buildup of the specific population. Future progress may be expected only when we have a clearer idea of the genetic architecture of drug response and of the multiple factors contributing to it. At that point, it will also be important to re-evaluate the limited associations between cognitive changes, on the one hand, and BCHE and CYP2D6 genotypes, on the other, which were identified in this study but which did not reach statistical significance.
References
Jonsson L, Wimo A (2009) The cost of dementia in Europe: a review of the evidence, and methodological considerations. Pharmacoeconomics 27:391–403
Dementia in Europe: Yearbook 2006 (2006). Alzheimer Europe. Available at: http://www.alzheimer-europe.org
Waldemar G, Dubois B, Emre M, Georges J, McKeith IG, Rossor M, Scheltens P, Tariska P, Winblad B (2007) Recommendations for the diagnosis and management of Alzheimer’s disease and other disorders associated with dementia: EFNS guideline. Eur J Neurol 14:e1–e26
Rabins PV, Blacker D, Rovner BW, Rummans T, Schneider LS, Tariot PN, Blass DM, McIntyre JS, Charles SC, Anzia DJ, Cook IA, Finnerty MT, Johnson BR, Nininger JE, Schneidman B, Summergrad P, Woods SM, Berger J, Cross CD, Brandt HA, Margolis PM, Shemo JP, Blinder BJ, Duncan DL, Barnovitz MA, Carino AJ, Freyberg ZZ, Gray SH, Tonnu T, Kunkle R, Albert AB, Craig TJ, Regier DA, Fochtmann LJ (2007) American Psychiatric Association practice guideline for the treatment of patients with Alzheimer’s disease and other dementias. Second edition. Am J Psychiatry 164:5–56
Ballard CG (2002) Advances in the treatment of Alzheimer’s disease: benefits of dual cholinesterase inhibition. Eur Neurol 47:64–70
Jann MW, Shirley KL, Small GW (2002) Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clin Pharmacokinet 41:719–739
Cacabelos R, Llovo R, Fraile C, Fernandez-Novoa L (2007) Pharmacogenetic aspects of therapy with cholinesterase inhibitors: the role of CYP2D6 in Alzheimer’s disease pharmacogenetics. Curr Alzheimer Res 4:479–500
Varsaldi F, Miglio G, Scordo MG, Dahl ML, Villa LM, Biolcati A, Lombardi G (2006) Impact of the CYP2D6 polymorphism on steady-state plasma concentrations and clinical outcome of donepezil in Alzheimer’s disease patients. Eur J Clin Pharmacol 62:721–726
Scacchi R, Gambina G, Moretto G, Corbo RM (2009) Variability of AChE, BChE, and ChAT genes in the late-onset form of Alzheimer’s disease and relationships with response to treatment with Donepezil and Rivastigmine. Am J Med Genet B Neuropsychiatr Genet 150B:502–507
Pilotto A, Franceschi M, D’Onofrio G, Bizzarro A, Mangialasche F, Cascavilla L, Paris F, Matera MG, Pilotto A, Daniele A, Mecocci P, Masullo C, Dallapiccola B, Seripa D (2009) Effect of a CYP2D6 polymorphism on the efficacy of donepezil in patients with Alzheimer disease. Neurology 73:761–767
Seripa D, Bizzarro A, Pilotto A, D’Onofrio G, Vecchione G, Gallo AP, Cascavilla L, Paris F, Grandone E, Mecocci P, Santini SA, Masullo C (2011) Role of cytochrome P4502D6 functional polymorphisms in the efficacy of donepezil in patients with Alzheimer’s disease. Pharmacogenet Genomics 21(4):225–320
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34:939–944
Folstein MF, Folstein SE, McHugh PR (1975) Mini-mental state. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12:189–198
Magni E, Binetti G, Bianchetti A, Rozzini R, Trabucchi M (1996) Mini-mental State Examination: a normative study in Italian elderly population. Eur J Neurol 3:1–5
Flockhart D (2007) Drug interactions: cytochrome P450 drug interaction table. Available at: http://medicine.iupui.edu/clinpharm/ddis/table.asp. Indiana University School of Medicine, version 5.0 released on 12 Jan 2009
Katz S (1983) Assessing self-maintenance: activities of daily living, mobility, and instrumental activities of daily living. J Am Geriatr Soc 31:721–727
Lawton MP, Brody EM (1969) Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist 9:179–186
Schipper HM (2011) Apolipoprotein E: implications for AD neurobiology, epidemiology and risk assessment. Neurobiol Aging 32(5):778–790
Sistonen J, Fuselli S, Levo A, Sajantila A (2005) CYP2D6 genotyping by a multiplex primer extension reaction. Clin Chem 51:1291–1295
Levo A, Koski A, Ojanpera I, Vuori E, Sajantila A (2003) Post-mortem SNP analysis of CYP2D6 gene reveals correlation between genotype and opioid drug (tramadol) metabolite ratios in blood. Forensic Sci Int 135:9–15
Koch W, Ehrenhaft A, Griesser K, Pfeufer A, Muller J, Schomig A, Kastrati A (2002) TaqMan systems for genotyping of disease-related polymorphisms present in the gene encoding apolipoprotein E. Clin Chem Lab Med 40:1123–1131
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS (2008) The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharmacol Ther 83:234–242
Bartels CF, Jensen FS, Lockridge O, van der Spek AF, Rubinstein HM, Lubrano T, La Du BN (1992) DNA mutation associated with the human butyrylcholinesterase K-variant and its linkage to the atypical variant mutation and other polymorphic sites. Am J Hum Genet 50:1086–1103
Podoly E, Shalev DE, Shenhar-Tsarfaty S, Bennett ER, Ben Assayag E, Wilgus H, Livnah O, Soreq H (2009) The butyrylcholinesterase K variant confers structurally derived risks for Alzheimer pathology. J Biol Chem 284:17170–17179
Tasker A, Ballard CG, Joachim C, Warden DR, Okello EJ, Perry RH, Khan N, Smith AD, Lehmann DJ, Perry EK (2008) Butyrylcholinesterase K variant associated with higher enzyme activity in the temporal cortex of elderly patients. Neurosci Lett 442:297–299
StatSoft (2005) STATISTICA ver. 7.1. Available at: StatSoft.Italia_srl
Aguglia E, Onor ML, Saina M, Maso E (2004) An open-label, comparative study of rivastigmine, donepezil and galantamine in a real-world setting. Curr Med Res Opin 20:1747–1752
Patterson CE, Todd SA, Passmore AP (2010) Effect of apolipoprotein E and butyrylcholinesterase genotypes on cognitive response to cholinesterase inhibitor treatment at different stages of Alzheimer’s disease. Pharmacogenomics J. doi:10.1038/tpj.2010.61
Bullock R, Bergman H, Touchon J, Gambina G, He Y, Nagel J, Lane R (2006) Effect of age on response to rivastigmine or donepezil in patients with Alzheimer’s disease. Curr Med Res Opin 22:483–494
Fuselli S, Dupanloup I, Frigato E, Cruciani F, Scozzari R, Moral P, Sistonen J, Sajantila A, Barbujani G (2004) Molecular diversity at the CYP2D6 locus in the Mediterranean region. Eur J Hum Genet 12:916–924
Sistonen J, Fuselli S, Palo JU, Chauhan N, Padh H, Sajantila A (2009) Pharmacogenetic variation at CYP2C9, CYP2C19, and CYP2D6 at global and microgeographic scales. Pharmacogenet Genomics 19:170–179
O’Brien KK, Saxby BK, Ballard CG, Grace J, Harrington F, Ford GA, O’Brien JT, Swan AG, Fairbairn AF, Wesnes K, del Ser T, Edwardson JA, Morris CM, McKeith IG (2003) Regulation of attention and response to therapy in dementia by butyrylcholinesterase. Pharmacogenetics 13:231–239
Blesa R, Bullock R, He Y, Bergman H, Gambina G, Meyer J, Rapatz G, Nagel J, Lane R (2006) Effect of butyrylcholinesterase genotype on the response to rivastigmine or donepezil in younger patients with Alzheimer’s disease. Pharmacogenet Genomics 16:771–774
Zanger UM, Raimundo S, Eichelbaum M (2004) Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn-Schmiedebergs Arch Pharmacol 369:23–37
Gaedigk A, Ryder DL, Bradford LD, Leeder JS (2003) CYP2D6 poor metabolizer status can be ruled out by a single genotyping assay for the -1584 G promoter polymorphism. Clin Chem 49:1008–1011
Raimundo S, Toscano C, Klein K, Fischer J, Griese EU, Eichelbaum M, Schwab M, Zanger UM (2004) A novel intronic mutation, 2988G > A, with high predictivity for impaired function of cytochrome P450 2D6 in white subjects. Clin Pharmacol Ther 76:128–138
Rodriguez-Antona C, Gurwitz D, de Leon J, Llerena A, Kirchheiner J, de Mesa EG, Ibarreta D (2009) CYP2D6 genotyping for psychiatric patients treated with risperidone: considerations for cost-effectiveness studies. Pharmacogenomics 10:685–699. doi:10.2217/pgs.09.15
Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, Zvartau-Hind ME, Hosford DA, Roses AD (2006) Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J 6:246–254
Borroni B, Colciaghi F, Pastorino L, Archetti S, Corsini P, Cattabeni F, Di Luca M, Padovani A (2002) ApoE genotype influences the biological effect of donepezil on APP metabolism in Alzheimer disease: evidence from a peripheral model. Eur Neuropsychopharmacol 12:195–200
Poirier J, Delisle MC, Quirion R, Aubert I, Farlow M, Lahiri D, Hui S, Bertrand P, Nalbantoglu J, Gilfix BM, Gauthier S (1995) Apolipoprotein E4 allele as a predictor of cholinergic deficits and treatment outcome in Alzheimer disease. Proc Natl Acad Sci USA 92:12260–12264
Farlow MR, Lahiri DK, Poirier J, Davignon J, Schneider L, Hui SL (1998) Treatment outcome of tacrine therapy depends on apolipoprotein genotype and gender of the subjects with Alzheimer’s disease. Neurology 50:669–677
Aerssens J, Raeymaekers P, Lilienfeld S, Geerts H, Konings F, Parys W (2001) APOE genotype: no influence on galantamine treatment efficacy nor on rate of decline in Alzheimer’s disease. Dement Geriatr Cogn Disord 12:69–77
Rigaud AS, Traykov L, Latour F, Couderc R, Moulin F, Forette F (2002) Presence or absence of at least one epsilon 4 allele and gender are not predictive for the response to donepezil treatment in Alzheimer’s disease. Pharmacogenetics 12:415–420
Farlow MR (2003) Clinical pharmacokinetics of galantamine. Clin Pharmacokinet 42:1383–1392
Acknowledgments
We gratefully thank Johanna Sistonen for helping with the CYP2D6 genotyping, Sara Raimondi and Giorgio Bertorelle for useful suggestions on data analysis, and Krisztina Vasarhelyi for language revision.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chianella, C., Gragnaniello, D., Maisano Delser, P. et al. BCHE and CYP2D6 genetic variation in Alzheimer’s disease patients treated with cholinesterase inhibitors. Eur J Clin Pharmacol 67, 1147–1157 (2011). https://doi.org/10.1007/s00228-011-1064-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-011-1064-x