
Abstract
Purpose
The aim of this investigation was to demonstrate that nonlinear mixed-effects population pharmacokinetic (PK) modeling can be used to evaluate data from studies of drug transport/excretion into human milk and hence to estimate infant exposure.
Methods
A sparse dataset from a previously published study of the use of oral tramadol for post-cesarean pain management in 75 lactating women was used. Milk and plasma samples were collected during days 2–4 of lactation, and tramadol and O-desmethyltramadol (ODT) concentration measurements in these samples were available. Absolute infant dose was obtained from the concentration measurements and estimated milk volume ingested, and expressed in micrograms per kilogram per day. Relative infant dose was calculated as a percentage of the absolute infant dose divided by the maternal dose (μg/kg/day). Nonlinear mixed-effects modeling was used to fit a population PK model to the data.
Results
The disposition of tramadol and ODT in plasma and the transition of these substances into milk were characterized by a five-compartment population PK mixture model with first-order absorption. The polymorphic ODT formation clearance in the plasma compartment was able to be characterized in both CYP2D6-poor and -extensive metabolizers. Milk creamatocrit was a significant covariate in ODT transfer between the plasma and milk compartments. The estimated relative infant doses in extensive and poor metabolizers, respectively, were 2.16 ± 0.57 and 2.60 ± 0.57% for tramadol, and 0.93 ± .20 and 0.47 ± 0.10% for ODT.
Conclusions
This study demonstrates that a population PK approach with sparse sampling of analytes in milk and plasma can yield quality information about the transfer process and that it also can be used to estimate the extent of infant exposure to maternal drugs via milk.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
For a number of years, our group has pursued research into the transfer of maternally ingested drugs into milk as a means of quantifying drug exposure in the breastfed infant. Drug concentration in milk is measured and infant dose estimated from the average concentration (Cavg), assuming an average milk intake of 0.15 L/kg/day [1]. The most robust studies have used a two-stage pharmacokinetic (PK) approach with rich datasets in small groups of patients or volunteers. Cavg is derived from measurements of area under the milk concentration–time curve (AUC), either after single doses or at steady-state [2]. However, contemporary PK drug studies in clinical trials often use a sparse sampling scheme and a population nonlinear mixed-effects modeling approach to derive the desired PK parameters, including AUC. This same methodology should also be applicable to studying the transfer of drugs into human milk, and in 2005 the U.S. Food and Drug Administration issued a draft guidance document for lactation studies that includes recommendations on the use of population PK protocols [3]. The aim of this investigation was to demonstrate the application of population PK modeling in evaluating data from studies of drug transport/excretion into human milk and hence to estimate infant exposure. The sparse dataset that we have used is from our previously published study of 75 lactating women who were treated with tramadol in the first few days after cesarean delivery of their infant [4]. Each woman contributed one plasma sample and several milk samples that were analyzed for tramadol and O-desmethyltramadol (ODT) content. These data were previously analyzed using a naïve data pooling approach, as they were not suited to a two-stage PK analysis.
Tramadol is widely used in the management of perioperative surgical pain, including in obstetrics after cesarean delivery. Both tramadol and its active O-desmethyl metabolite exert their analgesic effects in a stereospecific manner through two complementary and synergistic actions; (+)-tramadol and (−)-tramadol inhibit the neuronal uptake of serotonin and norepinephrine, respectively, while (+)-ODT acts as a μ-opioid receptor agonist and (−)-ODT is essentially inactive. [5–7]. The primary metabolic route via O-demethylation to produce ODT is catalyzed stereospecifically by cytochrome P450 2D6 (CYP2D6). For this reason, tramadol has been suggested as a CYP2D6 phenotyping probe [8, 9]. N–demethylation by CYP2B6 and -3A4 is also a significant metabolic pathway. Pharmacodynamically, (+)-ODT is important because of its high affinity for the μ-opioid receptor, compared to tramadol or N-desmethyltramadol [10, 11]. Given the polymorphic nature of CYP2D6 and the reduced neonatal renal blood flow, which is only 4% of cardiac output during the first 12 h post-partum compared to 25% in adults [12], and the ongoing development of drug-metabolizing enzymes in neonates [13], the inter-individual variability of tramadol and ODT transfer to milk could potentially give rise to significant exposure in breastfeeding infants.
Methods
Subjects and study design
The study design was reported previously [4]. Briefly, 75 postpartum mothers who were prescribed tramadol for pain management after cesarean delivery participated in the study. Mothers who had significant renal dysfunction, had decided not to breastfeed their infants, or had insufficient breast milk for analysis were excluded from the study. The study was approved by the local ethics committee, and participants gave written informed consent. Mothers taking prescribed 50 or 100 mg oral tramadol hydrochloride (equivalent to 167 μmol or 333 μmol of tramadol, respectively) four to six times daily were recruited, the majority on the second to fourth day after delivery. Maternal age, weight, concomitant medication, tramadol dose, and the approximate dates and times of all tramadol doses were recorded on the study day, as was the infant gestation period. Milk creamatocrit was also determined and fat content expressed as volume percentage [14]. Briefly, to measure creamatocrit, milk was vortexed for 1 min, drawn into a micro-hematocrit tube and the tube sealed at one end. It was then centrifuged at 8000 g for 5 min to separate the milk fat and aqueous layers. The lengths of the milk fat and total fat plus aqueous were measured with Vernier calipers and fat content expressed as a percentage of the total length. One blood sample (5 mL, heparinized) and two to three milk samples (2–3 mL) in equal aliquots from both breasts were collected following the fourth or fifth consecutive oral tramadol dose. Sampling times for each patient followed a randomization scheme of 1-h blocks over the dosing interval (exact time of each sample recorded). The schedule blocks were designed to provide an even sampling distribution across the dose interval.
Measurement of rac-tramadol and rac-ODT concentrations in plasma and milk
Quantification of rac-tramadol and rac-ODT concentrations was carried out by high-performance liquid chromatography (HPLC) as previously published [4]. The intra-day and inter-day relative standard deviations for the assay of tramadol (range 190–7590 nmol/L) and ODT (range 200–2000 nmol/L) in both milk and plasma were all <7.4%, as previously reported [4]. Accuracy of the method for tramadol in milk was 99.8 ± 8.1, 98.4 ± 2.6, and 100.2 ± 1.0% at 190, 1900, and 7590 nmol/L respectively, while the accuracy for ODT in milk was 100.8 ± 13.3, 99.7 ± 4.3, and 101.4 ± 3.1% at 200,1000, and 2000 nmol/L, respectively. Accuracy for tramadol in plasma was 97.1 ± 6.9 and 92.8 ± 3.1 at 190 and 7590 nmol/L, respectively, while for that for ODT was 109.4 ± 8.3 and 92.5 ± 5.3% at 200 and 2000 nmol/L, respectively. The recovery of tramadol from milk was 75.4 ± 6.6 and 80.8 ± 5.8% at 190 and 7590 nmol/L, respectively, while that of ODT from milk was 75.4 ± 4.5 and 75.0 ± 5.4% at 200 and 2000 nmol/L, respectively. The recovery of both analytes from milk was unaffected by creamatocrits ranging from 3.6 to 12.5%. The recovery of tramadol from plasma was 79.1 ± 6.5 and 84.8 ± 1.4% at 190 and 7590 nmol/L, respectively, while the recovery of ODT from plasma was 75.8 ± 2.2 and 87.8 ± 3.1% at 200 and 2000 nmol/L, respectively. The limits of quantitation and detection for tramadol in both milk and plasma were 57 and 30 nmol/L, respectively, while those for ODT were 40 and 20 nmol/L, respectively.
PK modeling
Log concentration versus time datasets for tramadol and ODT were analyzed by nonlinear mixed-effect modeling using NONMEM (ver. 6.2.0; ICON Development Solutions, Ellicott City, MD) with an Intel Visual FORTRAN 10.0 compiler. The subroutine within NONMEM was for user-defined linear mamillary models (ADVAN5 TRANS1). First-order conditional estimation (FOCE) with η−ε interaction was utilized for modeling. The minimum value of an objective function (OFV; a NONMEM-calculated global goodness-of-fit indicator equal to the −2 log-likelihood value of data) was used to choose suitable models during the model-building process. Unless otherwise specified, a difference in OFV of at least 6.63 (χ2 distribution with 1 df, P < 0.01) was considered to be statistically significant. For graphic model diagnosis, the R-based model-building aid Xpose 4.0 (http://www.r-project.org/) was used [15]. Secondary PK parameters were calculated using standard formulae [16].
Development of a base population PK model
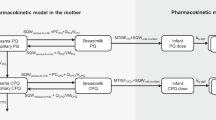
The practicalities of our experimental design limited us to one plasma data observation per patient. Preliminary modeling also indicated that over-parameterization was a potential problem when the plasma and milk datasets were combined into a single model (shown in Fig. 1); consequently, some structural model parameters had to be fixed. Data from the literature were used to fix the value for the absorption rate constant (kA) at 1.16 [17–19], the percentage of first-pass metabolism (FP) to ODT was set at 7.92% [17, 20], and the percentage of metabolic conversion of tramadol to ODT (CLM + first-pass metabolism; CL indicating clearance) for extensive metabolizers (MetabolismEM) was fixed at 30.9%; the weighed mean was calculated from a total of 331 subjects [8, 9, 20, 21]. A mixture model was used to define two subpopulations within the study population, namely, poor metabolizers (PM) and extensive metabolizers (EM). The proportion of PMs was set at 4% (Fraction = 0.04) in the mixture model, based on kernel density estimation of a linear frequency–distribution plot of plasma tramadol:ODT ratios which showed a clear antimode at 15, indicating that 4% of the population had PM status (data not shown). To allow semi-physiological models, where the drug is lost through the milk, to be investigated, the volumes of the milk compartments (V3 and V5) were both fixed at 125 mL, a physiologically based value calculated from the daily volume of milk consumed by infants aged approximately 1 week [22] and an average 67% emptying of the breast at each feed [23]. This also required the rate constants k24 and k35 to be fixed at a value of 1 to facilitate identifiability in the milk compartments.

Schematic representation of the pharmacokinetic compartments for the tramadol and O-desmethyltramadol (ODT) concentrations in plasma and breast milk after multiple oral administrations. See Table 1 for parameter definitions
The estimated structural parameters in the model were: CLtramadol, CLODT , MetabolismPM (total metabolism for PMs), V2, V3, k42, and k53. To mimic normal breastfeeding at days 2–4 after birth, we also investigated a semi-physiological model that emptied 67% of the breast milk compartments at 4-h intervals [23].
Allometric scaling was used on all plasma volume and clearance terms as follows; unless otherwise specified, volume terms were scaled with [ × (WT/70)1.0], while clearance terms were scaled with [ × (WT/70)0.75] [24]. As both between-subject variability (BSV) and residual error cannot be calculated simultaneously when only one observation exists per subject [25], two different models were tested for plasma components: one with no residual error and estimation of BSV, and the other with no BSV and estimation of residual error. As more than one milk concentration per patient existed, both BSV and residual error were estimated in the milk compartments. When residual error was estimated, an additive error model was used for the log-transformed data. As previously demonstrated, this would be expected to behave similarly to a proportional error model for untransformed data [26, 27].
Development of the final population PK model
The influence of the available covariates, namely, infant gestational age and milk creamatocrit, on the various model parameters was investigated. A linear relationship was tested for each covariate in the form: individual parameter value = population parameter value × [1 + percent effect on parameter x (covariate value for individual)/(average of covariate)]. Xpose was used to explore possible covariate relationships using the generalized additive modeling procedure function and inspection of correlation plots. Identified covariate relationships were evaluated within the NONMEM model using a stepwise, forwards and backwards covariate building process. Correlations among BSV terms were also investigated and, finally, weighted residuals (WRES) plots were assessed in arriving at the final model.
Model evaluation
A bootstrap procedure was performed by first using Perl speaks NONMEM (PsN; http://psn.sourceforge.net) to sample individuals from the original dataset with replacement and to generate 1000 new datasets, which were subsequently analyzed using NONMEM. The resulting parameters were then summarized as median and 2.5th and 97.5th percentiles [95% empirical confidence interval (CI)] to facilitate evaluation of the final model parameter estimates. In addition, to assess the predictive performance of the model, we also performed a visual predictive check (VPC) and numerical predictive check (NPC) stratified according to CYP2D6 EM or PM status for each observation compartment using PsN with 1000 replicate datasets simulated from the original. NPCs were assessed by comparing the actual with the expected number of data points within the 20, 40, 60, 80, 90, and 95% prediction intervals (PI). As only a small number were PMs, only the VPCs for EMs were evaluated by plotting the resulting 80% PI for tramadol and ODT with the original data.
Validation of infant dose derived from the model
The steady-state area under the curve (AUCss) values for tramadol and its metabolite were simulated for EM and PM subjects (1500 new datasets for each) from the population models described above and using the body weight and creamatocrit characteristics of the original dataset for a 100 mg dose of tramadol hydrochloride (333 μmol tramadol) administered at 6-h intervals. The AUCss for each analyte in their respective compartments was calculated using the linear trapezoidal rule on simulated concentrations estimated at 6-min intervals across the dose interval at steady-state. The Cavg was computed as the individual AUCss divided by the dose interval (6-h). Absolute infant dose (nmol/kg/day) was calculated as the product of the milk Cavg and an average infant milk intake of 0.15 L/kg/day [1]. Relative infant dose was calculated as the absolute infant:maternal dose (nmol/kg/day calculated using 70 kg, as per the original study [4]) and expressed as a percentage [1]. Post hoc Bayesian predicted individual estimates of CLM and CLT (total tramadol clearance; CLM + CLtramadol) for tramadol, and CLODT ODT were obtained for each simulated individual. The observed half-life (t½) of both tramadol and ODT were obtained for each simulated individual from the concentration–time curves.
Results
A total of 75, median (inter-quartile range, IQR) age 29 (25–32) years, all in early lactation (transitional milk on days 2–4 after birth) and receiving tramadol for post-cesarean pain management, were included in the analysis. Their median body weight at the time of study was 75 (67–91) kg. The median infant gestational age was 38.7 (38.2-39.2) weeks. In total, the women contributed 73 plasma samples and 214 milk samples for the study. The median milk creamatocrit was 4.4% (3.1–6.1%), with a range from 1 to 13.2%.
The simultaneous fit of a five-compartment model (including a dose/gut compartment) with first-order absorption and first-order elimination (Fig. 1) to the available concentration–time data sets (plasma and milk concentrations of tramadol and ODT vs. time) was successfully minimized. The absorption rate constant kA was fixed throughout the modeling process, as in the preliminary sensitivity analysis using only the plasma datasets, we were able to show that model parameters varied by less than 5% for kA values (range 0.6–1.6). We also carried out a sensitivity analysis (variability 20–40%) on the plasma parameter MetabolismEM. Apart from expected changes in CLPO, CLODT, and MetabolismPM, no other model parameters varied by more than 12%.
As indicated in the Methods, it was necessary to fix several parameters to achieve a satisfactory final model. Fixing V4, V5, k24, and k35 was necessary to avoid non-identification issues in the milk compartment as we chose a structure that enabled the investigation of semi-physiological models (i.e., one that had a volume parameter), while fixing kA and Fraction was necessary as our dataset had limited information to define these parameters and hence produce realistic parameter estimates.
The parameter estimates of the base and final models are summarized in Table 1. In the final model, residual error for the plasma compartments was 52 and 61% for tramadol and ODT, respectively. Although these values are high, they also incorporate BSV, which could not be estimated separately as there was only one plasma data point for each patient. A model with only BSV in the plasma compartments (and no residual error) was attempted; however, this produced substantial model mis-specification in the milk compartments. The residual error model was therefore chosen for model development. Residual error for the milk compartments was lower (35% for tramadol and 27% for ODT), as would be expected as it was possible to estimate BSV for the milk to plasma transfer rate constants k42 and k53.
The inclusion of 4-hourly 67% emptying of the milk compartments to mimic physiological feeding of a 3- to 4-day-old infant resulted in minimal changes to the final estimates of the model with no parameter changing more than 1% and no significant change in the OFV. Hence, this option was not included in the final model. Creamatocrit on k53 was the only covariate relationship that produced a significant (P < 0.01) decrease in OFV (6.867), with a 6.56% increase in k53 for each 1% increase in creamatocrit (i.e., as creamatocrit increases, the milk concentration of ODT decreases). The non-parametric bootstrap showed robust parameter estimates with a bias of less than 10 and 7% for the structural and random parameters, respectively. The population-predicted and individual-predicted plots versus observed plots for tramadol (Fig. 2a) and ODT (Fig. 2b) in milk showed some deviation from the expected line of fit at high drug concentrations (most evident in population predicted vs. observed). Weighted residuals vs. time for tramadol and ODT in plasma and milk are shown in Fig. 3. The VPCs for EMs in the plasma (Fig. 4a, b) and milk (Fig. 5 a, b) compartments did not show evidence of model misspecification. NPC checks performed for both EMs and PMs for each compartment showed good predictive performance, with the expected number of data points above and below all prediction intervals tested (data not shown).

Population- (open circle) and individual-predicted (filled circle) tramadol vs. observed data for tramadol (a) and ODT (b) concentrations (nmol/L) in breast milk for the final model. The lines of identity are also shown

Weighted residuals vs. time for tramadol in plasma (a) and milk (b) and ODT in plasma (c) and milk (d)

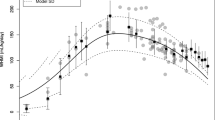
Visual predictive check of plasma concentration data (nmol/L on log10 scale) for tramadol (a) and ODT (b) in extensive metabolizers (EMs) showing observed concentrations vs. time after dose (open circles), with their 10th, 50th, and 90th percentiles (solid lines). Gray-shaded area 95% confidence intervals for the simulated 10th, 50th and 90th percentiles, using the final model

Visual predictive check of milk concentration data (nmol/L on log10 scale) for tramadol (a) and ODT (b) in EMs showing observed concentrations vs. time after dose (open circle), with their 10th, 50th and 90th percentiles (solid lines). Gray-shaded area 95% confidence intervals for the simulated 10th, 50th and 90th percentiles, using the final model
Table 2 shows simulated secondary PK parameters (separately for the EM and PM phenotypes) for tramadol (AUCss, Cavg, CLM, CLtramadol, t½) and ODT (AUCss, Cavg, CLODT, t½) in plasma, and AUCss and Cavg,for both tramadol and ODT in milk. For tramadol, mean CLM was 57% lower in PM than in EM subjects. Mean tramadol t½ and ODT t½ were 16 and 8% longer, respectively, in PMs. As expected, the milk:plasma ratios estimated from the AUCss data were not dependent on phenotype, with means of 1.9 for tramadol and 3.2 for ODT. Absolute and relative infant doses were simulated from the milk Cavg data, assuming an average maternal dose of 100 mg tramadol HCl given four times daily to a 70-kg woman. Absolute (and relative) infant doses in EMs and PMs were 412 ± 90 nmol/kg/day (2.16 ± 0.47%) and 496 ± 108 nmol/kg/day (2.60 ± 0.57%), respectively, for tramadol, and 178 ± 39 nmol/kg/day (0.93 ± 0.20%) and 90 ± 20 nmol/kg/day (0.47 ± 0.10%), respectively, for ODT. Relative infant doses [mean (range)] for tramadol plus ODT (expressed as tramadol equivalents) were 3.1% (1.82–8.52%) in EMs and 3.10% (1.81–8.43%) in PMs. Closer inspection of the simulation results revealed that irrespective of EM/PM status, none of the 1500 simulated datasets would result in a relative infant dose estimate of pharmacologically active drug that exceeded 10% of the maternal weight-adjusted dose.
Discussion
The disposition of rac-tramadol and rac-ODT in breast milk and plasma were fitted successfully to a first-order absorption and five-compartment mixed-effects model as an indirect means of evaluating exposure in breastfeeding infants. The simultaneous modeling of the two analytes in the plasma matrix and in the breast milk presented a number of challenges, particularly with the plasma dataset where there was only one observation for each patient. Given the complexity of the model (the sparse datasets available, 2 analytes and 2 matrices each, and a CYP2D6 phenotypic variation in tramadol metabolism), it was necessary to fix a number of model parameters using data from the literature. Nevertheless, we were able to fit an additive residual error for log-transformed data to both the plasma and milk compartments, while BSV could only be estimated for parameters related to the milk compartments. In the final model, we were only able to support one compartment for each analyte in its respective matrix. Previous PK models of plasma tramadol and ODT used a two-compartment model, with elimination from the central compartment [18, 28, 29]. Creamatocrit was the only significant covariate in the model. Increased milk lipid content was associated with decreased retention of ODT in the milk.
Breastfeeding leads to a small permanent loss of the drug from the body. We tried representing this loss with a function that partially emptied (67%) the breasts at 4-h intervals. There were minimal changes to the final parameter estimates (all <1%) and no significant change in the OFV. Hence, this semi-physiological representation was not included in the final model. The result also demonstrates that the clearance of tramadol and ODT by excretion in milk does not significantly influence overall drug clearance.
The mean simulated PK parameters and Cavg for plasma tramadol and ODT were comparable to published values [8, 30]. The final estimate of metabolic conversion to ODT in PMs was some 43% of that in EMs. The AUCss data in both milk and plasma reflect this difference, with PMs having lower mean milk and plasma ODT exposure than EMs. Nevertheless, if the mean AUCss for tramadol and ODT is summed in either phenotype, overall exposure to the pharmacologically active drug in milk (infant) or plasma (mother) is similar.
Our plasma PK data are comparable to those reported from previous studies. The model estimated that the volume of distribution for tramadol in plasma (332 L) was similar to the estimates of 306 L (β phase) and 168 L (1-compartment population PK model) after the oral administration of tramadol to patients undergoing surgery and to healthy volunteers [17, 31]. Similarly, the CLT (31.1 and 35.9 L/h) was in the same range as values reported after oral tramadol administration (19.6–42.6 L/h) [17, 31]. The mean simulated t½ estimates from our model in EMs and PMs for tramadol (7.1 and 8.3 h, respectively) and ODT (11.0 and 11.9 h, respectively) were similar to the range of means previously reported in healthy volunteer subjects, i.e., 4.3–8.8 h [8, 32] for tramadol and 5.3–11.9 h [8, 30, 33] for ODT. Our t½ estimate for tramadol was similar to the 7.2 h reported in women given the drug for pain control during labor, but that for ODT was significantly longer than the 5.5 h in their report [12]. The difference with ODT may be due to the use of non-compartmental analysis of pooled data in the latter study and/or because our subjects were some 4 days post-delivery, which is when pregnancy-induced changes in drug clearance are rapidly returning to normal values [34].
The mean tramadol Cavg in milk was 2749 nmol/L for EMs and 3305 nmol/L for PMs. Similarly, the mean ODT Cavg in milk was 1187 nmol/L for EMs and 602 nmol/L for PMs. These values were in close agreement with those derived by simple naïve pooling of the primary concentration data in our initial naïve pooled analysis of this dataset (2839 nmol/L for tramadol and 784 nmol/L for ODT) [4]. Not surprisingly, our mean estimates of relative infant dose in EM and PM subjects of 2.16 and 2.6% for tramadol, and 0.93 ± 0.20 and 0.47 ± 0.10% for ODT, respectively, were also in agreement with those reported in our previous publication where relative infant doses for the mixed population were 2.24% for tramadol 0.64% for ODT [4]. As none of the total relative infant doses (in tramadol equivalents) calculated from the simulated data were higher than the suggested 10% safety limit [1], it is unlikely that an infant would be exposed to relative doses higher than this recommended maximum. The mean milk:plasma ratios of 1.9 for tramadol and 3.2 for ODT in the present analysis were slightly different to those of our previous analysis (2.2 and 2.8, respectively) [4]. This difference is most likely due to the ratios in the original analysis being estimated from paired single point concentrations in milk and plasma, while in our present modeling design, they were derived from simulated AUCss data separately estimated for plasma and milk. The higher distribution of ODT into milk compared with tramadol seen with both analyses cannot be explained readily by their logDpH7 ratios of 0.23:1 and 1.07:1, respectively [35]. These ratios indicate that at a milk pH of around 7.2, lipid content should favor the transfer of tramadol over ODT. Hence, other transfer mechanisms are likely to be responsible for our observations, a conclusion that is supported by the covariate analysis which revealed that ODT transfer into milk decreased as lipid content increased.
Thus, our study clearly demonstrates that a population PK approach with sparse sampling of milk and/or plasma can be applied to estimating infant exposure to maternal drugs via breast milk. Our approach still allows milk Cavg to be used in the derivation of absolute and relative infant dose but, importantly, it also offers the ability to investigate BSV and also between-occasion variability (BOV) when the experimental design permits. In addition, our modeling of the tramadol dataset allowed us to evaluate infant dose differences in the CYP2D6 EM and PM subgroups as well as assess the influence of the covariates gestational age and creamatocrit. There is also the potential for understanding the transfer of a drug between the plasma and milk compartments in terms of the rate constants that control distribution. The sparse sampling design in a larger patient group than usually studied is also practical when dealing with the lactating mother, who has many and diverse responsibilities that could limit her participation in rich dataset experimental designs. In our analysis, we had to fix a number of parameters, both to provide identifiability and also due to an inability to estimate some parameters sufficiently. With only one plasma sample for each subject, the plasma dataset presented some modeling challenges. However, reliable estimates of structural parameters can still be obtained in this setting [25].
The downside of the population approach is that a large sample size is needed, and multiple plasma and milk concentration measurements are optimal for each patient in order to support the estimation of a large number of model parameters and be able to fully investigate BOV and BSV. From a practical point of view, we have found no difficulty in obtaining up to four milk samples per subject in such studies, but collecting more than one plasma sample per subject is problematic.
In our previous clinical study of neonate exposure to tramadol via milk at 2–4 days of age, we concluded that short-term maternal use of tramadol was compatible with breastfeeding. The present study using the same dataset shows that nonlinear mixed-effects modeling can be used to arrive at the same conclusion, with the added benefit of establishing population variability and covariate effects. Our conclusion regarding safety in lactation is conservative for two reasons. First, in estimating infant dose via milk we used an average milk intake of 0.15 L/kg/day, which probably overestimates milk production at days 2–4 of age by around 50% [36]. Second, our mean absolute infant dose via milk (154 μg/kg/day for tramadol plus its metabolite) is only 3.6% of the recommended therapeutic maintenance dose for tramadol in infants of 40 weeks post-conceptual age (4.3 mg/kg/day). Exposure to tramadol via breastmilk in older infants is also likely to be “safe” given that tramadol clearance in the first few days after birth (around 40 weeks of post-conceptual age) is some 73% of adult values, rising to 84% at 44 weeks and to adult levels by age 1 year [29]. Nevertheless, caution is warranted in individual infants, as both tramadol and ODT are pharmacologically active, the CYP2D6 polymorphism is an important covariate of tramadol clearance in the first few months of life [37], and there is evidence indicating that the clearance of ODT matures more slowly than that of tramadol [29].
In conclusion, we have shown that a sparse sampling data collection approach combined with population PK modeling can be used to describe the disposition of tramadol and ODT in plasma and milk and to subsequently estimate infant exposure to the drug via milk. In our view, this general approach is broadly suited to studies of drug disposition in milk and to the estimation of drug exposure in the breastfed infant. Nevertheless, careful pre-planning of the design of such studies is necessary to ensure that sampling strategies and covariate data are appropriately matched to the anticipated level of model complexity.
References
Bennett PN (1996) Use of the monographs on drugs. In: Bennett PN (ed) Drugs and human lactation, 2de edn. Elsevier, Amsterdam, pp 67–74
Ilett KF, Hackett LP (2007) Study design and data analysis for assessing drug transfer into milk. In: Hale TW, Hartmann PE (eds) Hale & Hartmann’s textbook of human lactation, 1st edn. Hale Publishing, Amarillo, pp 611–626
PK in Pregnancy Working Group of the Pregnancy Labeling Task Force. Guidance for industry. Clinical lactation studies—study design, data analysis, and recommendations for labeling. Draft February 2005. Uhl, K. and Stifano, T. M. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072097.pdf. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER). Accessed: 6 May 2010
Ilett KF, Paech MJ, Page-Sharp M et al (2008) Use of a sparse sampling study design to assess transfer of tramadol and its O-desmethyl metabolite into transitional breast milk. Br J Clin Pharmacol 65:661–666
Raffa RB, Friderichs E, Reimann W et al (1993) Complementary and synergistic antinociceptive interaction between the enantiomers of tramadol. J Pharmacol Exp Ther 267:331–340
Raffa RB, Friderichs E, Reimann W, Shank RP, Codd EE, Vaught JL (1992) Opioid and nonopioid components independently contribute to the mechanism of action of tramadol, an ‘atypical’ opioid analgesic. J Pharmacol Exp Ther 260:275–285
Grond S, Sablotzki A (2004) Clinical pharmacology of tramadol. Clin Pharmacokinet 43:879–923
Pedersen RS, Damkier P, Brosen K (2006) Enantioselective pharmacokinetics of tramadol in CYP2D6 extensive and poor metabolizers. Eur J Clin Pharmacol 62:513–521
Pedersen RS, Damkier P, Brosen K (2005) Tramadol as a new probe for cytochrome P450 2D6 phenotyping: a population study. Clin Pharmacol Ther 77:458–467
Gillen C, Haurand M, Kobelt DJ, Wnendt S (2000) Affinity, potency and efficacy of tramadol and its metabolites at the cloned human mu-opioid receptor. Naunyn Schmiedebergs Arch Pharmacol 362:116–121
Lai J, Ma SW, Porreca F, Raffa RB (1996) Tramadol, M1 metabolite and enantiomer affinities for cloned human opioid receptors expressed in transfected HN9.10 neuroblastoma cells. Eur J Pharmacol 316:369–372
Claahsen-van der Grinten HL, Verbruggen I, van den Berg PP, Sporken JM, Kollee LA (2005) Different pharmacokinetics of tramadol in mothers treated for labour pain and in their neonates. Eur J Clin Pharmacol 61:523–529
Allegaert K, Van den Anker JN, Verbesselt R et al (2005) O-demethylation of tramadol in the first months of life. Eur J Clin Pharmacol 61:837–842
Lucas A, Gibbs JA, Lyster RL, Baum JD (1978) Creamatocrit: simple clinical technique for estimating fat concentration and energy value of human milk. Br Med J (Clin Res Ed) 1:1018–1020
Jonsson EN, Karlsson MO (1999) Xpose-an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed 58:51–64
Wagner JG (1975) Fundamentals of clinical pharmacokinetics, 1st edn. Drug Intelligence Publications, Hamilton
Lintz W, Barth H, Osterloh G, Schmidt-Bothelt E (1986) Bioavailability of enteral tramadol formulations. 1st communication: capsules. Arzneimittelforschung 36:1278–1283
Garcia-Quetglas E, Azanza JR, Sadaba B, Munoz MJ, Gil I, Campanero MA (2007) Pharmacokinetics of tramadol enantiomers and their respective phase I metabolites in relation to CYP2D6 phenotype. Pharmacol Res 55:122–130
Garcia-Quetglas E, Azanza JR, Cardenas E, Sadaba B, Campanero MA (2007) Stereoselective pharmacokinetic analysis of tramadol and its main phase I metabolites in healthy subjects after intravenous and oral administration of racemic tramadol. Biopharm Drug Dispos 28:19–33
Lintz W, Erlacin S, Frankus E, Uragg H (1981) Biotransformation of tramadol in man and animal (author’s transl). Arzneimittelforschung 31:1932–1943
Paar WD, Poche S, Gerloff J, Dengler HJ (1997) Polymorphic CYP2D6 mediates O-demethylation of the opioid analgesic tramadol. Eur J Clin Pharmacol 53:235–239
James RJ, James A, Drewett RF, Cheetham TD (2007) Milk intake and feeding behavior in the first week of life and its relationship to cord blood ghrelin, leptin, and insulin concentrations. Pediatr Res 62:695–699
Kent JC, Mitoulas LR, Cregan MD, Ramsay DT, Doherty DA, Hartmann PE (2006) Volume and frequency of breastfeedings and fat content of breast milk throughout the day. Pediatrics 117:e387–e395
Anderson BJ, Holford NH (2008) Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol 48:303–332
Ette EI, Kelman AW, Howie CA, Whiting B (1995) Analysis of animal pharmacokinetic data: performance of the one point per animal design. J Pharmacokinet Biopharm 23:551–566
Nielsen EI, Viberg A, Lowdin E, Cars O, Karlsson MO, Sandstrom M (2007) Semimechanistic pharmacokinetic/pharmacodynamic model for assessment of activity of antibacterial agents from time-kill curve experiments. Antimicrob Agents Chemother 51:128–136
Li J, Jameson MB, Baguley BC, Pili R, Baker SD (2008) Population pharmacokinetic-pharmacodynamic model of the vascular-disrupting agent 5,6-dimethylxanthenone-4-acetic acid in cancer patients. Clin Cancer Res 14:2102–2110
Garrido MJ, Habre W, Rombout F, Troconiz IF (2006) Population pharmacokinetic/pharmacodynamic modelling of the analgesic effects of tramadol in pediatrics. Pharm Res 23:2014–2023
Allegaert K, Anderson BJ, Verbesselt R et al (2005) Tramadol disposition in the very young: an attempt to assess in vivo cytochrome P-450 2D6 activity. Br J Anaesth 95:231–239
Fliegert F, Kurth B, Gohler K (2005) The effects of tramadol on static and dynamic pupillometry in healthy subjects–the relationship between pharmacodynamics, pharmacokinetics and CYP2D6 metaboliser status. Eur J Clin Pharmacol 61:257–266
Gan SH, Ismail R, Wan Adnan WA, Zulmi W, Jelliffe RW (2004) Population pharmacokinetic modelling of tramadol with application of the NPEM algorithms. J Clin Pharm Ther 29:455–463
Malonne H, Sonet B, Streel B et al (2004) Pharmacokinetic evaluation of a new oral sustained release dosage form of tramadol. Br J Clin Pharmacol 57:270–278
Fliegert F, Kurth B, Gohler K (2006) Impact of the CYP2D6 metaboliser status on the pupillary response of tramadol-a commentary. Eur J Clin Pharmacol 62:77–78
Anderson GD (2005) Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin Pharmacokinet 44:989–1008
Chemical Abstract Services (2010). SciFinder Scholar. American Chemical Society, Washington D.C. Accessed 24 Mar 2010
Neville MC, Keller R, Seacat J et al (1988) Studies in human lactation: milk volumes in lactating women during the onset of lactation and full lactation. Am J Clin Nutr 48:1375–1386
Allegaert K, Van den Anker JN, de Hoon JN et al (2008) Covariates of tramadol disposition in the first months of life. Br J Anaesth 100:525–532
Acknowledgments
This study was supported by a research project grant from the Australian and New Zealand College of Anaesthetists.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Salman, S., Sy, S.K.B., Ilett, K.F. et al. Population pharmacokinetic modeling of tramadol and its O-desmethyl metabolite in plasma and breast milk. Eur J Clin Pharmacol 67, 899–908 (2011). https://doi.org/10.1007/s00228-011-1023-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-011-1023-6