
Abstract
The role of vascular endothelium in cardiovascular disorders is well recognized. Mature endothelial cells contribute to the repair of endothelial injury, but they only have a limited capacity to do so. This has led to growing interest and further investigation into circulating endothelial progenitor cells (EPCs) and their role in vascular healing, repair, and postnatal neovascularization. The current perception of vascular health is that of a balance between ongoing injury and resultant vascular repair, mediated at least in part by circulating EPCs. Circulating EPCs play an important role in accelerating endothelialization at areas of vascular damage, and EPC enumeration is a viable strategy for assessing reparative capacity. Recent studies have shown that EPCs are affected both in number and function by several cardiovascular risk factors as well as various cardiovascular disease states, such as hypertension, hypercholesterolemia, and coronary artery disease. The present review summarizes the most relevant studies on the effects of cardiovascular drugs on vascular function and EPCs, focusing on their mechanisms of action.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Endothelial dysfunction, atherosclerosis and cardiovascular disease
Atherosclerosis is a progressive and complex disease, characterized by thickening of the arterial wall, with focal complications in different vascular beds [1]. Risk factors and a genetic predisposition together seem to induce inflammatory processes that lead to cell damage and impair regeneration within the vessel wall [2]. However, despite intense efforts to determine the pathogenesis of atherosclerosis, this process remains poorly understood.
Several experimental and clinical studies have demonstrated that the endothelium, situated at the interface between blood flow and the vascular wall, plays a crucial role in the regulation of vascular tone and structure [1, 3]. For several years, the endothelial monolayer had been considered to be a simple barrier. It has become evident, however, that the endothelium is a complex organ, with paracrine and autocrine function, which provides a “first line” physiological defense against atherosclerosis [4]. A healthy endothelium is now considered to be a major regular of vascular homeostasis, regulating the vascular tone and structure and paying a key role in angiogenesis and inflammation.
Endothelial dysfunction is an early event in the pathogenesis of atherosclerosis. Mechanisms that can contribute to endothelial dysfunction are oxidative stress, upregulation of adhesion molecules, increase in the inflammatory response and protrombotic state. Vasoactive peptides, such as angiotensin II and endothelin-1, hypercholesterolemia, hyperhomocysteinemia, and hyperglycemia may participate in these mechanisms. Apoptosis of endothelial cells can also be implicated in endothelial dysfunction [5].
Oxidative stress can inhibit the three major endothelium-dependent vasodilator pathways, i.e., nitric oxide (NO), prostacyclin, and endothelium-derived hyperpolarizing factor and seems to be a first alteration responsible for triggering endothelial dysfunction in cardiovascular diseases. The reduced bioavailability of NO, the impairment in prostanoid synthesis, including prostacyclin, thromboxane A2, and/or isoprostanes, as well as the increased release of endothelin-1 can individually or in association contribute to the initiation and progression of the impairment of endothelial function [6].
A recently discovered pathway of prostacyclin signaling involves the activation of peroxisome proliferator-activated receptors (PPARs). These receptors seem to have very important roles in the beneficial endothelial effects of prostacyclin. In fact, this endothelium-derived vasodilator seems to have angiogenic properties that are dependent on its specific action on the PPAR signaling pathway [7].
Nitric oxide is a pivotal endothelium-derived substance. It is synthesized from the substrate L-arginine via endothelial NO synthase (eNOS) and plays a crucial role in vasorelaxation, inhibition of leukocyte-endothelial adhesion, vascular smooth muscle cell migration and proliferation, and platelet aggregation [8]. A defect in NO production or its activity has been proposed as a major mechanism of endothelial dysfunction, thus contributing to atherosclerosis [9].
A growing body of evidence shows that deregulation of the endothelial NO pathway, which can cause endothelial dysfunction, is not only associated with all major cardiovascular risk factors, such as hypercholesterolemia, diabetes, hypertension, and smoking, but that it also has a profound predictive value for future atherosclerotic disease progression [10, 11]. Therefore, the dysfunctional eNOS/NO pathway is considered to be a hallmark of endothelial dysfunction [9].
Despite experimental evidence demonstrating the contribution of endothelial eNOS/NO impairment in the pathogenesis of atherosclerosis, no single underlying mechanism can fully explain endothelial dysfunction. This may simply be due to the fact that atherosclerosis is a complex disease process and that multiple regulatory mechanisms are involved in endothelial NO bioactivity.
Endothelial progenitor cells and vascular repair
Circulating endothelial progenitor cells (EPCs) derived from bone marrow were isolated for the first time in 1997 [12]. Following the publication of this initial report by Asahara et al., growing interest has focused on gaining an understanding of the mobilization, homing, and function of EPCs under several conditions and also on its potential as a new strategy in regenerative medicine.
Bone marrow-derived EPCs contribute to the re-endothelialization of injured vessels and ischemia-induced neovascularization, and they improve endothelial function [13, 14]. Therefore, circulating EPCs may represent an important endogenous repair mechanism by which the body maintains the integrity of the endothelium monolayer [12, 15, 16]. Several clinical studies have shown that the number and function of EPCs are impaired in some pathological conditions. These findings suggest that it could be very important to estimate EPCs levels and to increase their bioactivity by appropriate interventions [13].
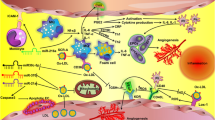
EPCs can be mobilized into the circulation in response to angiogenic growth factors, chemokines, and cytokines released following various stimuli, such as vascular trauma [17]. Granulocyte macrophage-colony stimulating factor (GM-CSF) seems to amplify EPC mobilization, recruiting them into the site of new blood vessel formation in severely ischemic tissues [18]. Hypoxia-inducible factor-1 (HIF-1), a central transcriptional regulator of hypoxia-specific gene expression, induces the expression of signaling factors, such as stromal-derived factor (SDF-1) and vascular endothelial growth factor (VEGF), in EPCs or the endothelium and facilitates adhesion of these progenitor cells to the ischemic endothelium [19]. SDF-1, also known as CXCL12, through interaction with its receptor CXCR4 (also known as CD184), modulates angiogenesis as well as hematopoiesis [2].
Of note, EPCs are believed to exert their function by two main strategies: activating locally the endothelial cells and/or differentiating into mature endothelial cells that integrate the damaged vessels. To do this, EPCs must home to “angiogenic active” sites and then adhere to the activated/damaged endothelial cells or to the extracellular matrix, thereby contributing to the endothelial repair process [20].
Recent clinical evidence supports the concept that several cardiovascular risk factors (hypertension, diabetes, hypercholesterolemia, and smoking) as well as various cardiovascular diseases are associated with EPC impairment, both in number and function [21]. Moreover, reduced EPC levels seem to be correlated with endothelial dysfunction and with an increased risk of cardiovascular events, compatible with the concept that impaired EPC-mediated vascular repair promotes the progression of vascular disease [22]. Importantly, it has been suggested that reduced levels of circulating EPCs represent a cellular marker that independently predicts outcome in patients with vascular disease, and the level of circulating EPCs has been proposed as a surrogate index of cumulative cardiovascular risk [2].
At the present time, EPC quantification is largely performed by two methods: (1) fluorescence-activated cell sorting (FACS) analysis of total blood cells or circulating mononuclear cells and (2) culture assay of blood-derived mononuclear cells. Circulating EPCs are characterized by the expression of CD133, CD34, and vascular endothelial growth factor receptor-2 (VEGFR2) [23]. Therefore, they can be identified and quantified based on the expression of these cell surface markers. In several studies, the marker combinations of CD34+KDR+, CD34+CD133+, and CD34+CD133+KDR+ have been the most frequently used markers for identifying human EPCs [24, 25].
It would be very interesting to develop pharmacological approaches that promote vascular repair and prevent endothelial cell apoptosis, thereby protecting the structural and functional integrity of the endothelium. Thus, within the context of promoting the health of the vascular system, it would be very beneficial to identify pharmacological interventions that could increase circulating EPCs levels and improve their bioactivity.
Stimulation of EPCs: a new putative effect of several cardiovascular drugs
Both experimental and human studies have demonstrated that the number of functionally active EPCs is regulated not only by various angiogenic cytokines and cardiovascular risk factors, but also by some interventions, including lifestyle modification (aerobic exercise, body weight loss, and smoking cessation) and by pharmacological intervention [15, 26].
EPCs exist in very small numbers, especially in the circulating blood of adults, where they only account for approximately 0.01% of all cells [17].
Several experimental and even clinical studies have demonstrated the impact of various pharmacological agents on the number and function of EPCs (Table 1). This impact could be of therapeutic relevance because persistent stimulation of EPCs by targeted pharmacological intervention could, at least theoretically, repair endothelial injury and prevent progression of atherosclerotic vascular disease in patients at risk.
In this review we summarize recent data on the effects of cardiovascular drugs on mobilization and functional activity of EPCs, with an attempt to focus on the mechanisms of action involved.
Statins
Endothelial dysfunction is frequently seen in hypercholesterolemic patients [3]. Unfortunately, a growing body of evidence indicates that hypercholesterolemia not only exerts a direct harmful effect on the endothelium but also indirectly reduces both the availability and functionality of EPCs, thus limiting EPC-mediated vascular repair [27].
Low-density lipoproteins (LDLs) play an active role in the onset and progression of atherosclerosis [28]. The impairment of endothelium-dependent vascular relaxation secondary to a decrease in NO bioavailability is one of the early deleterious effects produced by high plasma levels of LDLs [29]. It has also been shown that chronic hypercholesterolemia favors a proapoptotic status of vascular endothelial cells. The mechanism responsible for this effect has been extensively analyzed and seems to be mediated by reactive oxygen species (ROS) [30, 31].
The effectiveness of different 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) to reduce cholesterol levels and prevent cardiovascular morbidity and mortality has been consistently demonstrated [29]. A more recent development has been the demonstration that statins also exert benefits not related to the reduction of cholesterol levels; these are known as pleiotropic effects [32]. In addition to their lipid-lowering property, statins are also able to reduce vascular inflammation [33], decrease platelet aggregation and thrombus deposition [34], and increase endothelium-derived NO production [35]. Thus, although statins were initially designed to lower LDL-cholesterol levels, they have become established drugs in the treatment of coronary artery disease (CAD) even in patients with normal LDL levels, due to their pleiotropic effects [32]. In fact, increasing evidence supports profound, positive effects of statins on endothelial cell function through both cholesterol-dependent and cholesterol-independent effects [36, 37].
It is widely recognized that statins increase NO production by several mechanisms, including the upregulation of eNOS mRNA and protein levels and the restoration of eNOS activity reduced by oxLDLs or atherogenic native LDLs [38, 39]. Moreover, recent studies have shown that statins also exert a beneficial effect on angiogenesis and EPC bioactivity, both mechanisms that could also be associated with the significant improvement of endothelial function produced by these drugs [25, 29, 40, 41]. Therefore, augmentation of circulating EPCs by statin therapy may significantly contribute to the stimulation of neovascularization that follows tissue ischemia.
Previous data suggest that the modulation of EPC kinetics after statin treatment is unrelated to the decrease in serum LDL-cholesterol levels [25]. Landemesser et al. have demonstrated that similar reductions in LDL-cholesterol with simvastatin and ezetimibe in patients with chronic heart failure resulted in different effects on endothelial function and EPCs. There was a marked increase in functionally active EPCs after 4 weeks of statin treatment, whereas ezetimibe therapy had no effect on EPCs [42]. However, the exact mechanisms behind these serum lipid-independent effects of statins on EPCs kinetics are still not well understood.
Statin therapy and angiogenesis: mechanisms of action
Several clinical studies have clearly shown that statin therapy improves angiogenesis and EPC bioactivity. Here, we present the most relevant data on these effects and discuss the potential intracellular mechanisms involved.
Accumulating evidence suggests that several statins (rosuvastatin [43], simvastatin [42, 44, 45], atorvastatin [25, 46–48], fluvastatin [49], and cerivastatin [50]) mobilize EPCs, induce proliferation and differentiation, improve migratory function, have anti-apoptotic effects over EPCs, and increase EPC homing. However, studies with pravastatin have suggested that this statin is not as efficient as others in improving the number and function of EPCs [46, 51]. This observation, however, should be further investigated.
Several experiments establish a novel role for statins, analogous to that described for EPC-modulating cytokines, in the regulation of postnatal neovascularization [25, 41]. Statins seem to be a major stimuli for bone marrow-derived EPCs mobilization, thus improving vasculogenesis and re-endothelization [25, 41]. This effect on EPC activity may represent a novel pleiotropic effect of statin therapy [52].
There is increasing evidence for statin treatment increasing the number of circulating EPCs, based on FACS detection of CD34+/KDR+ cells or as assessed in vitro by culture assays of early EPCs. Vasa et al. showed that atorvastatin increased the number of circulating EPCs in patients with stable CAD. Moreover, this effect was as early as 1 week after the initiation of atorvastatin therapy and had risen to approximately threefold at 3–4 weeks after its introduction [25].
Dimmeler et al. demonstrated that the increase in the number of EPCs by atorvastatin is at least equipotent as the prototypic angiogenic cytokine VEGF, which is known to promote EPC mobilization and differentiation [40]. These researchers concluded that it was unlikely that the effects of statins were mediated via the upregulation of angiogenic growth factors. It would appear that statins augment circulating levels of bone marrow hematopoietic precursor cells and directly induce the differentiation of these precursor cells into EPCs. Thus, statins may influence not only the mobilization of hematopoietic progenitor cells, as has been shown for VEGF, but they may also modulate differentiation [40].
The effects of statins on eNOS appear to be critical for EPC mobilization and proliferation. Landmesser et al. showed a marked improvement in EPC mobilization and myocardial neovascularization after atorvastatin treatment in wild-type mice, while it had no effect in eNOS-deficient mice (eNOS−/−) [47]. Furthermore, in wild-type animals treated with cerivastatin, the inhibition of eNOS by nitro-L-arginine-methyl ester prevented the formation of angiotubes and collateral growth in response to ischemia, suggesting that eNOS is essential for statin to be able to enhance blood flow recovery after ischemia [50, 53].
Accumulating evidence suggests that EPCs treated with statins not only show an increased ability to form colonies and to improve proliferation [41, 54], but also demonstrate enhanced migratory capacity [25, 41, 48].
With respect to the statin-mediated delay of EPC senescence, the upregulation of the telomere-capping protein telomere repeat-binding factor TRF2 in EPCs (that prevents telomerase dysfunction) rather than an increase in telomere length is now thought to explain how statins favorably affect senescence in human EPCs [48, 54]. Human telomeres are bound by two double-strand telomere binding proteins, TRF1 and TRF2. The removal of TRF2 triggers apoptosis or senescence [55]. Premature senescence of cultured cells can be triggered by oxidative stress, leading to telomere dysfunction [56, 57]. Recent experimental and clinical evidence supports the possibility that telomere dysfunction can occur because of the loss of TRF2, implying a role of telomere “uncapping” rather than telomere shortening as an explanation for senescence [58, 59]. Spyridopoulos et al. have demonstrated that the delay of EPC premature senescence produced by statins is independent of mean telomere length [48]. In this study, exogenous TRF2 reproduced the statin effect, and a dominant-interfering mutation blocked this senescence delay, implicating TRF2 in this process [48]. The beneficial effects of statins on telomere function have also been related to ability of statins to enhance the migratory capacity of EPCs. It is possible that the induction of TRF2 by statins leads to “juvenation” of EPCs because replicative senescence is delayed. Therefore, these younger cells would migrate faster than older cells primarily due to their age characteristics [48].
There is emerging evidence suggesting that the phosphatidylinositol 3-kinase and Akt-dependent signaling pathway (PI3K/Akt) plays a critical role in statin-induced neovascularization and improvement in EPC viability [40]. In mature endothelial cells, it has been shown that the kinase Akt acts downstream of the angiogenic growth factors VEGF and angiopoietin, promoting endothelial cell survival [60]. The activation of Akt leads to the inhibition of endothelial cell apoptosis, stimulates endothelial NO synthesis, and mediates VEGF-induced endothelial cell migration [60, 61]. Akt has also been shown to be essential for EPC migration induced by VEGF and for the differentiation of EPCs [61]. Moreover, recent studies have identified Akt as a target for statins in their role as a modifier of EPC kinetics [40, 44]. It seems that statins rapidly activate Akt signaling in EPCs, enhance phosphorylation of the endogenous Akt substrate eNOS, inhibit apoptosis, and accelerate the formation of vascular structures in vitro in an Akt-dependent manner [41, 44]. Of note, the promotion of EPC proliferation, migration, and cell survival in vitro via the Akt signaling pathway by statins has been confirmed by functional blocking with dominant-negative Akt overexpression [41].
Finally, the increased adhesiveness towards endothelial cells reported in statin-treated EPCs, which could increase EPC homing at sites of vascular injury, seems to rely on the upregulation of endothelial integrin subunits α5, β1, αv and β5 [45].
In conclusion, multiple studies support that the improvement of EPC functions and neovascularization promoted by statins could contribute to the clinical benefit of these drugs and that these effects are—at least in part—independent of their lipid-lowering properties. However, several questions remain unanswered and warrant further investigation: Which mechanisms are involved in the release of EPCs by statin treatment? Is this release caused by enhanced proliferation and differentiation of committed stem cells within the bone marrow, or is it due to an increased release of a preexisting pool? What are the exact mechanisms governing these events? What makes endothelial cells attach to the injured vessel wall? How is this modulated by statins?
In fact, despite intense efforts to determine the exact mechanisms whereby statins improve EPCs function, their mechanism of action remains poorly understood.
Renin–angiotensin–aldosterone system active agents
The renin–angiotensin system (RAS) [41] is a circulating hormonal system that regulates blood pressure and flow. It is recognized that angiotensin II (Ang II) contributes to the development of atherosclerosis and promotes cardiovascular remodeling through its induction of cardiac hypertrophy, inflammation, apoptosis, and fibrosis through its action on the angiotensin type 1 receptor (AT1) [62]. Furthermore, AT1-receptor activation causes superoxide radical release via the NAPDH oxidase pathway, thereby increasing oxidative stress in the vascular wall, which leads to endothelial dysfunction [62, 63]. In contrast, angiotensin type 2 receptors (AT2) promote vasodilatation and are responsible for antifibrotic, anti-apoptotic, and anti-inflammatory effects [64].
AT2 receptors are highly expressed in the developing fetus, but they decline rapidly after birth. In the adult, the AT2-receptor is predominantly expressed in the brain and adrenals, being expressed in lower levels elsewhere. Therefore, angiotensin-dependent functions are primarily the result of AT1-receptor activation and, to a lesser extent, AT2-receptor activation [63]. Further research to assess the potential clinical benefits of specifically targeting the AT2-receptor could be of great value.
Ang II is formed by the activation of a cascade of enzymatic conversions from angiotensin I by angiotensin converting enzyme (ACE). Alternative proteases are present in the human heart (e.g., chymase), which produce Ang II independently of ACE [65]. ACE-inhibitors reduce the formation of Ang II and lead to the accumulation of NO and bradykinin [64]. The reduced stimulation of both AT1- and AT2-receptors is partially offset by an increase in AT1-receptor stimulation of the Ang II formed by the alternative pathway, potentially contributing to the “angiotensin escape phenomenon” observed in patients on chronic ACE-inhibitor treatment [64].
Recent evidence suggests that other angiotensin peptide fragments, including Ang-(1–7) and Ang-(3–8) (known as Ang IV), may bind to non-AT1 receptor subtypes [e.g., AT2, AT4, and Ang (1–7)/mas receptor] to counter-regulate the effects mediated via the AT1. Ang-(1–7) is formed directly from Ang I via the action of several tissue-specific endopeptidases or from Ang II via ACE. It interacts with the G-protein-coupled receptor mas and opposes the actions of Ang II by stimulating the release of prostacyclin and NO [67].
The Ang II metabolite, Ang IV, preferentially binds to a novel angiotensin binding site, which shows a poor affinity for Ang II (the AT4-receptor). A growing list of physiological functions has been attributed to the AT4-receptor, including an increase of blood flow in various vascular beds, antagonism of Ang II-induced hypertrophic changes, and modulation of diastolic function. Moreover, Ang IV has been shown to reverse endothelial dysfunction [66].
Both ACE-inhibitors and angiotensin II receptor blockers (ARBs), by increasing Ang I availability, elevate Ang-(1–7), which may contribute to the antihypertensive effects of these drugs.
Blockade of the RAS would be expected to improve endothelial function. There is a strong rationale for blockade of the RAS early in the cardio–reno–vascular continuum in order to prevent adverse events in patients with cardio-vascular disease.
Murohara et al. have reported that the Ang II–AT1-receptor pathway plays important roles in angiogenesis associated with ischemia and tumor [68, 69]. Moreover, Imanishi et al. have demonstrated that Ang II accelerates EPC senescence by a gp91phox-mediated increase of oxidative stress, resulting in EPCs dysfunction [70].
Spironolactone
In addition to its negative effects attributable to elevation of the blood pressure, aldosterone has been suggested to contribute to vascular damage by directly acting on the vasculature. Moreover, aldosterone can cause swelling of the endothelial cells and induce proinflammatory molecules in peripheral blood mononuclear cells [71, 72]. More recently, it has been demonstrated that aldosterone inhibits the formation of bone marrow-derived progenitor cells, at least partly, by attenuating VEGFR-2 expression and subsequent Akt signaling and that this effect of aldosterone was attenuated by cotreatment with spironolactone [73].
ACE-inhibitors
Although modulation of the RAS, by inhibition of ACE or through blockage of Ang II type 1 receptors, reduces mortality rates and major non-fatal cardiovascular events in high-risk patients, [74], the underlying mechanisms behind these beneficial effects have not been fully elucidated. There is increasing evidence suggesting that one of the pleiotropic effects of ACE-inhibitors on the cardiovascular system involves modulation of the number and functional activity of EPCs [75]. Moreover, this seems to be a class effect since an improvement in EPC biology has been demonstrated for several ACE-inhibitors (enalapril, ramipril, perindopril, and quinapril).
Treatment with enalapril has shown to benefit neovascularization in a murine hindlimb ischemia model. This ACE-inhibitor seems to augment circulating EPC levels and appears to influence EPCs through stimulating mobilization rather than maturation. In response to ischemic stress, enalapril, transiently and significantly increased the concentration of circulating SDF-1α but reduced its concentration in the bone marrow, suggesting that reduced binding of EPCs to SDF-1α in bone marrow may contribute to the release and mobilization of EPCs after ACE-inhibitor therapy [75]. The concentration gradient of plasma SDF-1α between the bone marrow and the peripheral blood may, at least in part, result from increased bone marrow activation of dipeptidylpeptidase IV (DPP IV; CD26), a cell surface endopeptidase that cleaves chemokines, such as SDF-1α [75]. Thus, the beneficial effects of enalapril seem to be at least in part due to its ability to alter the CD26/ DPP IV system, and SDF-1α seems to be critical in the mechanism through which enalapril modulates EPC mobilization [75].
Likewise, Min et al. have demonstrated that treatment with ramipril for 4 weeks increased circulating EPCs levels in patients with stable CAD [76]. Moreover, the increased number of EPCs was paralleled by an enhancement of the migratory, proliferative, adhesive, and in vitro vasculogenesis capacity of isolated EPCs [76]. In this work, the investigators suggested that the modulation of EPC kinetics after the ramipril treatment was unrelated to the decrease in blood pressure levels [76]. Moreover, as they observed that ramipril increased NO levels, they speculated that the activation of the bradykinin B2-receptor pathway by ACE-inhibitors may contribute to the observed effects of ramipril on the functional improvement of EPCs [76].
Another study demonstrated that the addition of quinapril and metoprolol to EPC therapy induced neovascularization and reduced the number of apoptotic cardiomyocytes. However, the mechanisms by which this positive effect occurred remain unclear [77].
Finally, perindopril is a long-acting ACE-inhibitor that has been demonstrated to be able to increase the number of circulating EPCs and to re-establish the ability of bone marrow mononuclear cells to differentiate into EPCs in a hindlimb ischemia model in spontaneously hypertensive rats, either alone or in combination with indapamide [78].
In conclusion, there is mounting evidence that ACE inhibition improves the biology of EPCs independently of a vasodilator or hemodynamic effect. However, again, many questions remain unsolved regarding the angiogenic effects of these widely prescribed cardiovascular drugs: What is the exact role of NO in ACE-inhibition-dependent vasculogenesis? Are all ACE-inhibitors equivalent regarding their angiogenic action? Would the magnitude of EPC improvement be similar in diabetic patients? What about other conditions? Would ACE-inhibitors be able to improve EPCs biology in patients with metabolic syndrome or heart failure?
Angiotensin II receptor blockers
Blockade of AT1 receptors results in the inhibition of a variety of deleterious effects. Furthermore, as circulating Ang II increases during treatment with AT1 blockers, the selective blockade of AT1 would be expected to result in the stimulation of unblocked AT2-receptors, thereby enhancing AT2-mediated vasodilatation and antiproliferative effects.
ARBs (olmesartan, irbesartan, losartan, candesartan, telmisartan) have consistently been reported to increase the number of EPCs [26, 79–82].
Bahlmann et al. demonstrated that Ang II subtype 1-receptor blockage (with olmesartan or irbersartan) increased the number of EPCs in patients with diabetes above that found in healthy subjects and that this effect was independent from their blood pressure-lowering action [26]. Similarly, Chen et al. demonstrated that the combined treatment with olmesartan and pravastatin increased the number of circulating EPCs in a murine model of balloon injury of the carotid arteries [79]. Additionally, studies in spontaneously hypertensive rats have shown that losartan and candesartan markedly increased the number and colony formation of EPCs and exerted a favorable effect on EPC migration, independently of their effects on blood pressure. The authors of these published studies suggested that the positive effects on EPC number and function are in part due to an antioxidant effect [81, 82].
Similarly as for statins, the so-called pleiotropic effects of ARBs are attracting the increasing interest in the cardiovascular community, including their antidiabetic and anti-inflammatory action [83, 84].
Some ARBs have also been shown to be selective PPAR-γ modulators, implicating an involvement in the metabolism, proliferation, and inflammation of cardiovascular cells [85]. Telmisartan may be especially effective in slowing down disease processes of the metabolic syndrome on account of its effect on PPAR-γ [85]. It has recently been demonstrated that telmisartan has a more potent anti-inflammatory effect than ramipril after an acute coronary syndrome. However, there are no differences between the two drugs in terms of the magnitude of improvement on EPC mobilization [80].
At the present time it is unknown whether the upregulation of progenitor cells by ARBs is linked mechanistically to the impact of these pharmacologic agents on endothelial function or whether it is just a parallel effect. Therapy with ARBs may provide a novel and effective therapeutic strategy for the repair of vascular injury. However, intracellular mechanisms involved in EPC stimulation by ARBs have yet to be explored. Further studies are required to determine the precise mechanisms of ARBs in modifying the number and function of EPCs.
Antianginals: ß-blockers, calcium antagonists, and nitrates
Multiple experimental models and a number of randomized, placebo-controlled, double-blind clinical trials have suggested anti-atherosclerosis effects for all of these antianginal agents [86]. How these effects are related to modification of EPC biology or to the documented reduction in adverse outcomes in patients with myocardial infarction, heart failure, or hypertension with ß-blockers is not clear.
Calcium antagonists: dihydropyridine calcium channel blockers
Most calcium antagonists have antioxidant effects and reduce experimental inflammatory cell invasion. Dihydropyridine-type calcium antagonists increase NO, block lipid peroxidation, and may be associated with a reduction in cardiovascular events [87].
Evidence that dihydropyridine calcium channel blockers affect the functional activity of EPCs was initially obtained with benidipine in culture studies. Treatment of murine mononuclear cells with this dihydropyridine calcium channel blocker was shown to increase the number of early EPCs after 7 days in culture, probably via the PI3K/Akt signaling pathway [88]. Additionally, nisoldipine, another L-type Ca2+ channel blocker, seems to induce the mobilization of EPCs in patients with essential hypertension independently of the blood pressure-lowering effect [89].
More recently, it has been demonstrated that nifedipine also improves the functional capacity of EPCs. This dihydropyridine calcium channel blocker enhances VEGF release from EPCs and improves the migratory capacity of cultured EPCs [90]. This study showed a nifedipine-dependent upregulation of manganese superoxide dismutase, which is known to confer resistance to oxidants, suggesting a role of the antioxidant system for the effects of nifedipine on EPCs [90].
It should be noted, however, that the majority of favorable results with calcium antagonists have been obtained from in vitro studies and that these need to be confirmed clinically. In addition, one could question whether these effects will persist in the context of cardiovascular disease, such as acute coronary syndromes and heart failure.
Beta blockers
It has recently been demonstrated that another group of cardiovascular drugs improves EPCs number and function—the beta blockers.
In spontaneously hypertensive rats, celiprolol has been shown to increase the number of circulating EPCs and to stimulate EPC colony formation and migration, while decreasing EPC senescence. This beta blocker inhibited oxidative stress in EPCs, suggesting that its beneficial effects in EPCs are mediated by its antioxidative properties [91].
Further studies with other beta blockers are required to determine if these benefits in EPC biology are exclusive of celiprolol or a class effect of all beta blockers.
Nitrates
Nitrate compounds have been used in the treatment of myocardial ischemia for more than 100 years. Their common mechanism is the release of NO, which is a major regulator of EPC mobilization, differentiation, and function [92]. Nevertheless, the majority of nitrates additionally stimulate the production of ROS, a process that is partly involved in the development of nitrate tolerance and may counteract the beneficial effects of NO on the endothelium [93].
In a small clinical trial, nitroglycerine treatment of healthy volunteers increased the level of circulating CD34 progenitor cells but also enhanced the susceptibility of expanded EPCs to apoptosis [94]. Moreover, ex vivo nitroglycerine exposure attenuated EPC differentiation, function, and survival [94].
More recently, Thum et al. demonstrated that different nitrates have a number of important effects on the circulating levels and function of EPCs [95]. Therapy with two different long-acting nitrates (nitroglycerine and isosorbide-5-dinitrate) increased the levels of circulating EPCs. However, functionally, there were strong differences between the tested nitrates, with isosorbide-5-dinitrate increasing ROS formation and impairing EPC function and nitroglycerine having favorable effects even on EPC function [95].
In one study, a considerable increase in oxidative stress was observed after treatment with certain long-acting nitrates, such as isosorbide-5-dinitrate [96]. In contrast, treatment with nitroglycerine did not cause tolerance and was not associated with evidence of increased ROS levels [95, 96]. Thum et al. concluded that the differences between the two studied long-acting nitrates in terms of induction of oxidative stress in EPCs may explain their differences in EPC biology [95].
In conclusion, further prospective studies are needed that determine the long-term effects of organic nitrates on number and function of EPCs.
Antiplatelet therapy: aspirin
Aspirin is widely used in the primary and secondary prevention of vascular disease. It is a cornerstone of therapy in acute coronary syndromes and has been shown to reduce atherosclerosis-related events in a multitude of clinical studies [97, 98]. Kharbanda et al. have reported a protective effect of aspirin against endothelial dysfunction [99].
Surprisingly, in a study with healthy humans, Chen et al. concluded that aspirin decreased the number of EPCs, decreased the proliferative, migratory, adhesive, and in vitro vasculogenesis capacity of EPCs, and decreased EPC production of iNOS in a concentration- and time-dependent manner [100]. However, recent in vivo observations have suggested that low-dose aspirin promotes the migration and adhesion and delays the onset of senescence of EPCs [101].
These studies seem to be contradictory as they provide evidence for both an inhibitory and a stimulating effect of aspirin on EPC biology. Therefore, the effects of aspirin on EPCs must be studied further.
Conclusion
Given the fact that EPCs contribute to postnatal neovascularization, an improved understanding of the regulation of EPCs could lead to new insights into the pathogenesis of vasculogenesis. Several cardiovascular drugs, such as statins, ACE-inhibitors, ARBs, and beta blockers have demonstrated a direct benefit on circulating EPCs that may contribute to their overall effects on the vascular wall. The augmentation of the number of EPCs through pharmacological modulation of the signaling pathways may be a novel strategy to improve neovascularization after ischemia and, thereby, provide a therapeutic concept for improving the numbers of EPCs in patients with cardiovascular diseases.
In conclusion, a more complete understanding of the complex factors regulating EPC biology is required in order to develop and investigate more focused clinical therapies.
References
Landmesser U, Hornig B, Drexler H (2004) Endothelial function: a critical determinant in atherosclerosis? Circulation 109:II-27–33
Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, Bohm M, Nickenig G (2005) Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med 353:999–1007
Deanfield JE, Halcox JP, Rabelink TJ (2007) Endothelial function and dysfunction: testing and clinical relevance. Circulation 115:1285–1295
Hadi H, Suwaidi J (2007) Endothelial dysfunction in diabetes mellitus. Vasc Health Risk Manag 3:853–876
Endemann DH, Schiffrin EL (2004) Endothelial dysfunction. J Am Soc Nephrol 15:1983–1992
Félétou M, Vanhoutte P (2006) Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture). Am J Physiol Heart Circ Physiol 291:H985–H1002
Pola R, Gaetani E, Flex A, Aprahamian T, Bosch-Marcé M, Losordo D, Smith R, Pola P (2008) Comparative analysis of the in vivo angiogenic properties of stable prostacyclin analogs: a possible role for peroxisome proliferator-activated receptors. J Mol Cell Cardiol 36:363–370
Davignon J, Ganz P (2004) Role of endothelial dysfunction in atherosclerosis. Circulation 109: III-27–32
Yang Z, Ming X-F (2006) Recent advances in understanding endothelial dysfunction in atherosclerosis. Clin Med Res 4:53–65
Lerman A, Zeiher AM (2005) Endothelial function: cardiac events. Circulation 111:363–368
Schachinger V, Britten MB, Zeiher AM (2000) Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 101:1899–1906
Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner J (1997) Isolation of putative progenitor endothelial cells for angiogenesis. Science 275:964–967
Umemura T, Higashi Y (2008) Endothelial progenitor cells: therapeutic target for cardiovascular diseases. J Pharmacol Sci 108:1–6
Wassmann S, Werner N, Czech T, Nickenig G (2006) Improvement of endothelial function by systemic transfusion of vascular progenitor cells. Circ Res 99:E74–E83
Kong D, Melo LG, Gnecchi M, Zhang L, Mostoslavsky G, Liew CC, Pratt RE, Dzau VJ (2004) Cytokine-induced mobilization of circulating endothelial progenitor cells enhances repair of injured arteries. Circulation 110:2039–2046
Werner N, Junk S, Laufs U, Link A, Walenta K, Bohm M, Nickenig G (2003) Intravenous transfusion of endothelial progenitor cells reduces neointima formation after vascular injury. Circ Res 93:e17–e24
Khoo C, Pozzilli P, Alison M (2008) Endothelial progenitor cells and their potential therapeutic applications. Regen Med 3:863–876
Takahashi T, Kalka C, Masuda H, Chen D, Silver M, Kearney M, Magner M, Isner JM, Asahara T (1999) Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med 5:434–438
Semenza GL (2006) Development of novel therapeutic strategies that target HIF-1. Expert Opin Ther Targets 10:267–280
Real C, Caiado F, Dias S (2008) Endothelial progenitors in vascular repair and angiogenesis: how many are needed and what to do? Cardiovasc Hematol Disord Drug Targets 8:185–193
Geft D, Schwartzenberg S, George J (2008) Circulating endothelial progenitor cells in cardiovascular disorders. Expert Rev Cardiovasc Ther 6:1115–1121
Yamahara K, Itoh H (2009) Potential use of endothelial progenitor cells for regeneration of the vasculature. Ther Adv Cardiovasc Dis 3:17–27
Hristov M, Weber C (2008) Endothelial progenitor cells in vascular repair and remodeling. Pharmacol Res 58:148–151
Peichev M, Naiyer AJ, Pereira D, Zhu Z, Lane WJ, Williams M, Oz MC, Hicklin DJ, Witte L, Moore MAS, Rafii S (2000) Expression of VEGFR-2 and AC133 by circulating human CD34+ cells identifies a population of functional endothelial precursors. Blood 95:952–958
Vasa M, Fichtlscherer S, Adler K, Aicher A, Martin H, Zeiher AM, Dimmeler S (2001) Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation 103:2885–2890
Bahlmann FH, de Groot K, Mueller O, Hertel B, Haller H, Fliser D (2005) Stimulation of endothelial progenitor cells: a new putative therapeutic effect of angiotensin II receptor antagonists. Hypertension 45:526–529
Chen J, Zhang F, Tao Q, Wang X, Zhu J, Zhu J (2004) Number and activity of endothelial progenitor cells from peripheral blood in patients with hypercholesterolaemia. Clin Sci (Lond) 107:273–280
Badimon L, Martínez-González J, Llorente-Cortés V, Rodríguez C, Padró T (2006) Cell biology and lipoproteins in atherosclerosis. Curr Mol Med 6:439–456
Rodríguez C, Slevin M, Rodríguez-Calvo R, Kumar S, Krupinski J, Tejerina T, Martínez-González J (2009) Modulation of endothelium and endothelial progenitor cell function by low-density lipoproteins: implication for vascular repair, angiogenesis and vasculogenesis. Pathobiology 76:11–22
Dimmeler S, Haendeler J, Galle J, Zeiher AM (1997) Oxidized low-density lipoprotein induces apoptosis of human endothelial cells by activation of CPP32-like proteases: a mechanistic clue to the `Response to Injury’ hypothesis. Circulation 95:1760–1763
Kume N, Kita T (2004) Apoptosis of vascular cells by oxidized LDL: involvement of caspases and LOX-1 and its implication in atherosclerotic plaque rupture. Circ Res 94:269–270
Ii M, Losordo D (2007) Statins and the endothelium. Vascul Pharmacol 46:1–9
Bustos C, Hernández-Presa M, Ortego M, Tuñón J, Ortega L, Pérez F, Díaz C, Hernández G, Egido J (1998) HMG-CoA reductase inhibition by atorvastatin reduces neointimal inflammation in a rabbit model of atherosclerosis. J Am Coll Cardiol 32:2057–2064
Lacoste L, Lam JYT, Hung J, Letchacovski G, Solymoss CB, Waters D (1995) Hyperlipidemia and coronary disease: correction of the increased thrombogenic potential with cholesterol reduction. Circulation 92:3172–3177
Laufs U, Liao JK (1998) Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem 273:24266–24271
Treasure CB, Klein JL, Weintraub WS, Talley JD, Stillabower ME, Kosinski AS, Zhang J, Boccuzzi SJ, Cedarholm JC, Alexander RW (1995) Beneficial effects of cholesterol-lowering therapy on the coronary endothelium in patients with coronary artery disease. N Engl J Med 332:481–487
Laufs U, La Fata V, Plutzky J, Liao JK (1998) Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation 97:1129–1135
Laufs U, Marra D, Node K, Liao JK (1999) 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing Rho GTPase-induced down-regulation of p27Kip1. J Biol Chem 274:21926–21931
Martinez-Gonzalez J, Raposo B, Rodriguez C, Badimon L (2001) 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition prevents endothelial NO synthase downregulation by atherogenic levels of native LDLs: balance between transcriptional and posttranscriptional regulation. Arterioscler Thromb Vasc Biol 21:804–809
Dimmeler S, Aicher A, Vasa M, Mildner-Rihm C, Adler K, Tiemann M, Rütten H, Fichtlscherer S, Martin H, Zeiher A (2001) HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest 108:391–397
Llevadot J, Murasawa S, Kureishi Y, Uchida S, Masuda H, Kawamoto A, Walsh K, Isner J, Asahara T (2001) HMG-CoA reductase inhibitor mobilizes bone marrow–derived endothelial progenitor cells. J Clin Invest 108:399–405
Landmesser U, Bahlmann F, Mueller M, Spiekermann S, Kirchhoff N, Schulz S, Manes C, Fischer D, de Groot K, Fliser D, Fauler G, Marz W, Drexler H (2005) Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation 111:2356–2363
Pirro M, Schillaci G, Romagno PF, Mannarino MR, Bagaglia F, Razzi R, Pasqualini L, Vaudo G, Mannarino E (2009) Influence of short-term rosuvastatin therapy on endothelial progenitor cells and endothelial function. J Cardiovasc Pharmacol Ther 14:14–21
Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K (2000) The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med 6:1004–1010
Walter DH, Rittig K, Bahlmann FH, Kirchmair R, Silver M, Murayama T, Nishimura H, Losordo DW, Asahara T, Isner JM (2002) Statin therapy accelerates reendothelialization: a novel effect involving mobilization and incorporation of bone marrow-derived endothelial progenitor cells. Circulation 105:3017–3024
Minami Y, Satoh M, Maesawa C, Takahashi Y, Tabuchi T, Itoh T, Nakamura M (2009) Effect of atorvastatin on microRNA 221 / 222 expression in endothelial progenitor cells obtained from patients with coronary artery disease. Eur J Clin Invest 39:359–367
Landmesser U, Engberding N, Bahlmann FH, Schaefer A, Wiencke A, Heineke A, Spiekermann S, Hilfiker-Kleiner D, Templin C, Kotlarz D, Mueller M, Fuchs M, Hornig B, Haller H, Drexler H (2004) Statin-induced improvement of endothelial progenitor cell mobilization, myocardial neovascularization, left ventricular function, and survival after experimental myocardial infarction requires endothelial nitric oxide synthase. Circulation 110:1933–1939
Spyridopoulos I, Haendeler J, Urbich C, Brummendorf TH, Oh H, Schneider MD, Zeiher AM, Dimmeler S (2004) Statins enhance migratory capacity by upregulation of the telomere repeat-binding factor TRF2 in endothelial progenitor cells. Circulation 110:3136–3142
Corsini A, Pazzucconi F, Pfister P, Paoletti R, Sirtori C (1996) Inhibitor of proliferation of arterial smooth-muscle cells by fluvastatin Lancet 348:1584
Sata M, Nishimatsu H, Suzuki E, Sugiura S, Yoshizumi M, Ouchi Y, Hirata Y, Nagai R (2001) Endothelial nitric oxide synthase is essential for the HMG-CoA reductase inhibitor cerivastatin to promote collateral growth in response to ischemia. FASEB J 15:2530–2532
Satoh M, Minami Y, Takahashi Y, Tabuchi T, Itoh T, Nakamura M (2009) Effect of intensive lipid-lowering therapy on telomere erosion in endothelial progenitor cells obtained from patients with coronary artery disease. Clin Sci (Lond) 116:827–835
Walter D, Zeiher A, Dimmeler S (2004) Effects of statins on endothelium and their contribution to neovascularization by mobilization of endothelial progenitor cells. Coron Artery Dis 15:235–242
Brouet A, Sonveaux P, Dessy C, Balligand J-L, Feron O (2001) Hsp90 ensures the transition from the early Ca2+−dependent to the late phosphorylation-dependent activation of the endothelial nitric-oxide synthase in vascular endothelial growth factor-exposed endothelial cells. J Biol Chem 276:32663–32669
Assmus B, Urbich C, Aicher A, Hofmann WK, Haendeler J, Rossig L, Spyridopoulos I, Zeiher AM, Dimmeler S (2003) HMG-CoA reductase inhibitors reduce senescence and increase proliferation of endothelial progenitor cells via regulation of cell cycle regulatory genes. Circ Res 92:1049–1055
Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T (1999) p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science 283:1321–1325
Spyridopoulos I, Isner J, Losordo D (2002) Oncogenic ras induces premature senescence in endothelial cells: role of p21(Cip1/Waf1). Basic Res Cardiol 97:117–124
Ben-Porath I, Weinberg R (2004) When cells get stressed: an integrative view of cellular senescence. J Clin Invest 113:8–13
Ferreira M, Miller K, Cooper J (2004) Indecent exposure: when telomeres become uncapped. Mol Cell 13:7–18
Karlseder J, Smogorzewska A, de Lange T (2002) Senescence induced by altered telomere state, not telomere loss. Science 295:2446–2449
Gerber H-P, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N (1998) Vascular endothelial growth factor regulates endothelial cell survival through the Phosphatidylinositol 3′-Kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem 273:30336–30343
Morales-Ruiz M, Fulton D, Sowa G, Languino LR, Fujio Y, Walsh K, Sessa WC (2000) Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt. Circ Res 86:892–896
Wassmann S, Nickenig G (2006) Pathophysiological regulation of the AT1-receptor and implications for vascular disease. J Hypertens Suppl 24:S15–S21
Nickenig G, Harrison DG (2002) The AT1-type angiotensin receptor in oxidative stress and atherogenesis: part I: oxidative stress and atherogenesis. Circulation 105:393–396
Werner C, Baumhäkel M, Teo K, Schmieder R, Mann J, Unger T, Yusuf S, Böhm M (2008) RAS blockade with ARB and ACE inhibitors: current perspective on rationale and patient selection. Clin Res Cardiol 97:418–431
Okunishi H, Miyazaki M, Toda N (1984) Evidence for a putatively new angiotensin II-generating enzyme in the vascular wall. J Hypertens 2:277–284
Vinh A, Widdop RE, Drummond GR, Gaspari TA (2008) Chronic angiotensin IV treatment reverses endothelial dysfunction in ApoE-deficient mice. Cardiovasc Res 77:178–187
Ferrario C (2006) Role of angiotensin II in cardiovascular disease therapeutic implications of more than a century of research. J Renin Angiotensin Aldosterone Syst 7:3–14
Sasaki K, Murohara T, Ikeda H, Sugaya T, Shimada T, Shintani S, Imaizumi T (2002) Evidence for the importance of angiotensin II type 1 receptor in ischemia-induced angiogenesis. J Clin Invest 109:603–611
Egami K, Murohara T, Shimada T, Sasaki K, Shintani S, Sugaya T, Ishii M, Akagi T, Ikeda H, Matsuishi T, Imaizumi T (2003) Role of host angiotensin II type 1 receptor in tumor angiogenesis and growth. J Clin Invest 112:67–75
Imanishi T, Hano T, Nishio I (2005) Angiotensin II accelerates endothelial progenitor cell senescence through induction of oxidative stress. J Hypertens 23:97–104
Oberleithner H, Ludwig T, Riethmuller C, Hillebrand U, Albermann L, Schafer C, Shahin V, Schillers H (2004) Human endothelium: target for aldosterone. Hypertension 43:952–956
Schiffrin EL (2006) Effects of aldosterone on the vasculature. Hypertension 47:312–318
Marumo T, Uchimura H, Hayashi M, Hishikawa K, Fujita T (2006) Aldosterone impairs bone marrow-derived progenitor cell formation. Hypertension 48:490–496
The OI (2008) Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 358:1547–1559
Wang C, Verma S, Hsieh I, Chen Y, Kuo L, Yang N, Wang S, Wu M, Hsu C, Cheng C, Cherng W (2006) Enalapril increases ischemia-induced endothelial progenitor cell mobilization through manipulation of the CD26 system. J Mol Cell Cardiol 41:34–43
Min TQ, Zhu CJ, Xiang WX, Hui ZJ, Peng SY (2004) Improvement in endothelial progenitor cells from peripheral blood by ramipril therapy in patients with stable coronary artery disease. Cardiovasc Drugs Ther 18:203–209
Boyle AJ, Schuster M, Witkowski P, Guosheng X, Seki T, Way K, Itescu S (2005) Additive effects of endothelial progenitor cells combined with ACE inhibition and {beta}-blockade on left ventricular function following acute myocardial infarction. J Renin Angiotensin Aldosterone Syst 6:33–37
You D, Cochain C, Loinard C, Vilar J, Mees B, Duriez M, Levy BI, Silvestre J-S (2008) Combination of the angiotensin-converting enzyme inhibitor perindopril and the diuretic indapamide activate postnatal vasculogenesis in spontaneously hypertensive rats. J Pharmacol Exp Ther 325:766–773
Chen M, Ichiki T, Ohtsubo H, Imayama I, Inanaga K, Miyazaki R, Sunagawa K (2007) Inhibition of balloon injury-induced neointimal formation by olmesartan and pravastatin in rats with insulin resistance. Hypertens Res 30:971–978
Porto I, Di Vito L, De Maria G, Dato I, Tritarelli A, Leone A, Niccoli G, Capogrossi M, Biasucci L, Crea F (2009) Comparison of the effects of ramipril versus telmisartan on high-sensitivity C-reactive protein and endothelial progenitor cells after acute coronary syndrome. Am J Cardiol 103:1500–1505
Yao E-H, Fukuda N, Matsumoto T, Kobayashi N, Katakawa M, Yamamoto C, Tsunemi A, Suzuki R, Ueno T, Matsumoto K (2007) Losartan improves the impaired function of endothelial progenitor cells in hypertension via an antioxidant effect. Hypertens Res 30:1119–1128
Yu Y, Fukuda N, Yao E-H, Matsumoto T, Kobayashi N, Suzuki R, Tahira Y, Ueno T, Matsumoto K (2008) Effects of an ARB on endothelial progenitor cell function and cardiovascular oxidation in hypertension. Am J Hypertens 21:72–77
Dahlöf B, Devereux R, Kjeldsen S, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, Lindholm L, Nieminen M, Omvik P, Oparil S, Wedel H, Group LS (2002) Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 359:995–1003
Fliser D, Buchholz K, Haller H, for the EToO, Pravastatin in I, Atherosclerosis I (2004) Antiinflammatory effects of angiotensin II subtype 1 receptor blockade in hypertensive patients with microinflammation. Circulation 110:1103–1107
Schupp M, Clemenz M, Gineste R, Witt H, Jr J, Helleboid S, Hennuyer N, Ruiz P, Unger T, Staels B, Kintscher U (2005) Molecular characterization of new selective peroxisome proliferator-activated receptor gamma modulators with angiotensin receptor blocking activity. Diabetes 54:3442–3452
Beattie MS, Shlipak MG, Liu H, Browner WS, Schiller NB, Whooley MA (2003) C-reactive protein and ischemia in users and nonusers of {beta}-blockers and statins: data from the heart and soul study. Circulation 107:245–250
Zanchetti A, Bond MG, Hennig M, Neiss A, Mancia G, Dal Palu C, Hansson L, Magnani B, Rahn K-H, Reid JL, Rodicio J, Safar M, Eckes L, Rizzini P, on behalf of the Ei (2002) Calcium antagonist lacidipine slows down progression of asymptomatic carotid atherosclerosis: principal results of the European Lacidipine Study on Atherosclerosis (ELSA), a randomized, double-blind, long-term trial. Circulation 106:2422–2427
Ando H, Nakanishi K, Shibata M, Hasegawa K, Yao K, Miyaji H (2006) Benidipine, a dihydropyridine-Ca2+ channel blocker, increases the endothelial differentiation of endothelial progenitor cells in vitro. Hypertens Res 29:1047–1054
Benndorf RA, Gehling UM, Appel D, Maas R, Schwedhelm E, Schlagner K, Silberhorn E, Hossfeld DK, Rogiers X, Böger R (2007) Mobilization of putative high-proliferative-potential endothelial colony-forming cells during antihypertensive treatment in patients with essential hypertension. Stem Cells Dev 16:329–338
Passacquale G, Desideri G, Croce G, Murgo S, Mancarelli MM, Zazzeroni F, Alesse E, Ferri C (2008) Nifedipine improves the migratory ability of circulating endothelial progenitor cells depending on manganese superoxide dismutase upregulation. J Hypertens 26:737–746
Yao E-H, Fukuda N, Matsumoto T, Katakawa M, Yamamoto C, Han Y, Ueno T, Kobayashi N, Matsumoto K (2008) Effects of the antioxidative [beta]-blocker celiprolol on endothelial progenitor cells in hypertensive rats. Am J Hypertens 21:1062–1068
Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, Zeiher AM, Dimmeler S (2003) Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med 9:1370–1376
Jung O, Marklund SL, Geiger H, Pedrazzini T, Busse R, Brandes RP (2003) Extracellular superoxide dismutase is a major determinant of nitric oxide bioavailability: in vivo and ex vivo evidence from ecSOD-deficient mice. Circ Res 93:622–629
DiFabio JM, Thomas GR, Zucco L, Kuliszewski MA, Bennett BM, Kutryk MJ, Parker JD (2006) Nitroglycerin attenuates human endothelial progenitor cell differentiation, function, and survival. J Pharmacol Exp Ther 318:117–123
Thum T, Fraccarollo D, Thum S, Schultheiss M, Daiber A, Wenzel P, Munzel T, Ertl G, Bauersachs J (2007) Differential effects of organic nitrates on endothelial progenitor cells are determined by oxidative stress. Arterioscler Thromb Vasc Biol 27:748–754
Jurt U, Gori T, Ravandi A, Babaei S, Zeman P, Parker JD (2001) Differential effects of pentaerythritol tetranitrate and nitroglycerin on the development of tolerance and evidence of lipid peroxidation: a human in vivo study. J Am Coll Cardiol 38:854–859
Antithrombotic Trialists’ Collaboration (2002) Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. Br Med J 324:71–86
Mehta J (1998) Salutary effects of aspirin in coronary artery disease are not limited to its platelet inhibitory effects. Clin Cardiol 21:879–884
Kharbanda RK, Walton B, Allen M, Klein N, Hingorani AD, MacAllister RJ, Vallance P (2002) Prevention of inflammation-induced endothelial dysfunction: a novel vasculo-protective action of aspirin. Circulation 105:2600–2604
Chen T, Chen J, Xie X (2006) Effects of aspirin on number, activity and inducible nitric oxide synthase of endothelial progenitor cells from peripheral blood. Acta Pharmacol Sin 27:430–436
Hu Z, Zhang F, Yang Z, Zhang J, Zhang D, Yang N, Zhang Y, Cao K (2008) Low-dose aspirin promotes endothelial progenitor cell migration and adhesion and prevents senescence. Cell Biol Int 32:761–768
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
António, N., Fernandes, R., Rodriguez-Losada, N. et al. Stimulation of endothelial progenitor cells: a new putative effect of several cardiovascular drugs. Eur J Clin Pharmacol 66, 219–230 (2010). https://doi.org/10.1007/s00228-009-0764-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-009-0764-y