
Abstract
Aim
The aim of the present study was to investigate the existence of time-dependent pharmacokinetics of artesunate (ARS) during 5 consecutive days of oral administration to 10 healthy Vietnamese subjects (aged 21–52 years and weighing 49–90 kg).
Methods
Each volunteer received 200 mg oral doses of ARS once daily for 5 consecutive days. Blood samples (3 ml each) were collected on days 1 and 5 at 0 (before dosing), 0.5, 1, 2, 3, 4, and 6 h after drug administration. During days 2, 3, and 4, the same volumes of blood were collected at 0, 1, 2, and 4 h after dosing. Plasma ARS and dihydroartemisinin (DHA) were measured using high-performance liquid chromatography–mass spectrometry (LC-MS).
Results
Results did not show evidence of time-dependency for ARS or the active plasma metabolite DHA. There were no differences in the concentrations of ARS and DHA at all sampling times on days 1 and 5. In addition, the pharmacokinetics of both compounds were similar on days 1 and 5. This finding confirms that the enzyme auto-induction in drug metabolism may not be characteristic of the endoperoxide sesquiterpene antimalarial group.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Artemisinin (ARN) is an endoperoxide sesquiterpene lactone antimalarial found in the wormwood Artemisia annua. Artesunate (ARS) and dihydroartemisinin (DHA) are ARN derivatives that possess more potent blood schizontocidal activity than their parent compound [1]. In addition, DHA is the active plasma metabolite of ARS. The pharmacokinetics of ARN and its derivatives have been investigated in several studies. Time-dependent pharmacokinetics of ARN has been reported in healthy volunteers [2, 3] and in malaria patients [4, 5, 6]. Auto-induction of ARN metabolism is thought to be the main cause of decreased drug bioavailability during treatment. A decline in plasma DHA concentrations during the 5-day treatment with oral ARS in patients with falciparum malaria has been reported [7]. Presumably an oral dose of ARS is mainly chemically hydrolyzed to DHA in the gut lumen and enzymatic metabolism only plays a minor role at most, and the 16.9% increased production rate of CYP2B6 by ARS can be explained by the inductive capacity of DHA [8]. Significantly lower AUC and increased CL/f were observed in patients with malaria during the convalescence phase following the administration of a 4-day oral dosing of DHA [9]. Specific metabolic pathways of these ARN derivatives have been forwarded as a hypothesis to explain these observations. The aim of the present study was to investigate the time-dependency of the pharmacokinetics of ARS when given orally for 5 consecutive days to healthy Vietnamese subjects.
Materials and methods
Subjects
Ten healthy male Vietnamese subjects aged 21–52 years and weighing 49–90 kg were recruited into the study. Sample size calculation was performed based on the data showing an increase in production rate of CYP2B6 by approximately 20% by ARS and DHA [8], and intra-individual variability of pharmacokinetic parameters (AUC and t1/2z) of DHA, which is the major active plasma metabolite of ARS [12]. A total of at least 10 subjects was therefore sufficient to detect a difference of 20% in AUC0–∞ with 90% power and coefficient of variation (CV) in pharmacokinetic parameters of 20% at a confidence level of 5%.
Biochemistry and hematology test results in all subjects were within the normal ranges. Subjects excluded from the study were those who were receiving medication or those who had a history of drug or alcohol abuse. Written informed consent was obtained from each subject prior to the study. The study protocol was approved by the ethics committee of Cho Ray Hospital, Ho Chi Minh City, Vietnam.
Study procedures
The study was conducted at the Cho Ray Hospital in Ho Chi Minh City, Vietnam, from December 2006 to June 2007. ARS tablets (50 mg) were obtained from Mekophar Chemical Pharmaceutical, Ho Chi Minh City, Vietnam (batch 07001FN). Each volunteer received 200-mg oral doses of ARS once daily for 5 consecutive days. Blood samples (3 ml each) were collected into sodium heparinized tubes on days 1 and 5 at 0 (before dosing), 0.5, 1, 2, 3, 4, and 6 h after each dosing. During days 2, 3, and 4, the same volumes of blood samples were collected at 0, 1, 2, and 4 h after each dosing. Plasma was separated immediately by centrifugation at 3,000 g for 15 min and stored at −70°C until analysis.
All patients were physically examined and adverse reactions during the study were recorded with the date and time at which they occurred and disappeared. These included gastrointestinal, central nervous, cardiovascular, dermatological effects, as well as other changes possibly attributable to the study drug. Adverse effects were assessed on the basis of non-suggestive questioning by the study investigators. Routine blood investigations (hematology and biochemistry) and urinalysis were performed prior to and at the end (2 days after last drug administration) of the study.
Drug assay
Standard powders of DHA and ARN (an internal standard) were gifts from the Central Pharmaceutical Factory N. 1, Ha Noi, Vietnam (99.8% purity). ARS standard powder was obtained from the intravenous ARS formulation (60 mg per vial; 99.5% purity), a marketing product of the Central Pharmaceutical Factory N. 1. High-performance liquid chromatography–grade methanol, acetonitrile, dichloromethane, and tert-methyl butyl ether were purchased from Merck. Glacial acetic acid was obtained from Quangzhou Chemical Factory, China. Deionized filtered water was from the Biochemistry Department at Cho Ray Hospital. Plasma from healthy volunteers used for calibration curves was provided from the blood bank of the Hematology Department, Cho Ray Hospital.
ARS, DHA, and ARN measurements were assessed using high-performance liquid chromatography–mass spectrometry (LC-MS) according to the method of Diem Thuy et al. [10]. Briefly, samples were prepared using liquid-liquid extraction with a mixture of dichloromethane:tert-methyl butyl ether (8:2, v/v) and 1 ml plasma. Chromatographic separations were performed using an Elipse XDB-C18 column (4.6 × 150 mm, 5 μm, Agilent Technologies, USA). The mobile phase consisted of a mixture of acetonitrile and 0.003 M glacial acetic acid (62:38, v/v). The mass spectrometer (Agilent 1000 Series Liquid Chromatography/Mass Spectrometer Detector Trap System, Agilent Technologies, USA) was operated using electrospray ionization in the positive ion mode. The fragment ions of ARS and DHA exhibited an m/z of 261, and the sum of both parent and daughter ion masses was used in drug quantification. Calibration curves were prepared by spiking 1 ml of blank plasma with various quantities of ARS or DHA (10, 20, 50, 100, 200, 400, 800, 1,600, and 3,200 ng) and a fixed amount of internal standard (ARN: 100 ng).
Quality control (QC) samples for ARS and DHA were made up in plasma using a stock solution separate from that used to prepare the calibration curve, at concentrations of 50, 800, and 3,200 ng/ml plasma (two samples at each concentration). Samples were aliquoted into cryovials, and stored frozen at −20 °C for use with each analytical run. The results of the QC samples provided the basis for accepting or rejecting the run. At least four of the six QC samples had to be within ±20% of their respective nominal value. Two of the six QC samples could be outside ±20% of their respective nominal value, but not at the same concentration. Concentrations of ARS and DHA were determined from regression lines of peak height ratios of ARS and DHA to ARN by using unweighted least-squares regression. The limits of quantification (LOQ) for ARS and DHA were 2 and 10 ng/ml, respectively. The intra-day variations in precision for both ARS and DHA were below 15%.
Pharmacokinetic analysis
Pharmacokinetic parameters were determined by noncompartmental analysis using ADAPT II, release 4.0 [11]. The maximum concentration (Cmax) and the time to reach maximum concentration (tmax) were obtained visually from the plasma concentration-time data. The terminal elimination rate constant (λz) was estimated by log-linear regression of at least three final plasma concentration-time data points. The elimination half-life (t1/2z) was calculated using the ratio 0.693/λz. The area under the curve from zero time to the last observed time (AUClast) was calculated using the linear trapezoidal rule for ascending data points and the log trapezoidal rule for descending data points. The area under the curve extrapolated from the last data point to infinity was estimated by dividing the concentration at the last time point by the elimination rate constant (AUCextra = Cj/λz). The area under the curve from time zero to infinity (AUC∞) was the sum of AUClast and AUCextra.
Statistical analysis
All quantitative data are presented as median (range) values, with the exception of the pooled concentration data of ARS and DHA on day 1 and day 5, which are presented as mean ± SD values. Comparisons between paired quantitative data were made using the Wilcoxon signed-rank test. Differences were considered significant at α = 0.05 for all tests.
Results
A summary of the baseline characteristics of the 10 Vietnamese healthy male volunteers is presented in Table 1.
ARS pharmacokinetics
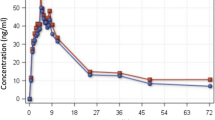
Table 2 shows the plasma concentrations of ARS and DHA following the administration of 200-mg oral doses of ARS once daily for 5 consecutive days. There were no statistically significant differences in the median ARS concentrations at sampling times of 1 and 2 h after drug administration over 5 days (Fig. 1). There were no significant differences in the ARS pharmacokinetic parameters between days 1 and 5 (Table 2). The t1/2z of ARS was 0.57 ± 0.16 h (mean ± SD, n = 20). The tmax of ARS varied between 0.5 and 1 h. The mean Cmax derived from the pooled data of days 1 and 5 was 65 ± 31 (n = 20) with a CV of 31.2%. Median Cmax values of ARS over 5 consecutive days varied between 34.7 and 97.1 ng/ml.

Plasma concentration-time profile of ARS in healthy volunteers following the administration of 200-mg oral doses of ARS once daily for 5 consecutive days. Plot data for day 1 (circles) and day 5 (squares) are presented as medians with 25th to 75th percentiles

Plasma concentration-time profile of DHA in healthy volunteers following the administration of 200-mg oral doses of ARS once daily for 5 consecutive days. Plot data for day 1 (circles) and day 5 (squares) are presented as medians with 25th to 75th percentiles
DHA pharmacokinetics
Table 2 presents plasma concentrations of the active metabolite DHA following administration of 200-mg oral doses of ARS once daily for 5 consecutive days. Median Cmax values of DHA over 5 consecutive days were in the range of 361–947 ng/ml. The inter-individual variation was high, with CV values varying from 34.3 to 131% for 0.5–6 h following drug administration. The highest CV values, ranging from 80 to 131%, were observed at sampling times of 0.5 and 1 h (Fig. 2).
The pharmacokinetic parameters of DHA on days 1 and 5 were statistically similar (Table 3). Pooled data for days 1 and 5 were used for the calculation of Cmax and t1/2z (n = 20). The Cmax of DHA was 654 ± 293 (mean ± SD) with a CV value reaching 44.8%. The t1/2z of DHA was 0.87 ± 0.24 h (mean ± SD). The tmax of DHA varied from 1 to 2 h.
Discussion
Results from the present study showed that there were no differences in concentrations of either the parent drug ARS or its active plasma metabolite DHA over 5 days in healthy volunteers receiving 200-mg oral doses of ARS once daily for 5 consecutive days. All pharmacokinetic parameters, i.e., tmax, Cmax, t1/2z, AUClast, and AUC0–∞ were similar between days 1 and 5. These findings suggest that the reduction in DHA concentrations on day 5 of a 5-day oral DHA monotherapy regimen observed in patients with uncomplicated falciparum malaria may not be due to auto-induction of DHA metabolism, but instead attributable to the restoration of drug-metabolizing enzymes during the convalescent phase of malaria infection [9]. This seems to uphold the theory that not all endoperoxide antimalarials are inducers of their own metabolism.
An observation of a decline in plasma DHA concentrations reported during the 5-day treatment with oral ARS in patients with malaria may be partly explained by the low sensitivity of the assay method used for drug analysis (LOQ 50 ng/ml) [7]. The increase in hepatic enzyme activities and hepatic blood flow as a consequence of the disease recovery [13, 14] could lead to the reduction in Cmax and AUC of ARS or DHA during the second half of the 5-day treatment period.
Nevertheless, there were limitations in the design of the present study. Five days may be a relatively short time interval to make a good in vivo test for possible enzyme induction. Although the sample size of 10 subjects was calculated based on rational grounds, an absence of an influence of enzyme induction on pharmacokinetics of ARS and DHA cannot be definitely concluded. The estimation of the terminal elimination rate constant was based only on three data points, and therefore an inaccurate estimation of λz could have a major influence on both AUC (and clearance)—parameters that are crucial for any conclusion about induction. The calculation of volume of distribution and total clearance was not possible without convincing data of a more or less complete conversion of ARS to DHA.
The observation that DHA and ARS failed to exhibit time-dependent pharmacokinetic characteristics is concordant with the hypothesis that the metabolic pathway of DHA may differ from ARN. In a study on 17 Vietnamese falciparum malaria patients treated with intravenous ARS [15], the active plasma metabolite DHA was found to be converted to α-DHA-β-glucuronide (α-DHA-G), which was excreted in urine. Human liver microsomes incubated with [12-3H]DHA and cofactors for either glucuronidation or cytochrome P450-catalyzed oxidation in vitro revealed that DHA was metabolized mainly via glucuronidation. The formation of α-DHA-G is catalyzed by UDP-glucuronosyltransferases (UGT) including UGT1A9 and UGT2B7 [15]. It should be realized that it cannot be excluded that UGT is an inducible enzyme [15]. In contrast, the in vitro metabolism of ARN has been shown to be cytochrome P450-dependent via a specific isoform, CYP2B6, and CYP2C19 is inducible by ARN [16, 17].
The conversion of ARS to DHA is most likely due to enzymatic (non-CYP450) ester hydrolysis and pH-dependent chemical hydrolysis [18]. However, the metabolism of another ARN derivative, arteether (ARE), to DHA by human liver microsomes has been shown to be mediated by CYP2B6, CYP3A4, and CYP3A5. CYP3A4 catalyzed the transformation of ARE to DHA at rates that were ten- and fivefold those found for CYP2B6 and CYP3A5, respectively [19]. In human liver, the amounts of CYP2B6 and CYP3A5 are approximately 0.7 and 20% of the amount of CYP3A4, respectively [20]. Therefore, CYP3A4 is the primary enzyme involved in the formation of DHA from ARE.
The susceptibility of P. falciparum parasites to antimalarial drugs tends to decrease over time. There have not been any reports of clinical resistance to ARN and its derivatives thus far. Resistance to these antimalarials, however, is highly likely to develop if they are used extensively as monotherapy regimens for malaria treatment. Due to the absence of pharmacokinetic time-dependency for both ARS and DHA, together with the assumption that the physiological status in the convalescent phase (5–7 days after treatment) in malaria patients is similar to that in healthy volunteers, the steady-state concentrations of ARS and DHA during the recovery phase of the disease may be used to monitor parasite sensitivity to these drugs in areas affected by malaria. The early detection of the development of drug resistance can therefore permit more careful dosage regimen management in treating falciparum malaria. Finally, the absence of time-dependent pharmacokinetics for ARS and DHA supports the clinical use of ARS or DHA in artemisinin-based combination therapies (ACTs) [21].
References
Li GQ, Guo XB, Fu LC, Jian HX, Wang XH (1994) Clinical trials of artemisinin and its derivatives in the treatment of malaria in China. Trans R Soc Trop Med Hyg 88(Suppl 1):S5–S6
Ashton M, Hai TN, Sy ND, Huong DX, Van Huong N, Niêu NT, Công LD (1998) Artemisinin pharmacokinetics is time-dependent during repeated oral administration in healthy male adults. Drug Metab Dispos 26:25–27
Ashton M, Gordi T, Trinh NH, Nguyen VH, Nguyen DS, Nguyen TN, Dinh XH, Johansson M, Le DC (1998) Artemisinin pharmacokinetics in healthy adults after 250, 500 and 1000 mg single oral doses. Biopharm Drug Dispos 19:245–250
Ashton M, Nguyen DS, Nguyen VH, Gordi T, Trinh NH, Dinh XH, Nguyen TN, Le DC (1998) Artemisinin kinetics and dynamics during oral and rectal treatment of uncomplicated malaria. Clin Pharmacol Ther 63:482–493
Alin MH, Ashton M, Kihamia CM, Mtey GJ, Björkman A (1996) Clinical efficacy and pharmacokinetics of artemisinin monotherapy and in combination with mefloquine in patients with falciparum malaria. Br J Clin Pharmacol 4:587–592
Hassan Alin M, Ashton M, Kihamia CM, Mtey GJ, Björkman A (1996) Multiple dose pharmacokinetics of oral artemisinin and comparison of its efficacy with that of oral artesunate in falciparum malaria patients. Trans R Soc Trop Med Hyg 90:61–65
Khanh NX, de Vries PJ, Ha LD, van Boxtel CJ, Koopmans R, Kager PA (1999) Declining concentrations of dihydroartemisinin in plasma during 5-day oral treatment with artesunate for falciparum malaria. Antimicrob Agents Chemother 43:690–692
Elsherbiny DA, Asimus SA, Karlsson MO, Ashton M, Simonsson US (2008) A model based assessment of the CYP2B6 and CYP2C19 inductive properties by artemisinin antimalarials: implications for combination regimens. J Pharmacokinet Pharmacodyn 10928:9084–9096
Diem Thuy LT, Ngoc Hung L, Danh PT, Na-Bangchang K (2008) Pharmacokinetics of a five-day oral dihydroartemisinin monotherapy regimen in patients with uncomplicated falciparum malaria. Drug Met Pharmacokinet (in press)
Diem Thuy LT, Ngoc Hung L, Danh PT, Na-Bangchang K (2008) Development and validation of a liquid chromatography-mass spectrometry method for the simultaneous quantification of artesunate and dihydroartemisininin human plasma (submitted)
d’ Argenio DZ, Schumitzky A (2003) ADAPT II, release 4: pharmacokinetic/pharmacodynamic system analysis software. http://bmsr.usc.edu/Software/ADAPT/ADAPT.html. Accessed 14 May 2008
Na-Bangchang K, Krudsood S, Silachamroon U, Molunto P, Tasanor O, Chalermrut K, Tangpukdee O, Kano S, Looareesuwan S (2004) The pharmacokinetics of oral dihydroartemisinin and artesunate in healthy Thai volunteers. Southeast Asian J Trop Med Pub Health 35:575–580
Batty KT, Ashton M, Ilett KF, Edwards G, Davis TM (1998) The pharmacokinetics of artemisinin (ART) and artesunate (ARTS) in healthy volunteers. Am J Trop Med Hyg 58:125–126
Molyneux ME, Looareesuwan S, Menzies IS, Grainger SL, Phillips RE, Wattanagoon Y, Thompson RP, Warrell DA (1989) Reduced hepatic blood flow and intestinal malabsorption in severe falciparum malaria. Am J Trop Med Hyg 40:470–476
Ilett KF, Ethell BT, Maggs JL, Davis TM, Batty KT, Burchell B, Binh TQ, Thu le TA, Hung NC, Pirmohamed M, Park BK, Edwards G (2002) Glucuronidation of dihydroartemisinin in vivo and by human liver microsomes and expressed UDP- glucuronosyltransferases. Drug Metab Dispos 30:1005–1012
Svensson US, Ashton M, Trinh NH, Bertilsson L, Dinh XH, Nguyen VH, Nguyen TN, Nguyen DS, Lykkesfeldt J, Le DC (1998) Artemisinin induces omeprazole metabolism in human beings. Clin Pharmacol Ther 64:160–167
Svensson US, Ashton M (1999) Identification of the human cytochrome P450 enzymes involved in the in vitro metabolism of artemisinin. Br J Clin Pharmacol 48:528–535
Olliaro PL, Nair NK, Sathasivam K, Mansor SM, Navaratnam V (2002) Pharmacokinetics of artesunate after single oral administration to rats. BMC Pharmacol 1:12–15
Grace JM, Aguilar AJ, Trotman KM, Peggins JO, Brewer TG (1998) Metabolism of beta-arteether to dihydroqinghaosu by human liver microsomes and recombinant cytochrome P450. Drug Metab Dispos 26:313–317
Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP (1994) Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther 270:414–423
Mutabingwa TK (2005) Artemisinin-based combination therapies (ACTs): best hope for malaria treatment but inaccessible to the needy! Acta Trop 95:305–315
Acknowledgements
The study was supported by Thammasat University and UNDP-UNICEF Special Program in Research and Training in Tropical Diseases. We thank Dr. Matthew J. Cheesman for editing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Diem Thuy, L.T., Ngoc Hung, L., Danh, P.T. et al. Absence of time-dependent artesunate pharmacokinetics in healthy subjects during 5-day oral administration. Eur J Clin Pharmacol 64, 993–998 (2008). https://doi.org/10.1007/s00228-008-0506-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-008-0506-6