
Abstract
Objective
Loperamide is biotransformed in vitro by the cytochromes P450 (CYP) 2C8 and 3A4 and is a substrate of the P-glycoprotein efflux transporter. Our aim was to investigate the effects of itraconazole, an inhibitor of CYP3A4 and P-glycoprotein, and gemfibrozil, an inhibitor of CYP2C8, on the pharmacokinetics of loperamide.
Methods
In a randomized crossover study with 4 phases, 12 healthy volunteers took 100 mg itraconazole (first dose 200 mg), 600 mg gemfibrozil, both itraconazole and gemfibrozil, or placebo, twice daily for 5 days. On day 3, they ingested a single 4-mg dose of loperamide. Loperamide and N-desmethylloperamide concentrations in plasma were measured for up to 72 h and in urine for up to 48 h. Possible central nervous system effects of loperamide were assessed by the Digit Symbol Substitution Test and by subjective drowsiness.
Results
Itraconazole raised the peak plasma loperamide concentration (Cmax) 2.9-fold (range, 1.2–5.0; P<0.001) and the total area under the plasma loperamide concentration-time curve (AUC0-∞) 3.8-fold (1.4–6.6; P<0.001) and prolonged the elimination half-life (t½) of loperamide from 11.9 to 18.7 h (P<0.001). Gemfibrozil raised the Cmax of loperamide 1.6-fold (0.9–3.2; P<0.05) and its AUC0-∞ 2.2-fold (1.0–3.7; P<0.05) and prolonged its t½ to 16.7 h (P<0.01). The combination of itraconazole and gemfibrozil raised the Cmax of loperamide 4.2-fold (1.5–8.7; P<0.001) and its AUC0-∞ 12.6-fold (4.3–21.8; P<0.001) and prolonged the t½ of loperamide to 36.9 h (P<0.001). The amount of loperamide excreted into urine within 48 h was increased 3.0-fold, 1.4-fold and 5.3-fold by itraconazole, gemfibrozil and their combination, respectively (P<0.05). Itraconazole, gemfibrozil and their combination reduced the plasma AUC0–72 ratio of N-desmethylloperamide to loperamide by 65%, 46% and 88%, respectively (P<0.001). No significant differences were seen in the Digit Symbol Substitution Test or subjective drowsiness between the phases.
Conclusion
Itraconazole, gemfibrozil and their combination markedly raise the plasma concentrations of loperamide. Although not seen in the psychomotor tests used, an increased risk of adverse effects should be considered during concomitant use of loperamide with itraconazole, gemfibrozil and especially their combination.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Loperamide is a peripherally acting μ opioid receptor agonist, which is widely used in the symptomatic treatment of diarrhoea. It is devoid of the typical central nervous system effects of opioids, because of a low oral bioavailability and low penetration through the blood-brain barrier [1, 2].
The exact oral bioavailability of loperamide in humans is unknown. Its absorption is thought to be incomplete, and it is supposed to be subject to considerable first-pass metabolism, because the plasma concentrations of unchanged loperamide are very low [1, 3]. The elimination half-life (t½) of loperamide is about 10 to 20 h [1, 3]. In vitro studies indicate that loperamide is biotransformed mainly by the cytochrome P450 (CYP) 2C8 and 3A4 enzymes to its main in vivo metabolite, N-desmethylloperamide [4]. In addition, loperamide is a substrate of the P-glycoprotein drug transporter [2]. Intestinal expression of the P-glycoprotein may partly explain the low oral bioavailability of loperamide, and its expression at the blood-brain barrier may explain the low penetration of loperamide to the central nervous system [2, 5].
Concomitant use of quinidine, a potent inhibitor of the P-glycoprotein [6], has resulted in opioid-like central nervous system effects after loperamide intake, associated with only marginal effects on the plasma concentrations of loperamide [7, 8]. On the other hand, ritonavir, an inhibitor of both the P-glycoprotein and CYP3A4 [9, 10], has increased the area under the plasma loperamide concentration-time curve (AUC) 2.7-fold without evidence for enhanced central nervous system effects [11]. The combination of the CYP2C8 inhibitor trimethoprim and CYP2C9 inhibitor sulfamethoxazole has raised the AUC of loperamide 1.9-fold [12].
The importance of CYP2C8 in drug metabolism has become increasingly recognized during the past few years [13]. Gemfibrozil is one of the most potent inhibitors of CYP2C8 in vivo and it has markedly raised the plasma concentrations of several CYP2C8 substrates; for example, repaglinide (8.1-fold increase in AUC) [14], cerivastatin (5.6-fold increase in AUC) [15], pioglitazone (3.2-fold increase in AUC) [16], and rosiglitazone (2.3-fold increase in AUC) [17]. In addition to inhibiting CYP2C8 [18, 19], in vitro studies have shown that gemfibrozil inhibits the hepatic organic anion transporting polypeptide 1B1 (OATP1B1) [20] and some UDP-glucuronosyltransferases [21]. Of note, gemfibrozil does not inhibit CYP3A4 [22] or the P-glycoprotein [23]. Itraconazole, on the other hand, is a potent inhibitor of CYP3A4 [24, 25] and the P-glycoprotein [26, 27], but it does not inhibit CYP2C8 either in vitro or in vivo [16, 28].
We hypothesized that inhibition of CYP2C8 by gemfibrozil, and CYP3A4 and P-glycoprotein by itraconazole might increase the plasma concentrations of loperamide. Therefore, we have investigated the effects of itraconazole, gemfibrozil and their combination on the pharmacokinetics and effects of loperamide in healthy subjects.
Materials and methods
Subjects
Twelve healthy volunteers (6 men, 6 women) participated in the study after giving written informed consent. Their mean (±SD) age was 21±2 years (range, 19–25 years), weight 70±14 kg (range, 48–95 kg) and height 176±8 cm (range, 158–184 cm). They were ascertained to be healthy by medical history, physical examination and routine laboratory tests before they were entered in the study. None was a tobacco smoker or used any continuous medication.
Study design
The study protocol was approved by the Ethics Committee for Studies in Healthy Subjects and Primary Care of the Helsinki and Uusimaa Hospital District and by the National Agency for Medicines. A randomized crossover study with four phases and a washout period of 4 weeks was carried out. The volunteers took 100 mg itraconazole (first dose 200 mg; Sporanox, Janssen-Cilag, Borgo San Michele, Italy), 600 mg gemfibrozil (Lopid, Parke-Davis, Freiburg, Germany), both itraconazole and gemfibrozil, or placebo orally twice daily at 0900 hours and 2100 hours for 5 days. After an overnight fast on day 3, they received their morning dose of itraconazole, gemfibrozil, their combination or placebo at 0800 hours. At 0900 hours, the volunteers ingested a single dose of 4 mg loperamide (two Imodium 2 mg capsules; Orion Pharma, Espoo, Finland; under licence of Janssen Pharmaceutica, Beerse, Belgium) with 150 ml water. The subjects were under direct medical supervision during the days of administration of loperamide. Naloxone for intravenous and intramuscular use was available in case of central nervous system depression, but was not needed.
Blood sampling and urine collection
On the days of administration of loperamide, a forearm vein of each subject was cannulated with a plastic cannula and kept patent with an obturator. Timed blood samples were drawn prior to and 1, 2, 3, 4, 5, 7, 9, 12, 24, 36, 48 and 72 h after the administration of loperamide. The blood samples (5–10 ml each) were taken into tubes that contained ethylenediaminetetraacetic acid. Plasma was separated within 30 min after blood sampling and stored at −70°C until analysis. All excreted urine was collected in the following fractions in relation to the administration of loperamide: −1–12 h, 12–24 h, 24–36 h and 36–48 h. The volume of the urine was measured and an aliquot of 5 ml from each fraction was stored at −70°C until analysis.
Determination of drug concentrations in plasma and urine
Loperamide and its N-desmethyl metabolite were extracted as previously described [29] and their concentrations were measured by use of an Applied Biosystems/MDS SCIEX Q-Trap liquid chromatography-tandem mass spectrometry system (Sciex Division of MDS, Foster City, Calif.). Chromatographic separation was achieved on a Symmetry C8 column (150×2.1 mm internal diameter, 3.5 μm particle size) (Waters, Milford, Mass.) protected by a Symmetry C8 guard column (10×2 mm internal diameter) (Waters). Mobile phase consisted of 5 mmol·l−1 ammonium formate (pH 4.0, adjusted with 99% formic acid) (A) and acetonitrile (B). The mobile phase gradient comprised 0 min at 65% A, 4 min to 50% A, 4 min to 0% A, 5 min at 0% A and 12 min at 65% A, giving a total chromatographic run time of 25 min. An aliquot of 5 μl was injected at a mobile phase flow rate of 180 μl/min. The mass spectrometer was operated in positive TurboIonSpray mode and the samples were analyzed via multiple reaction monitoring employing precursor ion to product ion for each analyte and internal standard (acetylloperamide). The ion transitions were m/z 477 to m/z 266 for loperamide, m/z 463 to m/z 252 for N-desmethylloperamide and m/z 519 to m/z 266 for acetylloperamide. The limit of quantification of plasma loperamide was 0.05 ng/ml and the inter-assay coefficient of variation (CV) was 7.8% at 0.1 ng/ml (n=8), 10.1% at 1.0 ng/ml (n=8) and 9.0% at 10 ng/ml (n=9). The limit of quantification of loperamide in urine was 0.1 ng/ml and the inter-assay CV was 5.4% at 10 ng/ml and 8.1% at 100 ng/ml (n=6). N-desmethylloperamide concentrations are given in arbitrary units (U/ml) relative to the ratio of the peak height of N-desmethylloperamide to that of the internal standard in the chromatogram. Authentic N-desmethylloperamide standard was not available for its absolute quantification, but the use of arbitrary units should not affect the conclusions regarding relative changes in kinetic data. A signal to noise ratio of 10:1 was used as determination limit and the CV for N-desmethylloperamide in plasma was 9.7% (n=15) and in urine 6.7% (n=8). Gemfibrozil and itraconazole did not interfere with the assay. Plasma gemfibrozil, itraconazole and hydroxyitraconazole concentrations were determined by HPLC [30–32]. The limit of quantification was 0.1 μg/ml for gemfibrozil and 10 ng/ml for itraconazole and hydroxyitraconazole. Day-to-day CV was below 7% for gemfibrozil (n=9) and below 6% for itraconazole and hydroxyitraconazole (n=6) at relevant concentrations.
Pharmacokinetics
The pharmacokinetics of loperamide and N-desmethylloperamide were characterized by peak concentration in plasma (Cmax), time to Cmax (tmax), areas under the plasma concentration-time curve (AUC0–72 and AUC0-∞), elimination half-life (t½), amount excreted into urine from 0 to 48 h (Ae), renal clearance (CLrenal) and the ratios of the Ae and plasma AUC0–72 of N-desmethylloperamide to that of loperamide, as appropriate. The pharmacokinetics of gemfibrozil, itraconazole and hydroxyitraconazole were characterized by Cmax and AUC0–13 on day 3 and the morning trough concentration on days 3 (C0,3d), 4 (C0,4d), 5 (C0,5d) and 6 (C0,6d). The terminal log-linear part of each concentration-time curve was identified visually. The elimination rate constant (ke) was determined by linear regression analysis of the log-linear part of the plasma drug concentration-time curve. The t½ was calculated by the equation t½=ln2/ke. The AUC values were calculated by a combination of the linear and log-linear trapezoidal rules with extrapolation to infinity, when appropriate, by division of the last measured concentration by ke. The CLrenal of loperamide was calculated by dividing the amount excreted into urine from 0 to 48 h by AUC0–48.
Psychomotor measurements
The effects of loperamide were assessed by subjective drowsiness and the Digit Symbol Substitution Test (DSST) before administration of loperamide and immediately after each blood sampling for up to 12 h. Before the first study phase, the volunteers were properly trained to perform the tests. In the DSST, the number of digits correctly substituted in 2 min was recorded. Subjective drowsiness was measured with a 100-mm-long horizontal visual analog scale (VAS). For both variables, the area under the effect-time curve from 0 to 12 h (AUEC0–12) was calculated by the linear trapezoidal rule. In addition, the maximum responses in both variables were recorded.
Statistical analysis
Results are expressed as mean values ±SD in the text and tables and as mean values ±SEM in the figures. The pharmacokinetic and pharmacodynamic variables of loperamide and N-desmethylloperamide after the four pretreatments were compared with repeated measures analysis of variance (ANOVA), followed by a posteriori testing with the paired t-test with the Bonferroni correction. The tmax values were compared with Friedman’s ANOVA followed by the Wilcoxon signed-rank test with the Bonferroni correction. Comparisons of the pharmacokinetic variables of gemfibrozil, itraconazole and hydroxyitraconazole between the gemfibrozil or itraconazole alone and their combination phase were made with the paired t-test. The Pearson correlation coefficient was used for investigation of relationships between the pharmacokinetic variables of gemfibrozil, itraconazole and hydroxyitraconazole and the extent of interaction with loperamide. All the data were analyzed with the statistical program Systat for Windows, version 6.0.1 (SPSS, Chicago, Ill.). Differences were considered statistically significant when P was less than 0.05.
Results
Pharmacokinetics of loperamide and N-desmethylloperamide
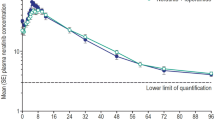
Treatment with itraconazole, gemfibrozil, as well as their combination resulted in significantly increased plasma concentrations of loperamide (Fig. 1, Table 1). Itraconazole raised the mean Cmax of loperamide 2.9-fold (range, 1.2-fold to 5.0-fold; P<0.001) and its AUC0-∞ 3.8-fold (1.4-fold to 6.6-fold; P<0.001) and prolonged the t½ of loperamide from 11.9 to 18.7 h (P<0.001). Gemfibrozil raised the Cmax of loperamide 1.6-fold (0.9-fold to 3.2-fold; P<0.05) and its AUC0-∞ 2.2-fold (1.0-fold to 3.7-fold; P<0.05) and prolonged its t½ to 16.7 h (P<0.01). The combination of itraconazole and gemfibrozil raised the Cmax of loperamide 4.2-fold (1.5-fold to 8.7-fold; P<0.001) and its AUC0-∞ 12.6-fold (4.3-fold to 21.8-fold; P<0.001) and prolonged the t½ of loperamide to 36.9 h (P<0.001). The longest observed t½ of loperamide in the itraconazole-gemfibrozil phase was 47.9 h. Itraconazole, gemfibrozil and their combination reduced the plasma AUC0–72 ratio of N-desmethylloperamide to loperamide by 65%, 46% and 88%, respectively (P<0.001) (Fig. 1, Table 1).

Mean±SEM plasma concentrations of loperamide (a) and N-desmethylloperamide (b) in 12 healthy volunteers. A single oral dose of 4 mg loperamide was given on day 3 of a 5-day treatment with 100 mg itraconazole (first dose 200 mg), 600 mg gemfibrozil, both itraconazole and gemfibrozil, or placebo twice daily. Insets depict the same data on a semi-logarithmic scale. Open circles indicate placebo phase; solid squares gemfibrozil phase; solid triangles itraconazole phase; solid stars itraconazole-gemfibrozil phase
Itraconazole, gemfibrozil and their combination increased the amount of loperamide excreted into urine 3.0-fold (P<0.001), 1.4-fold (P<0.05) and 5.3-fold (P<0.001) of the control, respectively. Gemfibrozil and the combination of gemfibrozil and itraconazole reduced the CLrenal of loperamide by 28% (P<0.05) and 34% (P<0.01), respectively, compared with placebo. The ratio of N-desmethylloperamide to loperamide in urine (from 0 to 48 h) was decreased by 64%, 31% and 86% by itraconazole, gemfibrozil and their combination, respectively (P<0.001).
Psychomotor effects of loperamide
No significant differences between the phases were seen in the effects of loperamide on performance in DSST or in subjective drowsiness (Fig. 2, Table 2).

Mean±SEM recording of subjective drowsiness (visual analog scale; VAS) (a) and results of Digit Symbol Substitution Test (DSST) (b) following a single oral dose of 4 mg loperamide on day 3 of a 5-day treatment with 100 mg itraconazole (first dose 200 mg), 600 mg gemfibrozil, both itraconazole and gemfibrozil, or placebo twice daily. Open circles indicate placebo phase; solid squares gemfibrozil phase; solid triangles itraconazole phase; solid stars itraconazole-gemfibrozil phase. For clarity, only some of the error bars are presented
Pharmacokinetics of gemfibrozil, itraconazole and hydroxyitraconazole
The plasma concentrations of itraconazole and hydroxyitraconazole were markedly lower during the itraconazole-gemfibrozil phase than during the itraconazole phase (Fig. 3, Table 3). Gemfibrozil reduced the mean Cmax and AUC0–13 of itraconazole by 57% and 54%, respectively (P<0.001), and those of hydroxyitraconazole by 37% and 41%, respectively (P<0.001). In one subject (no. 10), the plasma concentrations of itraconazole were below the limit of quantification (i.e., below 10 ng/ml) for up to 25 h after the intake of itraconazole at 0800 hours on day 3 and his morning trough itraconazole concentrations were very low on days 5 and 6. In the same subject, the AUC0-∞ of loperamide during the itraconazole-gemfibrozil phase was smaller than in any other subject.

Mean±SEM plasma concentrations of itraconazole (a), hydroxyitraconazole (b) and gemfibrozil (c) in 12 healthy volunteers on days 3 to 6 of a 5-day treatment with 100 mg itraconazole (first dose 200 mg), 600 mg gemfibrozil, both itraconazole and gemfibrozil, or placebo twice daily. Solid squares indicate gemfibrozil phase; solid triangles itraconazole phase; solid stars itraconazole-gemfibrozil phase
Itraconazole had no significant effect on the plasma concentrations of gemfibrozil on day 3, but the morning trough concentrations of gemfibrozil were slightly higher on days 5 and 6 during the itraconazole-gemfibrozil phase than during the gemfibrozil phase (Fig. 3, Table 4). No statistically significant correlations were evident between the pharmacokinetic variables of itraconazole, hydroxyitraconazole and gemfibrozil and the extent of their interaction with loperamide.
Discussion
This study showed that administration of normal therapeutic doses of either itraconazole or gemfibrozil alone markedly raised the plasma concentrations of loperamide. Itraconazole raised the mean AUC of loperamide 3.8-fold (in some subjects up to 6.6-fold), and gemfibrozil raised it 2.2-fold (up to 3.7-fold). Furthermore, when itraconazole and gemfibrozil were taken concomitantly, they acted synergistically to further raise the plasma concentrations of loperamide. Concomitant use of itraconazole and gemfibrozil raised the mean AUC of loperamide 12.6-fold, in some subjects even more than 20-fold. Although not seen in the DSST or subjective drowsiness, such increases in loperamide plasma concentrations may result in adverse effects, especially when loperamide is used at a higher dose or on a regular basis.
The findings that itraconazole and gemfibrozil alone, and particularly in combination, reduced the plasma N-desmethylloperamide to loperamide AUC0–72 ratio and the ratio of N-desmethylloperamide to loperamide in urine indicate that both itraconazole and gemfibrozil inhibited the biotransformation of loperamide to its main metabolite, N-desmethylloperamide. This is well in line with recent in vitro data showing that loperamide is mainly biotransformed by CYP3A4 and CYP2C8 [4]. It was estimated that, at the very low clinically observed plasma concentrations of loperamide, loperamide N-demethylation would be catalyzed by CYP3A4 (53%) to a slightly greater extent than by CYP2C8 (38%) [4]. The present in vivo results suggest that CYP3A4 and CYP2C8 contribute to the metabolism of loperamide in a similar order, also in vivo, in humans (i.e., itraconazole had a greater effect than gemfibrozil on the pharmacokinetics of loperamide). In addition, these data substantiate the role of gemfibrozil as a potent inhibitor of CYP2C8 in vivo. Because loperamide is a substrate [2] and itraconazole an inhibitor [26, 27] of the P-glycoprotein, inhibition of the P-glycoprotein in the gut wall could also have contributed to their pharmacokinetic interaction, by increasing the oral bioavailability of loperamide.
The finding that the combination of itraconazole and gemfibrozil synergistically raised the AUC of loperamide is similar to our earlier finding on their effect on the pharmacokinetics of repaglinide [14]. In that study, a similar treatment with gemfibrozil alone raised the AUC of repaglinide 8.1-fold, that with itraconazole 1.4-fold, while treatment with their combination raised the AUC of repaglinide 19.4-fold [14]. In another study, gemfibrozil alone raised the AUC of pioglitazone 3.2-fold, but itraconazole had no effect on the pharmacokinetics of pioglitazone and did not alter the effects of gemfibrozil on pioglitazone [16]. Thus, an interaction study using the probe inhibitors itraconazole and gemfibrozil and their combination appears to be a useful tool for studying the relative roles of CYP3A4 and CYP2C8 in the in vivo metabolism of drugs.
Gemfibrozil alone and together with itraconazole reduced the renal clearance of loperamide in the present study. Similarly, in a previous study, gemfibrozil has reduced the renal clearance of pravastatin [33]. Although loperamide is a substrate of the P-glycoprotein, and P-glycoprotein is expressed in the kidney [5], inhibition of the P-glycoprotein by gemfibrozil is unlikely to explain its effects on loperamide renal clearance, because (at least the parent) gemfibrozil does not inhibit the P-glycoprotein [23]. Moreover, itraconazole, a well-established inhibitor of the P-glycoprotein [26, 27], had no significant effect on the renal clearance of loperamide. Thus, inhibition of a transporter other than P-glycoprotein in the kidney may contribute to the interaction between gemfibrozil and loperamide.
During the itraconazole-gemfibrozil phase, the plasma concentrations of itraconazole and hydroxyitraconazole were reduced on average by about 50% compared with those during the itraconazole phase. This finding has also been observed in two previous studies [14, 16], and may be explained by a reduction in the oral bioavailability of itraconazole caused by gemfibrozil, or by displacement of itraconazole and hydroxyitraconazole from plasma proteins by gemfibrozil, leading to increased clearance or volume of distribution. Interestingly, in the present study, the plasma concentrations of itraconazole were very low (i.e., below the lower limit of quantification) in one subject during the itraconazole-gemfibrozil combination phase. His plasma itraconazole concentrations were within those of other subjects during the itraconazole phase. The mechanisms underlying this individual finding are unknown, but genetic variability in intestinal drug transporters may be involved.
In a previous study, concomitant use of quinidine (a single 600 mg dose) has resulted in a reduced respiratory response to carbon dioxide inhalation 30 min after administration of a single, rather high, 16 mg oral dose of loperamide [7]. This suggests that inhibition of the P-glycoprotein at the blood-brain barrier by quinidine enhanced the penetration of loperamide to the brain. This effect was associated with only marginal effects on the pharmacokinetics of loperamide, but the plasma concentrations of loperamide were measured for up to 4 h only [7]. In another study, ritonavir, also an inhibitor of the P-glycoprotein as well as CYP3A4, did not enhance the central nervous system effects of 16 mg oral loperamide, as measured by pupil diameter, cold pressor test and transcutaneous measurements of PCO2 and PO2, despite a 2.7-fold increase in the plasma AUC of loperamide [11]. In our study, treatment with either gemfibrozil, itraconazole, or their combination did not result in increased drowsiness or reduced performance in the DSST after intake of 4 mg loperamide in spite of several-fold increases in the plasma concentrations of loperamide. This may be partly explained by the use of a low dose of loperamide or by a lack of effect of itraconazole on the penetration of loperamide to the brain, or both. Within the first 12 h after loperamide ingestion during the itraconazole-gemfibrozil phase, the plasma concentrations of the active N-desmethyl metabolite of loperamide were roughly half of those during the placebo phase, which may have reduced possible differences in the pharmacodynamic measurements. Furthermore, the tests used may not have been sensitive enough to detect central opioid effects after a low dose of loperamide. The low, although clinically relevant, dose of loperamide was chosen because of safety reasons; unexpectedly strong pharmacodynamic effects might have been caused by the combination of different interaction mechanisms (inhibition of CYP3A4, CYP2C8 and P-glycoprotein in intestinal wall, liver and blood-brain barrier).
In an analysis of 216 Poison Center reports on loperamide ingestions, loperamide overdose (up to 0.94 mg/kg) was associated with related symptoms in about 30% of the patients [34]. The most common symptoms were drowsiness and vomiting. No life-threatening symptoms or fatalities occurred in that series, but one patient was reported to have developed coma [34]. In our study, combination of itraconazole and gemfibrozil increased the mean AUC of loperamide 12-fold and in some subjects even more than 20-fold. The mean t½ of loperamide was prolonged from 11.9 h to 36.9 h by the combination of itraconazole and gemfibrozil (the longest t½ of loperamide during the itraconazole-gemfibrozil phase was 47.9 h). Furthermore, the plasma concentration profile of N-desmethylloperamide suggests slow elimination of this metabolite during the itraconazole-gemfibrozil phase (Fig. 1b). N-desmethylloperamide is partially excreted into urine and partially further metabolized via demethylation (to didesmethylloperamide) and glucuronidation [11]. Our results suggest that CYP3A4 and CYP2C8 are involved in the further metabolism of N-desmethylloperamide and indicate that the parent loperamide and its active N-desmethyl metabolite may accumulate during prolonged use of loperamide with inhibitors of CYP3A4 and CYP2C8. Thus, adverse effects may occur in some patients using gemfibrozil, itraconazole, or their combination concomitantly with loperamide, in particular if the patient uses a high dose of loperamide (the maximum recommended daily dose of loperamide is 16 mg) or uses it on a regular basis.
Polypharmacy is common particularly in elderly patients, and the concomitant use of various drugs that inhibit different CYP enzymes or the P-glycoprotein is not uncommon. The finding that the AUC of loperamide was synergistically increased by the combination of the CYP3A4 inhibitor, itraconazole, and the CYP2C8 inhibitor, gemfibrozil, highlights the importance of conducting drug interaction studies with concomitant administration of inhibitors of different enzymes, when a drug has more than one major elimination pathway. It is reasonable to assume that also other inhibitors of CYP3A4 and CYP2C8 can have a synergistic inhibitory effect on the metabolism of loperamide.
In conclusion, itraconazole, gemfibrozil and particularly their combination markedly raise the plasma concentrations of loperamide, indicating that both CYP3A4 and CYP2C8 are important in the metabolism of loperamide in humans. Although not disclosed by the psychomotor tests used in this study with a single 4 mg oral dose of loperamide, an increased risk of adverse effects should be considered during concomitant use of loperamide with itraconazole, gemfibrozil and especially their combination.
References
Killinger JM, Weintraub HS, Fuller BL (1979) Human pharmacokinetics and comparative bioavailability of loperamide hydrochloride. J Clin Pharmacol 19:211–218
Dagenais C, Graff CL, Pollack GM (2004) Variable modulation of opioid brain uptake by P-glycoprotein in mice. Biochem Pharmacol 67:269–276
Doser K, Meyer B, Nitsche V, Binkert-Graber P (1995) Bioequivalence evaluation of two different oral formulations of loperamide (Diarex Lactab vs Imodium capsules). Int J Clin Pharmacol Ther 33:431–436
Kim KA, Chung J, Jung DH, Park JY (2004) Identification of cytochrome P450 isoforms involved in the metabolism of loperamide in human liver microsomes. Eur J Clin Pharmacol 60:575–581
Fromm MF (2004) Importance of P-glycoprotein at blood-tissue barriers. Trends Pharmacol Sci 25:423–429
Fromm MF, Kim RB, Stein CM, Wilkinson GR, Roden DM (1999) Inhibition of P-glycoprotein-mediated drug transport: A unifying mechanism to explain the interaction between digoxin and quinidine. Circulation 99:552–557
Sadeque AJ, Wandel C, He H, Shah S, Wood AJ (2000) Increased drug delivery to the brain by P-glycoprotein inhibition. Clin Pharmacol Ther 68:231–237
Skarke C, Jarrar M, Schmidt H, Kauert G, Langer M, Geisslinger G, Lötsch J (2003) Effects of ABCB1 (multidrug resistance transporter) gene mutations on disposition and central nervous effects of loperamide in healthy volunteers. Pharmacogenetics 13:651–660
Gutmann H, Fricker G, Drewe J, Toeroek M, Miller DS (1999) Interactions of HIV protease inhibitors with ATP-dependent drug export proteins. Mol Pharmacol 56:383–389
von Moltke LL, Greenblatt DJ, Grassi JM, Granda BW, Duan SX, Fogelman SM, Daily JP, Harmatz JS, Shader RI (1998) Protease inhibitors as inhibitors of human cytochromes P450: high risk associated with ritonavir. J Clin Pharmacol 38:106–111
Tayrouz Y, Ganssmann B, Ding R, Klingmann A, Aderjan R, Burhenne J, Haefeli WE, Mikus G (2001) Ritonavir increases loperamide plasma concentrations without evidence for P-glycoprotein involvement. Clin Pharmacol Ther 70:405–414
Kamali F, Huang ML (1996) Increased systemic availability of loperamide after oral administration of loperamide and loperamide oxide with cotrimoxazole. Br J Clin Pharmacol 41:125–128
Totah RA, Rettie AE (2005) Cytochrome P450 2C8: substrates, inhibitors, pharmacogenetics and clinical relevance. Clin Pharmacol Ther 77:341–352
Niemi M, Backman JT, Neuvonen M, Neuvonen PJ (2003) Effects of gemfibrozil, itraconazole and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia 46:347–351
Backman JT, Kyrklund C, Neuvonen M, Neuvonen PJ (2002) Gemfibrozil greatly increases plasma concentrations of cerivastatin. Clin Pharmacol Ther 72:685–691
Jaakkola T, Backman JT, Neuvonen M, Neuvonen PJ (2005) Effects of gemfibrozil, itraconazole and their combination on the pharmacokinetics of pioglitazone. Clin Pharmacol Ther 77:404–414
Niemi M, Backman JT, Granfors M, Laitila J, Neuvonen M, Neuvonen PJ (2003) Gemfibrozil considerably increases the plasma concentrations of rosiglitazone. Diabetologia 46:1319–1323
Wang JS, Neuvonen M, Wen X, Backman JT, Neuvonen PJ (2002) Gemfibrozil inhibits CYP2C8-mediated cerivastatin metabolism in human liver microsomes. Drug Metab Dispos 30:1352–1356
Kajosaari LI, Laitila J, Neuvonen PJ, Backman JT (2005) Metabolism of repaglinide by CYP2C8 and CYP3A4 in vitro: effect of fibrates and rifampicin. Basic Clin Pharmacol Toxicol 97:249–256
Shitara Y, Hirano M, Sato H, Sugiyama Y (2004) Gemfibrozil and its glucuronide inhibit the organic anion transporting polypeptide 2 (OATP2/OATP1B1:SLC21A6)-mediated hepatic uptake and CYP2C8-mediated metabolism of cerivastatin: analysis of the mechanism of the clinically relevant drug-drug interaction between cerivastatin and gemfibrozil. J Pharmacol Exp Ther 311:228–236
Prueksaritanont T, Zhao JJ, Ma B, Roadcap BA, Tang C, Qiu Y, Liu L, Lin JH, Pearson PG, Baillie TA (2002) Mechanistic studies on metabolic interactions between gemfibrozil and statins. J Pharmacol Exp Ther 301:1042–1051
Backman JT, Kyrklund C, Kivistö KT, Wang JS, Neuvonen PJ (2000) Plasma concentrations of active simvastatin acid are increased by gemfibrozil. Clin Pharmacol Ther 68:122–129
Kivistö KT, Zukunft J, Hofmann U, Niemi M, Rekersbrink S, Schneider S, Luippold G, Schwab M, Eichelbaum M, Fromm MF (2004) Characterisation of cerivastatin as a P-glycoprotein substrate: studies in P-glycoprotein-expressing cell monolayers and mdr1a/b knock-out mice. Naunyn Schmiedebergs Arch Pharmacol 370:124–130
Jurima-Romet M, Crawford K, Cyr T, Inaba T (1994) Terfenadine metabolism in human liver. In vitro inhibition by macrolide antibiotics and azole antifungals. Drug Metab Dispos 22:849–857
Olkkola KT, Backman JT, Neuvonen PJ (1994) Midazolam should be avoided in patients receiving the systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther 55:481–485
Wang EJ, Lew K, Casciano CN, Clement RP, Johnson WW (2002) Interaction of common azole antifungals with P glycoprotein. Antimicrob Agents Chemother 46:160–165
Jalava KM, Partanen J, Neuvonen PJ (1997) Itraconazole decreases renal clearance of digoxin. Ther Drug Monit 19:609–613
Ong CE, Coulter S, Birkett DJ, Bhasker CR, Miners JO (2000) The xenobiotic inhibitor profile of cytochrome P4502C8. Br J Clin Pharmacol 50:573–580
Ganssmann B, Klingmann A, Burhenne J, Tayrouz Y, Aderjan R, Mikus G (2001) Simultaneous determination of loperamide and its demethylated metabolites in plasma and urine by high-performance liquid chromatography-atmospheric-pressure ionization mass spectrometry. Chromatographia 53:656–660
Hengy H, Kölle EU (1988) Determination of gemfibrozil in plasma by high performance liquid chromatography. Arzneimittelforschung 35:1637–1639
Remmel RP, Dombrovskis D, Canafax DM (1988) Assay of itraconazole in leukemic patient plasma by reversed-phase small-bore liquid chromatography. J Chromatogr 432:388–394
Allenmark S, Edebo A, Lindgren K (1990) Determination of itraconazole in serum with high-performance liquid chromatography and fluorescence detection. J Chromatogr 532:203–206
Kyrklund C, Backman JT, Neuvonen M, Neuvonen PJ (2003) Gemfibrozil increases plasma pravastatin concentrations and reduces pravastatin renal clearance. Clin Pharmacol Ther 73:538–544
Litovitz T, Clancy C, Korberly B, Temple AR, Mann KV (1997) Surveillance of loperamide ingestions: an analysis of 216 Poison Center reports. J Toxicol Clin Toxicol 35:11–19
Acknowledgements
We thank Mr. Mikko Neuvonen, MSc, Mr. Jouko Laitila, Mrs. Kerttu Mårtensson, Mrs. Eija Mäkinen-Pulli and Mrs. Lisbet Partanen for skillful assistance in performing this study and drug analyses. This study was supported by grants from the Helsinki University Central Hospital Research Fund and Sigrid Juselius Foundation. Experiments comply with the current laws, inclusive of ethics approval.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Niemi, M., Tornio, A., Pasanen, M.K. et al. Itraconazole, gemfibrozil and their combination markedly raise the plasma concentrations of loperamide. Eur J Clin Pharmacol 62, 463–472 (2006). https://doi.org/10.1007/s00228-006-0133-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-006-0133-z