
Abstract
Objective
To study the influence of CYP3A4 inhibition by ketoconazole on the disposition of venlafaxine in individuals with different CYP2D6 pheno- and genotypes.
Methods
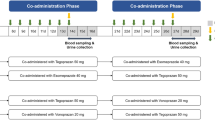
In an open two-phase study, 21 healthy volunteers with known CYP2D6 pheno- and genotype [14 extensive metabolisers (EMs), 7 poor metabolisers (PMs)] were given a single oral dose of venlafaxine (50 mg to EMs and 25 mg to PMs). Plasma and urine levels of venlafaxine and its three metabolites were measured and the pharmacokinetics of venlafaxine were determined. After a 2-week washout period, subjects were treated for 2 days with ketoconazole (100 mg twice daily) starting 1 day before the administration of venlafaxine; and the same parameters as for the administration of venlafaxine only were measured.
Results
Data were evaluated from 20 subjects (14 EMs and 6 PMs) who completed the study. The dose-corrected AUC of venlafaxine was on average 2.3 times higher (P<0.01) and that of its active metabolite O-desmethylvenlafaxine 3.4 times lower (P<0.0001) in PMs than EMs. There was a good correlation between the debrisoquine metabolic ratio and the ratio between the AUC of venlafaxine and that of O-desmethylvenlafaxine (Rs=0.93, P<0.002). The majority of subjects showed higher plasma levels of venlafaxine and O-desmethylvenlafaxine upon co-administration of ketoconazole. AUC of venlafaxine significantly increased by 36% and that of O-desmethylvenlafaxine by 26% (P<0.01). Cmax values increased by 32% and 18%, respectively. The elimination half-life of venlafaxine was unaltered. Three of the PMs displayed marked increases in AUC (81, 126 and 206%) and Cmax (60, 72, 119%) of venlafaxine while the other three showed small or no changes.
Conclusions
Ketoconazole consistently affected the disposition of venlafaxine in EMs of debrisoquine while the response in PMs was erratic. The precise mechanisms underlying this interaction remain to be elucidated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Venlafaxine is a combined serotonin and norepinephrine re-uptake inhibitor used in the treatment of depression and generalised anxiety disorder. Venlafaxine undergoes extensive first-pass metabolism following oral administration. Studies using liver microsomes and expressed enzymes have shown that venlafaxine is metabolised mainly by cytochrome P 450 2D6 (CYP2D6) to its active major metabolite O-desmethylvenlafaxine (O-DM-V) and by CYP3A4 to the inactive minor metabolite N-desmethylvenlafaxine (N-DM-V) [1, 2]. However, other enzymes such as CYP2C19 and CYP2C9 have also been reported to be involved in the metabolism of venlafaxine [1, 2, 3]. O-DM-V and N-DM-V are further metabolised to N,O-didesmethylvenlafaxine (N,O-DDM-V).
CYP2D6 exhibits genetic polymorphism with interindividual differences in metabolic activity [4]. Individuals carrying two mutated alleles coding for no functional enzyme are classified as poor metabolisers (PMs), while those who carry at least one functional CYP2D6 allele are extensive metabolisers (EMs) [5, 6, 7]. Major differences in the pharmacokinetics of venlafaxine in PMs relative to EMs have been documented [8, 9]. CYP3A4 is involved in the metabolism of many drugs and inhibition or induction of this enzyme frequently results in drug–drug interactions. However, it has been suggested that concomitant use of venlafaxine and inhibitors of CYP3A4 would not cause any important changes in the disposition of venlafaxine since CYP3A4-mediated N-demethylation is a minor pathway in the metabolism of venlafaxine [9]. Although not empirically investigated, CYP3A4 might be of greater quantitative importance in individuals lacking functional CYP2D6 genes, i.e. PMs, predisposing them to clinically significant interactions between venlafaxine and CYP3A4 inhibitors.
The aim of the present work was to test this hypothesis by investigating the influence of CYP3A4 inhibition by ketoconazole on the disposition of venlafaxine in individuals with different CYP2D6 pheno- and genotypes.
Material and methods
Study population and design
Twenty-one Swedish healthy Caucasian volunteers (10 males and 11 females), aged between 23 years and 47 years participated in the study. Fourteen were EMs of debrisoquine [debrisoquine metabolic ratio (MRdeb) 0.13–4.97] and seven were PMs (MRdeb 63–260). Seven of the EMs carried the *1/*1 CYP2D6 genotype, six were *1/*4 and one was *1/*5. Six PMs were homozygous for the CYP2D6*4 allele and one was homozygous for CYP2D6*3. Except for oral contraceptives (four subjects, three EMs and one PM) and single doses of mild analgesics, no drugs other than the study drugs were allowed.
The study was approved by the ethics committee at Karolinska Institutet, Huddinge University Hospital. Written informed consent was obtained from all study participants.
Study part I
Subjects received a single oral dose of venlafaxine (Efexor, Wyeth Lederle Nordiska AB, Stockholm, Sweden) in the morning after overnight fasting. The dose was pheno/genotype adjusted. Thus, EMs received 50 mg venlafaxine whereas PMs were given 25 mg, except for one subject who received 50 mg venlafaxine on the first occasion. Venous blood samples (15 ml) were obtained immediately before and 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 24, 36 and 48 h after drug administration. Plasma was separated by centrifugation and stored at 20°C until analysed. Urine was collected up to 48 h after venlafaxine dose. Urine volumes were recorded and aliquots stored at 20°C until analysed.
Study part II
After a 2-week wash-out period, oral treatment with ketoconazole was commenced. Subjects received 100 mg ketoconazole (Fungoral, Janssen-Cilag AB, Stockholm, Sweden) in the morning and evening on day 1. On day 2, subjects received ketoconazole (100 mg) and venlafaxine (25 mg or 50 mg depending on the phenotype) 1 h apart in the morning and 100 mg ketoconazole in the evening. Blood and urine samples were collected as in study part I. The sample taken 4 h after the administration of ketoconazole, i.e. 3 h after that of venlafaxine, was used for ketoconazole analysis.
Analytical methods
Determination of CYP2D6 phenotype and genotype
CYP2D6 phenotype was determined using the debrisoquine urinary metabolic ratio (MR) calculated as the ratio between the recovery of debrisoquine and that of 4-hydroxydebrisoquine in an 8-h urine collection after the oral administration of 10 mg debrisoquine (Debrisoquine, Cambridge Laboratories, United Kingdom) [4, 7].
Determination of CYP2D6 genotype was carried out in leukocyte DNA by allele-specific PCR amplification for the defective alleles CYP2D6*3 and 4, as described [6, 7]. CYP2D6*5 was identified by restriction fragment length polymorphism (RFLP) assay with the restriction endonuclease EcoRI, yielding a 13-kb fragment in case of CYP2D6 gene deletion [10]. Alleles identified as neither CYP2D6*3, *4 nor *5 were classified as CYP2D6*1.
Analysis of venlafaxine and its metabolites, O-desmethylvenlafaxine, N-desmethylvenlafaxine, and N,O-didesmethylvenlafaxine in plasma and urine
Metoprolol (internal standard, 25 μg/ml), saturated potassium carbonate solution (100 μl) and diisopropylether (3 ml) were added to the plasma and urine samples (500 μl). The mixture was extracted by shaking for 10 min in a 12-ml polypropylene tube and centrifuged at 300 rpm for 10 min. The upper phase was decanted after freezing of the water phase in a cooling bath. The organic phase was extracted for 5 min with 150 μl hydrochloric acid, centrifuged, and the organic phase was discarded. The acid phase (50 μl) was injected into the HPLC column.
The chromatographic column was a Zorbax SB-C18 column, 7.5×4.6 mm; 3.5-μm particles (ChromTech AB, Hägersten, Sweden). The UV detector was operated at 220 nm. The eluent consisted of 18% acetonitrile and 82% buffer containing 0.75% ammonia and 0.7% phosphoric acid in water, pH 3.5. The flow rate was 1.0 ml/min. Retention times for venlafaxine, O-DM-V, N-DM-V and N,O-DDM-V were 8.52, 2.42, 7.32 and 2.16 min, respectively.
For validation of the method, calibration curves were constructed on three separate days and controls run in pentaplicates on each day. The nominal values for venlafaxine and N,O-DDM-V controls were 300, 1000 and 2000 nmol/l; for the other two metabolites—O-DM-V and N-DM-V, control values were 450, 1500 and 3000 nmol/l. The recovery of venlafaxine and metabolites varied between 93.2% and 97%. The within-assay coefficient of variation was better than 8.5% for all control levels and the deviation better than 12.5%. The corresponding between-assay values were better than 6.9% and 12.5%, respectively. The limit of quantification was 20 nmol/l for venlafaxine and N,O-DDM-V, and 30 nmol/l for O-DM-V and N-DM-V.
Analysis of ketoconazole in plasma
Ketoconazole levels were quantified using a modification [11] of the method described by Riley and James [12]. The limit of quantification was 0.5 μmol/l and the between-day coefficient of variation was 7.2% at a concentration of 1.0 μmol/l.
Pharmacokinetic analysis
Area under the plasma concentration–time curve (AUC0–inf), oral clearance (CL/F), plasma peak concentration (Cmax), and terminal half-life (t1/2) were calculated using WinNonlin software [13]. The pharmacokinetics model was a one-compartment model with first-order absorption and elimination. AUC and Cmax values of PMs were dose corrected, i.e. multiplied by two since PMs received only half of the venlafaxine dose given to EMs. The urinary recovery of venlafaxine and its metabolites was calculated as percentage of the given dose.
Statistics
The data was analysed using paired t-test (with and without ketoconazole) or unpaired t-test (EMs vs PMs) with the level of statistical significance set at 0.05. Log-transformation of data was applied when a marked asymmetry (skewness) was observed. Correlations were assessed using Spearman's rank test.
Results
All subjects completed the study except one PM who discontinued participation after the first venlafaxine administration due to side effects (described below). Data from this subject was consequently excluded from statistical and pharmacokinetic analyses. Data from all subjects was used in the safety assessment.
Pharmacokinetics
The pharmacokinetic parameters of venlafaxine before and after pre-treatment with ketoconazole are shown in Table 1. Following co-administration of ketoconazole, the AUC of venlafaxine increased significantly in EMs and in the total group. Also, a pronounced increase was seen in three PM subjects (mean increase +138%), while no increase was seen in the other three. Similar changes were seen in Cmax and CL/F, while the terminal half-life remained unaltered.
The pharmacokinetic parameters of O-DM-V were affected in a way similar to that of venlafaxine (Table 2). Following ketoconazole co-administration, AUC and Cmax of O-DM-V increased significantly in EMs and in the total group. In the PMs, there was a non-significant trend towards higher values for both parameters. The elimination half-life of O-DM-V was unaffected by ketoconazole treatment.
The concentrations of N-DM-V and N,O-DDM-V were frequently below the lowest quantifiable level, making AUC calculations unreliable. As shown in Table 2, peak plasma levels were low relative to those of venlafaxine and O-DM-V. There were no significant changes in the Cmax of either metabolite following ketoconazole administration.
Urine data from one PM subject was excluded from statistical analyses due to incomplete urine collection. In the remaining subjects, changes in plasma AUC of venlafaxine and its metabolites were not accompanied by similar changes in the urinary excretion of these substances (data not shown). The average total (venlafaxine plus metabolites) urine venlafaxine recovery among the 19 subjects was 50% (range 30–76%) of the administered dose following venlafaxine-only administration and 48% (range 34–61%) following co-administration of ketoconazole. Co-administration of ketoconazole brought about a 14% decrease (P<0.01) in total (venlafaxine plus metabolites) urine recovery in EMs, whereas the recovery was unaffected in PMs (data not shown). The decrease observed in EMs was mainly due to a 31% reduction in the amount of excreted N,O-DDM-V (P<0.001) and 9% reduction in that of O-DM-V (P<0.05). The excretion of these metabolites was unchanged in PMs.
As shown in Table 1 and Table 2, the average value of dose-corrected AUC of venlafaxine was significantly larger (6496 nmol/l h) in PMs relative to EMs (2771 nmol/l h) following the administration of venlafaxine only (P<0.01), while the AUC of O-DM-V AUC was smaller in PMs than EMs (2249 vs 7564 nmol/l h, P<0.0001). The average terminal half-life of venlafaxine was longer in PMs (10.6 h) than EMs (5.2 h) (P<0.001). There was a good correlation between debrisoquine MR and the AUC of venlafaxine in EMs (Rs=0.78, P<0.002) as well as in the whole group (Rs=0.77, P<0.002). Debrisoquine MR was also highly correlated to the ratio between the AUC of venlafaxine and that of O-DM-V, determined in the absence of ketoconazole (Rs=0.93, P<0.001 in the whole group, Rs=0.90, P<0.002 in EMs) (Fig. 1).

Correlation between debriosoquine metabolic ratio and the ratio between AUC of venlafaxine and that of O-DM-V
Plasma ketoconazole concentrations 4 h after ketoconazole administration varied between 1.2 μmol/l and 6.4 μmol/l. There was no correlation between the individual ketoconazole concentrations and the effects on the pharmacokinetic parameters of venlafaxine and its metabolites.
Safety
All subjects except one, an EM, experienced one or more adverse effects during the study. The most common side effect was nausea, observed in 16 of 21 subjects. Tiredness was reported in seven subjects, dry mouth in five, flush in two, and accommodation disturbances in two. Adverse effects were most pronounced 0.5–3 h after venlafaxine administration, coinciding with the plasma concentration peaks of venlafaxine and O-DM-V. Three PMs vomited within 30–45 min following venlafaxine-only administration and five subjects (three EMs and two PMs) vomited 33–60 min after administration of ketoconazole plus venlafaxine.
Discussion
The metabolism of venlafaxine has been shown to be largely dependent on CYP2D6 that catalyses the formation of O-DM-V. CYP3A4-mediated N-demethylation of venlafaxine is considered to be a minor pathway [1, 2]. Our study confirms the major role of CYP2D6 in the O-demethylation of venlafaxine in vivo. Venlafaxine AUC and the ratio between the AUCs of venlafaxine and O-DM-V correlated highly with the debrisoquine MR. One previous study has shown a relationship between the CYP2D6 genotype and steady-state plasma concentration ratios between O-DM-V and venlafaxine [8]. O-DM-V is considered to inhibit the re-uptake of serotonin and noradrenaline with a potency similar to that of the parent compound. Therefore, it has been assumed that the CYP2D6-dependent interindividual differences in venlafaxine metabolism would be of limited clinical relevance [2]. In the study of Veefkind et al., no significant differences were found in the sum of the serum levels of venlafaxine and O-DM-V among the different CYP2D6 genotypes [8]. However, there were only three PM subjects in that study. In a report based on four patient cases, Lessard et al. have suggested that decreased CYP2D6 activity might predispose to cardiovascular toxicity of venlafaxine [9]. Further studies on larger patient populations are required to elucidate whether the CYP2D6 polymorphism is of importance for the clinical effects of venlafaxine.
Because of the minor quantitative role of the CYP3A4-mediated N-demethylation pathway, only PMs of debrisoquine have been thought to be at a potentially higher risk of adverse effects when co-treated with CYP3A4-inhibiting substances such as ketoconazole. Our results indicate that this is not the case. Indeed, the majority of the subjects showed higher plasma levels of venlafaxine upon co-administration of ketoconazole irrespective of CYP2D6 phenotype (Table 1). In EMs, the increase in AUC of venlafaxine was accompanied by a slight decrease in oral clearance although the elimination half-life remained unaltered. Further, treatment with ketoconazole did not affect the ratio between venlafaxine and O-DM-V in either group (data not shown). Also, the terminal half-life of O-DM-V remained unaltered upon treatment with ketoconazole (data not shown). Taken together, these findings suggest that ketoconazole primarily affects the bioavailability of venlafaxine rather than its elimination. The lack of effect of ketoconazole on the elimination of venlafaxine is not unexpected since CYP3A4 is not a major route of elimination in EMs. However, the apparent effect on the bioavailability is a novel finding that has not been previously reported. Of particular interest is the finding that treatment with ketoconazole led to an increase in the levels of O-DM-V in most subjects. Since O-DM-V is pharmacologically active, the interaction with ketoconazole is potentially significant from a clinical point of view.
The effect of ketoconazole on the pharmacokinetics of venlafaxine in PMs was inconsistent. As shown in Table 3, three subjects exhibited a marked increase in the AUC and Cmax of venlafaxine, together with a major decrease in oral clearance. In two of these subjects, t1/2 was prolonged upon co-administration of ketoconazole. In these subjects, ketoconazole seems to affect the bioavailability as well as the elimination of venlafaxine; the latter probably mediated via CYP3A4 inhibition. In three of the PMs the effect of ketoconazole on the oral clearance of venlafaxine was more pronounced than that in EMs. However, in two PMs, CL/F remained unaltered by ketoconazole treatment. The reason for this discrepancy is not self-evident. Nausea and vomiting are well-known, concentration-dependent adverse effects of venlafaxine and, in our study, almost all subjects developed nausea upon treatment although only four actually vomited after drug intake. Vomiting potentially affects the absorption of venlafaxine and might lead to erroneous estimations of Cmax and AUC. However, the fact that most EMs did not vomit speaks in favour of a real effect of ketoconazole on the bioavailability of venlafaxine. Further, the most pronounced effect of ketoconazole on the AUC and Cmax of venlafaxine was observed in a PM who did not vomit after drug intake. Four subjects used oral contraceptives, but they were evenly distributed between the CYP2D6 pheno-/genotype groups and the use of oral contraceptives would not be expected to have any significant effects on the pharmacokinetics of venlafaxine. The mechanism by which ketoconazole seems to affect the bioavailability of venlafaxine is unclear. The possible inhibitory effects of ketoconazole on enzymes other than CYP3A4 (such as CYP2C enzymes) as well as its known inhibitory effect on P-glycoprotein leading to increased absorption of venlafaxine in the gut are of interest and merit further studies.
References
Fogelman SM, Schmider J, Venkatakrishnan K, von Moltke LL, Harmatz JS, Shader RI, Greenblatt DJ (1999) O- and N-demethylation of venlafaxine in vitro by human liver microsomes and by microsomes from cDNA-transfected cells: effect of metabolic inhibitors and SSRI antidepressants. Neuropsychopharmacology 20:480–490
Otton SV, Ball SE, Cheung SW, Inaba T, Rudolph RL, Sellers EM (1996) Venlafaxine oxidation in vitro is catalysed by CYP2D6. Br J Clin Pharmacol 41:149–156
Fukuda T, Nishida Y, Zhou Q, Yamamoto I, Kondo S, Azuma J (2000) The impact of the CYP2D6 and CYP2C19 genotypes on venlafaxine pharmacokinetics in a Japanese population. Eur J Clin Pharmacol 56:175–180
Mahgoub A, Idle JR, Dring DG, Lancaster R, Smith RL (1977) Polymorphic hydroxylation of debrisoquine in man. Lancet 2:584–586
Skoda RC, Gonzalez FJ, Demierre A, Meyer UA (1988) Two mutant alleles of the human cytochrome P450 db1 gene (P450,II, D1) associated with genetically deficient metabolism of debrisoquine and other drugs. Proc Natl Acad Sci U S A 85:5240–5243
Heim M, Meyer UA (1990) Genotyping of poor metabolisers of debrisoquine by allele-specific PCR amplification. Lancet 336:529–532
Dahl ML, Johansson I, Porsmyr-Palmertz M, Ingelman-Sundberg M, Sjöqvist F (1992) Analysis of the CYP2D6 gene in relation to debrisoquine and desipramine hydroxylation in Swedish population. Clin Pharmacol Ther 51:12–17
Veefkind AH, Haffmans PM, Hoencamp E (2000) Venlafaxine serum levels and CYP2D6 genotype. Ther Drug Monit 22:202–208
Lessard E, Yessine MA, Hamelin BA, O'Hara G, LeBlanc J, Turgeon J (1999) Influence of CYP2D6 activity on the disposition and cardiovascular toxicity of the antidepressant agent venlafaxine in humans. Pharmacogenetics 9:435–443
Johansson I, Lundqvist E, Bertilsson L, Dahl M-L, Sjöqvist F, Ingelman-Sundberg M (1993) Inherited amplification of an active gene in the cytochrome P450 CYP2D locus as a cause of ultrarapid metabolism of debrisoquine. Proc Natl Acad Sci U S A 90:11825–11829
Böttiger Y, Tybring G, Götharsson E, Bertilsson L (1997) Inhibition of the sulphoxidation of omeprazole by ketoconazole in poor and extensive metabolizers of S-mephenytoin. Clin Pharmacol Ther 62:384–391
Riley CM, James MO (1986) Determination of ketoconazole in plasma, liver, lung and adrenal of the rat by high-performance liquid chromatography. J Chromatogr 377:287–294
WinNonlin reference guide, Version 3.0. 1998–1999 Pharsight Corporation, Mountain view, CA, USA
Acknowledgement
We thank Tommy Petterson and Anneli Wahlberg for excellent help. This work is supported by the Swedish Research Council (project 3902) and Karolinska Institutet. J. Lindh is supported by the Swedish Foundation for Strategic Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lindh, J.D., Annas, A., Meurling, L. et al. Effect of ketoconazole on venlafaxine plasma concentrations in extensive and poor metabolisers of debrisoquine. Eur J Clin Pharmacol 59, 401–406 (2003). https://doi.org/10.1007/s00228-003-0627-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-003-0627-x