
Abstract
Indo-Pacific lionfish (Pterois volitans and P. miles) recently invaded Western Atlantic waters, rapidly spreading through the Caribbean and Gulf of Mexico (GoM). Previous genetic analyses using the mitochondrial d-loop determined that populations in the Western North Atlantic (NA) region have up to nine haplotypes, whereas Caribbean populations contain four of the North Atlantic haplotypes. The genetic composition of GoM populations, reported here for the first time, could lend insight into the pathway of dispersal into the GoM and better understanding of the biogeography of this recent invader. Here, we determined the genetic composition of lionfish throughout the GoM and compared haplotype composition to Caribbean and North Atlantic regions. We found that GoM samples contained only three d-loop haplotypes that are common in the Caribbean and North Atlantic. The genetic structure differed significantly among the three regions (AMOVA:Φ CT = 0.062; p = 0.001), but we found no differences between locations within regions (AMOVA:Φ SC = 0.005; p = 0.092). The composition of GoM samples most closely matches the composition of Caribbean samples indicating that Caribbean populations are the likely source of the GoM populations. As each region was successively invaded, a drop in haplotype diversity and changes in haplotype frequencies occurred indicating dispersal limitation across basin boundaries and founder effects within each basin. The lack of differentiation within regions indicates rapid population growth and unfettered dispersal within basins after initial colonization. We find no evidence of secondary invasions within samples. With well-established populations, the probability of detecting a secondary invasion is minuscule.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Two lionfishes, the Pacific red lionfish (Pterois volitans) and the devil firefish (P. miles), have established alien, invasive populations in the Western Atlantic Ocean as a result of releases from the ornamental pet trade (Hamner et al. 2007; Betancur et al. 2011). Lionfishes are believed to have first established populations in Miami, Florida, USA, and after a lag period, quickly established populations along the eastern seaboard of the USA. The Bahamas were exclusively colonized by P. volitans in 2004 (Freshwater et al. 2009), and the spread of lionfish has rapidly accelerated with their establishment throughout much of the Caribbean Sea by 2009 (Betancur et al. 2011). Most recently, P. volitans has spread to the Gulf of Mexico (GoM) (Betancur et al. 2011) and south of the Amazon outflow in Brazil (Ferreira et al. 2015).
Studying the genetic composition of these recently established lionfish can give insight into the genetic relationships among populations, the routes of invasion into new areas, the restrictions to gene flow and range expansion. Several studies have now investigated the population genetics of P. volitans in several locations in the invaded range and have discovered nine unique haplotypes in the mitochondrial control region d-loop (Freshwater et al. 2009; Betancur et al. 2011; Toledo-Hernández 2014; Butterfield et al. 2015). To date, there have been 37 d-loop haplotypes discovered for P. volitans from the native range in Indonesia and the Philippines (Freshwater et al. 2009), indicating that a founder effect has occurred in the invaded range. None of the nine haplotypes observed in the invaded range have been found in populations in the native range. Samples from North Atlantic populations, consisting of North Carolina, Bermuda and Bahamas combined, contain all nine haplotypes (H01-09, Betancur et al. 2011). By comparison, the Caribbean population samples contain a subset of four haplotypes, which are all found in the North Atlantic (H01–H04, Butterfield et al. 2015). The composition of the GoM lionfish is still unknown.
There is ample evidence to believe that lionfish will exhibit gene flow restrictions between the Caribbean and the GoM, and even within the GoM. Despite the capability of invasive lionfish to spread widely, it is also clear that there are semipermeable barriers to dispersal, as evidenced by the genetic founder effects that occurred as the lionfish invaded the Caribbean (Betancur et al. 2011). Similar dispersal restrictions could exist for the GoM. Additionally, several species exhibit genetic structure across the southern tip of Florida from the GoM to the Atlantic (Gold and Richardson 1998). Florida is not a strict barrier for all species; however, some species exhibit considerable levels of shared genetic identity between the Atlantic and GoM, a signature indicative of contemporary gene flow (Gold and Richardson 1998). Within the northern GoM, there are also observed biogeographic breaks near Mobile Bay, Alabama, and the outflow of the Mississippi River (Portnoy and Gold 2012). Alternatively, there may have been secondary introductions that could drive genetic discontinuities among locations, as is suggested for the Caribbean (Butterfield et al. 2015). Investigation of the genetic structure of GoM lionfish in comparison with those in other parts of the Atlantic will advance our understanding of gene flow and dispersal in the Western Atlantic region and the most likely invasion routes.
Here, we present the first description of the genetic composition and diversity of lionfish in the GoM. We compare the patterns of genetic composition and structure to samples from the Caribbean and North Atlantic. We infer the most likely route of invasion to the GoM and possible barriers to gene flow among locations in the GoM and elsewhere. We discuss the observed biogeographic patterns from studies of the mitochondrial control region in this species, and we address the hypothesis that there may have been a secondary introduction of lionfish into the Caribbean.
Methods
Sampling
Lionfish were collected from nine sample sites in Texas, Mississippi and Florida. Samples were collected 100 miles off the Texas coastline at three locations (East Flower Garden Bank, West Flower Garden Bank and Stetson Bank; Online Resource 1) in the Flower Garden Banks National Marine Sanctuary (FGBNMS). At the FGBNMS, lionfish were collected by permitted divers on SCUBA using pole spears between 2011 and 2013 under FGBNMS permits (FGBNMS-2009-001, FGBNMS-2011-002, and FGBNMS-2014-001). Whole fish were frozen aboard the NOAA R/V MANTA and taken to the laboratory where tissue samples were taken from the soft dorsal fin. Lionfish from Mississippi and West Florida were collected by divers armed with spears from one oil production platform (VK-385) offshore and south of Mobile Bay and two small artificial reef sites offshore northwest Florida and three sites offshore southwest Florida consisting of natural or artificial habitat (Online Resource 1). Fishes were kept on ice, and tissue samples were taken from the pectoral fin by researchers in the Department of Coastal Sciences at the University of Southern Mississippi (USM). In addition to samples from the GoM, samples were also collected from Tiger Rock, in Bocas del Toro, Panama, to increase the sample size from Panama previously reported by Butterfield et al. (2015). The Panamanian samples were collected by divers on SCUBA using pole spears. The fish were kept on ice, and tissue samples were taken from the pectoral fin. All tissues from the FGBNMS were preserved in 95 % ethanol; all tissues from USM were preserved in RNAlater. All samples were shipped to Texas A&M University–Corpus Christi (TAMUCC) for subsequent processing and analysis.
The frequencies of nine haplotypes were collected from 14 locations in the North Atlantic and Caribbean from previously published studies (Freshwater et al. 2009; Betancur et al. 2011; Toledo-Hernández 2014; Butterfield et al. 2015) to compare with the GoM samples. The sequences for all P. volitans haplotypes from the native and invaded range were collected from GenBank (Freshwater et al. 2009; GenBank accession numbers FJ516407–FJ516454).
DNA analysis
Tissue samples were stored at −80 °C until DNA extraction using Qiagen DNeasy blood and tissue kits (Qiagen, Valencia, California, USA). Double-stranded DNA was quantified by time-resolved fluorescence using a SpectraMax® M Series Multi-Mode Microplate Reader (Molecular Devices LLC, California, USA). A 679 base-pair region of the mitochondrial control region (d-loop) was amplified from genomic DNA (5 ng) in 10 µL reactions using the following PCR conditions: 1× BioMixTM (Bioline, Massachusetts, USA), which includes 2.5 mM MgCl2; 0.5 mg/μL Bovine SerumAlbumin (BSA), and 10 μM each LionA_H (5′-CCA TCT TAA CAT CTT CAG TG-3′) and LionB_L (5′-CAT ATC AAT ATG ATC TCA GTAC-3′) by denaturing DNA at 94 °C for 3 min, followed by 32 cycles of denature for 30 s at 94 °C, annealing for 30 s at 48 °C, and extension for 45 s at 72 °C, followed by 5 min extension at 72 °C, and held at 12 °C until placed in the freezer (modified from Freshwater et al. 2009). PCR products were cleaned using ExoSap-IT® (Affymetrix, California, USA), fluorescently quantified and then sequenced following the BigDye Terminator v3.1 Cycle Sequencing Kit protocol using Applied Biosystems 3730xl genetic analyzer (Life Technologies Corporation, California, USA) in the TAMUCC Genomics Core Laboratory (http://genomics.tamucc.edu). Sequences were aligned and edited in Geneious 7 (Biomatters, Ltd., Auckland, New Zealand).
Statistical analyses
Exact tests (Raymond and Rousset 1995) were used to identify differences in haplotype frequencies among the nine sample sites within locations in the GoM using Arlequin 3.5 (1,000,000 Markov chain steps and 100,000 burn-in steps). There were no significant differences in haplotype frequencies among samples (e.g., EFGB, WFGB, Stetson Bank) within locations (e.g., EFGB, WFGB, Stetson Bank in Texas); therefore, samples within locations were pooled within four locations (Texas, Mississippi, N. Florida, S. Florida).
Genetic differentiation among regions (N. Atlantic, Caribbean, GoM) and among locations within regions was tested with an analysis of molecular variance (AMOVA, Excoffier et al. 1992) using a simple pairwise genetic distance model in Arlequin 3.5. Pairwise Φ CT estimates for all regions and pairwise Φ SC for all locations within regions were estimated, and significance was tested using 10,000 permutations. Differences in haplotype frequencies among locations were tested as described above.
Migrate (Beerli and Palczewski 2010) was used to infer migrations rates, but convergence among replicated runs could not be obtained. It should also be noted that given the recent invasion and non-equilibrium status of alien lionfish populations, the assumptions of the Migrate model are violated. For these reasons, we do not present the Migrate results.
Results
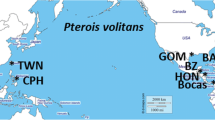
Three haplotypes were identified in 188 samples from the Gulf of Mexico as belonging to P. volitans; all three haplotypes were observed at each location (Online Resource 2, and Fig. 1). We found no individuals with P. miles haplotypes. The three GoM haplotypes (H01, H02 and H04; sensu Freshwater et al. 2009) are a subset of the four haplotypes sampled in the Caribbean Sea, which are themselves a subset of the nine haplotypes in the North Atlantic. The most frequently observed haplotype in the GoM was H02 (60.1 %) followed by H01 (27.7 %), and the least frequent was H04 (12.2 %). The frequencies of H01 and H02 are similar to the Caribbean region but differ markedly from the North Atlantic (Online Resource 2, and Fig. 1). Haplotype H04 was more frequent in the GoM samples than in both the Caribbean and North Atlantic samples. Additionally, three haplotypes (H01, H02, H04) were found among the 46 samples from Bocas del Toro, Panama; the rare H03 was not found in these samples.

Map of the sample locations from this study; Butterfield et al. (2015); Toledo-Hernández (2014); Betancur-R et al. (2011) and; Freshwater et al. (2009). Pie charts indicate the proportions of haplotype frequencies within each sample. Solid lines indicate location of a strong dispersal restriction between; a North Atlantic and Caribbean and, b North Atlantic and Gulf of Mexico. Dashed line indicates location of a weak dispersal restriction between c Caribbean and Gulf of Mexico
AMOVA results indicated that a significant amount of genetic variation is explained by differences among regions (Φ CT = 0.062; p = 0.001), and to a lesser extent among locations within regions (Φ SC = 0.005; p = 0.092; Table 1). Using orthogonal a priori contrasts within the AMOVA model to dissect the genetic structuring among regions, the N. Atlantic was significantly differentiated from the other regions investigated (Φ = 0.075; p = 0.0022) and the GoM was very weakly differentiated from the Caribbean (Φ = 0.0098; p = 0.060).
Pairwise Φ ST estimates and exact tests revealed significant differences among samples in each of the different regions, but no differences among samples within regions were detected (p < 0.0072, FDR = 0.05, Online Resource 3). Additionally, the sample from Panama (this study and Butterfield et al. 2015) was not found to differ significantly with any location within the Caribbean region as was previously reported by Butterfield et al. (2015).
Discussion
The new data presented here support the hypothesis that the lionfish invaded the GoM from populations in the Caribbean, and there may be slight gene flow restrictions between the Caribbean and GoM. Pterois volitans invaded the GoM from Caribbean populations which, themselves, originated in the North Atlantic. Betancur et al. (2011) concluded that the Caribbean was colonized from the N. Atlantic, and the Caribbean was colonized by lionfish before the GoM (Schofield 2009). The GoM lionfish were most genetically similar to the Caribbean, in that both regions lacked the rare haplotypes H05–H09 which have been previously found in the North Atlantic region. The GoM was also substantially genetically differentiated from N. Atlantic. The most parsimonious conclusion is that there is no detectable direct gene flow to the GoM from the North Atlantic.
It is likely that any gene flow between the GoM and North Atlantic regions would be strongly unbalanced, with GoM larvae dispersing into the North Atlantic region. If P. volitans were to invade the GoM from the North Atlantic, their weakly motile larvae would have to swim against the Florida Current, in the Straits of Florida. Pterois volitans is a reef-associated species, the adults of which show particularly strong site fidelity and a low movement rate (Akins et al. 2014); therefore, range expansion likely results from larval dispersal. It is apparent from our data that lionfish are not dispersing into the GoM from the North Atlantic along the Florida shelf.
Similar to invasive lionfish, a variety of native species exhibit population genetic discontinuities at the southern tip of Florida including hermit crabs (Young et al. 2002), a loliginid squid (Herke and Foltz 2002) and estuarine dependent fishes (Gold and Richardson 1998). However, some species including a different loliginid squid (Herke and Foltz 2002), reef-associated fishes (Gold and Richardson 1998; Karlsson et al. 2009) and pelagic fishes (Gold and Richardson 1998; Broughton et al. 2002) do not show a hard genetic break here. The life histories of the species and historical gene flow may explain why gene flow appears restricted for some species and not others (Gold and Richardson 1998). Indeed, the “historical” demography of the invasive lionfish is largely responsible for the observed pattern of genetic structure and diversity, where the location of the original introduction has much higher genetic diversity than locations colonized much later.
A weak genetic founder effect is evident in the GoM lionfish samples indicating that there is a gene flow restriction between the GoM and Caribbean. Despite extensive sampling, the GoM samples lacked the rare haplotype H03 which is present in three locations in the Caribbean. There were also differences in the frequencies of the three haplotypes that were shared among the regions (i.e., genetic structure). There has been relatively little work comparing populations of organisms between the Caribbean and the northern GoM. This is probably due, in part, to major differences in the fish fauna between the two regions. However, one study on blacktip sharks (Carcharhinus limbatus) showed evidence of reduced gene flow between Caribbean and GoM populations (Keeney et al. 2005). Blacktip sharks have very different life histories than teleostean fishes like P. volitans. The sharks do not have dispersive pelagic larvae; in fact, small-bodied sharks like this are typically restricted to the continental shelf, and adults (often males) will disperse along the coastline. More studies will be necessary to better understand the nature of larval dispersal and gene flow restrictions between the Caribbean and GoM for marine organisms.
Interpretation through a population genetic lens
The haplotype frequencies presented here indicate not only the locations of potential gene flow restrictions, and they are also indicative of rapid population growth. In interpreting the haplotype frequencies of a recent invasion of alien species, such as P. volitans, it is critical to realize that the system is not at equilibrium with respect to migration, mutation and genetic drift. Genes are flowing from the North Atlantic (nine haplotypes) into the Caribbean, and yet only four of the nine haplotypes have been observed in the Caribbean. A similar scenario is playing out between the Gulf of Mexico and the Caribbean. This pattern can be understood in the context of the genetic bottlenecks associated with founder effect, subsequent genetic drift, rapid population expansion and too little time for the system to reach equilibrium.
During a founder event, a subset of a population colonizes a new area, and these events are classically defined by a reduction in genetic diversity and changes in allele frequency, which are determined by the number of founding individuals and random chance. Genetic drift, the process by which haplotype frequencies fluctuate randomly from generation to generation, is mathematically defined in the Wright–Fisher Model (Wright 1937; Fisher and Ford 1950; Crow and Kimura 1970). Following this model, genetic drift and thus the rate at which allele frequencies change, is extremely rapid in small populations and slow in large populations. Founding populations of lionfish in the Caribbean and Gulf of Mexico were likely to harbor a subset of the genetic diversity in the colonizing population and likely experienced accelerated genetic drift. Rapid growth of the founding population would reduce genetic drift and the proportion of migrants, thereby establishing a genetic discontinuity, and thus explaining the present pattern of genetic structure in lionfish.
The genetic pattern described here in lionfish has been more generally described as gene surfing, where certain genes ride the wave of population expansion and others may not (Hallatschek et al. 2007; Hallatschek and Nelson 2008; Excoffier and Ray 2008). Whether or not genetic diversity attenuates with the wave of population expansion is a function of the ability of founder populations to overcome Allee effects, grow rapidly with little migration and thus “pull” the wave forward. Where Allee effects are negligible, the colonization wave front is pulled, and genetic diversity can decrease due to founder effect, as observed among the lionfish populations in geographic regions delineated here. Hallatschek and Nelson (2008) further model “pushed” colonization waves as those where Allee effects are great and migration pushes the front forward with negligible founder effect. While vast tracts of nearshore habitat have been colonized by lionfish without attenuation of genetic diversity, it seems unlikely that Allee effects are strong in most of the invaded range except between the narrow regions of genetic discontinuity, especially given the rate of advance of the colonization wave over the Caribbean and then the Gulf of Mexico. Perhaps the initial colonization wave across the North Atlantic region, which developed over decades, was due at least in part to Allee effects. We suggest, however, that a tsunami of lionfish migration propagated through the Caribbean and again in the Gulf of Mexico, “pushing” all genetic surfers in their path. The addition of variable migration rates to gene surfing theory is likely a useful one.
An alternative hypothesis that the differences in allele frequencies among the Caribbean and North Atlantic are due to secondary introduction sources in the Caribbean is unsupported (Butterfield et al. 2015). We do not dispute that alternative introduction sources are possible or even likely. We do dispute that these secondary sources have contributed to the present pattern of population genetic structure. The seemingly high proportion of the rare H03 haplotype in five samples from Panama is the primary evidence presented for a secondary introduction (Butterfield et al. 2015). Our additional 46 samples from Bocas del Toro revealed that samples from Panama did not appear to be different from the rest of the Caribbean. Rather, the vast majority of lionfish in the Caribbean and the GoM are most likely to have originated solely from the North Atlantic. First, the observed haplotype pattern is consistent with a genetic bottleneck caused by founder effect and followed by rapid population growth. Second, a secondary introduction of lionfish in the Caribbean or GoM is extremely unlikely to have involved the same haplotypes as are already present in the North Atlantic because lionfish are far more genetically diverse in their native range and pet stores than in their non-native range. There are only nine lionfish haplotypes in the Atlantic Ocean in a sample of 1294 fish, while there are 38 haplotypes in a sample of 70 lionfish collected from sites in West Indonesia and Philippines, obtained from the aquarium trade (Freshwater et al. 2009). Lastly, the timing of the spread of the lionfish to various locations in the North Atlantic and then finally into the Caribbean is consistent with a colonization of the Caribbean solely from the North Atlantic without any indication of a second colonization source (Schofield 2009). At this point of the invasion in the North Atlantic, Caribbean, and GoM, it will be very difficult for any additional haplotypes to attain a high enough frequency to be detected in a sample due to the large size of the population and slow genetic drift, barring selection for a new haplotype.
Conclusion
Understanding the patterns of genetic differentiation among populations of P. volitans will help us to better understand how the invasion is progressing. Identifying barriers to gene flow in this species can inform our understanding of biogeography in the regions of study and predict the possible outcome of future invasions, which can aid marine managers in their decision making. It is not presently feasible to eradicate P. volitans from its new, invaded range; however, understanding patterns of gene flow and larval dispersal can inform control of this species and improve population models that estimate population growth of P. volitans in invaded populations (e.g., Morris et al. 2011). P. volitans has a high dispersal potential and has spread rapidly throughout the Caribbean and Gulf of Mexico. A better understanding of the propensity of this species for dispersal, such as the average dispersal distance, the propensity for self-recruitment and identifying barriers to dispersal, can help inform whether local control efforts can be effective in maintaining low densities in critical habitats.
References
Akins JL, Morris JA, Green SJ (2014) In situ tagging technique for fishes provides insight into growth and movement of invasive lionfish. Ecol Evol 4:3768–3777. doi:10.1002/ece3.1171
Beerli P, Palczewski M (2010) Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 185:313–326. doi:10.1534/genetics.109.112532
Betancur RR, Hines A, Acero PA, Ortí G, Wilbur AE, Freshwater DW (2011) Reconstructing the lionfish invasion: insights into Greater Caribbean biogeography. J Biogeogr 38:1281–1293. doi:10.1111/j.1365-2699.2011.02496.x
Broughton RE, Stewart LB, Gold JR (2002) Microsatellite variation suggests substantial gene flow between king mackerel (Scomberomorus cavalla) in the western Atlantic Ocean and Gulf of Mexico. Fish Res 54:305–316. doi:10.1016/s0165-7836(01)00275-2
Butterfield JSS et al (2015) Wide-ranging phylogeographic structure of invasive red lionfish in the Western Atlantic and Greater Caribbean. Mar Biol 162:773–781. doi:10.1007/s00227-015-2623-y
Crow JF, Kimura M (1970) An introduction to population genetics theory. The Blackburn Press, Caldwell
Excoffier L, Ray N (2008) Surfing during population expansions promotes genetic revolutions and structuration. Trends Ecol Evol 23:347–351
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Ferreira CE, Luiz OJ, Floeter SR, Lucena MB, Barbosa MC, Rocha CR, Rocha LA (2015) First record of invasive lionfish (Pterois volitans) for the Brazilian Coast. PLoS ONE 10:e0123002. doi:10.1371/journal.pone.0123002
Fisher RA, Ford B (1950) The Sewall Wright effect. Heredity 4:117–119
Freshwater WD et al (2009) Mitochondrial control region sequence analyses indicate dispersal from the US East Coast as the source of the invasive Indo-Pacific lionfish Pterois volitans in the Bahamas. Mar Biol 156:1213–1221. doi:10.1007/s00227-009-1163-8
Gold J, Richardson L (1998) Mitochondrial DNA diversification and population structure in fishes from the Gulf of Mexico and Western Atlantic. J Hered 89:404–414
Hallatschek O, Nelson DR (2008) Gene surfing in expanding populations. Theor Popul Biol 73:158–170
Hallatschek O, Hersen P, Ramanathan S, Nelson DR (2007) Genetic drift at expanding frontiers promotes gene segregation. Proc Natl Acad Sci USA 104:19926–19930
Hamner RM, Freshwater DW, Whitfield PE (2007) Mitochondrial cytochrome b analysis reveals two invasive lionfish species with strong founder effects in the western Atlantic. J Fish Biol 71:214–222. doi:10.1111/j.1095-8649.2007.01575.x
Herke S, Foltz D (2002) Phylogeography of two squid (Loligo pealei and L. plei) in the Gulf of Mexico and northwestern Atlantic Ocean. Mar Biol 140:103–115. doi:10.1007/s002270100680
Karlsson S, Saillant E, Gold JR (2009) Population structure and genetic variation of lane snapper (Lutjanus synagris) in the northern Gulf of Mexico. Mar Biol 156:1841–1855. doi:10.1007/s00227-009-1217-y
Keeney DB, Heupel MR, Hueter RE, Heist EJ (2005) Microsatellite and mitochondrial DNA analyses of the genetic structure of blacktip shark (Carcharhinus limbatus) nurseries in the northwestern Atlantic, Gulf of Mexico, and Caribbean Sea. Mol Ecol 14:1911–1923. doi:10.1111/j.1365-294X.2005.02549.x
Morris JA, Shertzer KW, Rice JA (2010) A stage-based matrix population model of invasive lionfish with implications for control. Biol Invas 13:7–12. doi:10.1007/s10530-010-9786-8
Portnoy DS, Gold JR (2012) Evidence of multiple vicariance in a marine suture-zone in the Gulf of Mexico. J of Biogeogr 39:1499–1507. doi:10.1111/j.1365-2699.2012.02699.x
Raymond M, Rousset F (1995) An exact test for population differentiation. Evolution 49:1280–1283
Schofield P (2009) Geographic extent and chronology of the invasion of non-native lionfish. Aquat Invas. doi:10.3391/ai.2009.4.3
Toledo-Hernández C (2014) Population ecology and genetics of the invasive lionfish in Puerto Rico. Aquat Invas 9:227–237. doi:10.3391/ai.2014.9.2.12
Wright S (1937) The distribution of gene frequencies in populations. Proc Natl Acad Sci USA 23:307–320
Acknowledgments
We thank the FGBNMS staff for help with collections and samples from the FGBNMS, and G. Palmer, A. Downey-Wall and J. Selwyn for collections and samples from Panama.
Funding
This study was funded by start up funds provided to JDH by Texas A&M University–Corpus Christi.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national and/or institutional guidelines for the care and use of animals were followed. (IACUC protocol #05-14).
Additional information
Responsible Editor: T. Reusch.
Reviewed by Undisclosed experts.
This article is part of the Topical Collection on Invasive Species.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Johnson, J., Bird, C.E., Johnston, M.A. et al. Regional genetic structure and genetic founder effects in the invasive lionfish: comparing the Gulf of Mexico, Caribbean and North Atlantic. Mar Biol 163, 216 (2016). https://doi.org/10.1007/s00227-016-2981-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00227-016-2981-0