
Abstract
Globally, sharks are under enormous pressure from fishing efforts. One such species is the silky shark, Carcharhinus falciformis, which occurs in all the Earth’s tropical oceans and is captured in large numbers in pelagic fisheries. Regionally, the silky shark is listed as Vulnerable to Near Threatened by the International Union for the Conservation of Nature due to high levels of direct and bycatch exploitation. Despite major conservation concerns about this species, little is known about its genetic status and level of demographic or evolutionary connectivity among its regional distributions. We report a genetic assessment of silky sharks sampled across a major portion of the species’ global range. We sequenced the complete mitochondrial DNA control region from 276 individuals taken from the western Atlantic and Indo-Pacific Oceans and the Red Sea. Overall, haplotype and nucleotide diversities were relatively large (0.93 ± 0.01 and 0.61 ± 0.32 %, respectively). Nucleotide diversity in Indo-Pacific sharks, however, was significantly lower and about half that in Atlantic sharks. Strong phylogeographic partitioning occurred between ocean basins. Furthermore, shallow but significant pairwise statistical differentiation occurred among most regional samples within the Indo-Pacific, but not the western Atlantic. Overall, at least five mitochondrial DNA populations of silky sharks were identified globally. Despite historically large population sizes, silky sharks appear to be isolated on relatively small spatial scales, at least in the Indo-Pacific, indicating that conservation and management efforts will need to be exerted at relatively small scales in a pelagic and highly vagile species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Widely distributed marine species are often composed of geographically localized, genetically differentiated populations despite presumed high levels of vagility (e.g., dolphins, Andrews et al. 2010; lobster, Chow et al. 2011; yellow tang, Eble et al. 2011; fiddler crab, Aoki and Wada 2013). These distinct regional populations commonly are delineated by known dispersal barriers such as the East Pacific Barrier, the Isthmus of Panama (but see Marko 2002) and the southern tip of Africa and South America (Briggs and Bowen 2012). Within these regions, however, populations are generally fairly well connected, commonly thought to be a consequence of highly dispersive planktonic larval phases often influenced by oceanic current patterns. Unlike most widely distributed marine fish and invertebrate species, however, sharks do not possess planktonic life stages and their dispersal (which can lead to genetic connectivity) therefore occurs only by active movements of juveniles and adults. Despite this apparent limitation for long-distance dispersal via ocean currents, sharks often display widespread distributions, with many large species occurring globally.
Many shark species are heavily overfished regionally (Ferretti et al. 2008; Camhi et al. 2009; Ward-Paige et al. 2010; Pérez-Jiménez 2014). As sharks tend to be slow growing and late to mature, overfishing can profoundly, negatively impact population persistence. When globally distributed species actually are comprised of local, isolated breeding groups, overfishing can lead to regional extirpation. Information on connectivity among breeding groups within shark species over their geographic range, therefore, is essential to inform management and conservation efforts, including tracking of the fin trade (Chapman et al. 2009; Shivji 2010).
Assaying the degree of population subdivision in sharks is often done by determining the levels and geographical distribution patterns of DNA variation. Most of the studies conducted thus far have detected a wide range of population genetic differentiation, ranging from little subdivision globally to highly structured populations even within an ocean basin. Commonly, these studies reveal that population structure in sharks broadly corresponds to the type of habitat favored by individual species. For example, sharks occupying mainly open-ocean habitats (i.e., beyond the continental shelf; pelagic) have thus far displayed either no differentiation (basking shark, Hoelzel et al. 2006) or population fragmentation detectable only over inter-ocean basin scales (shortfin mako shark, Schrey and Heist 2003; whale shark, Castro et al. 2007; Schmidt et al. 2009). In contrast, sharks with preferences for mostly coastal habitats or only occasionally found in pelagic waters, tend to show population structuring on much smaller, albeit variable, geographic scales (e.g., blacktip shark, Keeney et al. 2005; Keeney and Heist 2006; scalloped hammerhead shark, Duncan et al. 2006; Chapman et al. 2009; Nance et al. 2011; lemon sharks, Schultz et al. 2008; gray nurse shark, Ahonen et al. 2009; sandbar shark, Portnoy et al. 2010; bull shark, Karl et al. 2011; Atlantic nurse shark, Karl et al. 2012a; spinner shark, Geraghty et al. 2013; dusky shark, Geraghty et al. 2014; spot-tail shark, Giles et al. 2014). Complicating the a priori inference of shark population structure based on species habitat type, however, is the broad spectrum of habitat use, life histories and movement patterns displayed by even closely related species. Given the variable scale of population relationships detected in the published studies, it is clear that deriving a general picture of evolutionary dispersal history to explain the current widespread distribution of many large sharks will require assessing phylogeographic structure in more species (Giles et al. 2014). It is also likely that the detailed population structure information required for properly managing commercially exploited sharks will require assessment by individual species of interest.
The silky shark, Carcharhinus falciformis, is a circumglobally distributed, tropical and subtropical species. It is essentially pelagic in habitat, with adults and older juveniles found in deep waters just off continental and insular shelves but also commonly in open-ocean waters (Bonfil 2008; Bonfil et al. 2009). In contrast, silky shark newborns and early juveniles, at least in the western Atlantic, are found in deeper waters on the continental shelf, suggesting geographically localized nursery grounds (Yokota and Lessa 2006; Bonfil 2008). To date, there is no evidence of female philopatry in silky sharks to nursery grounds.
As widely distributed, large-bodied sharks that utilize deep shelf and open-ocean habitats, it is thought that silky sharks are capable of long-distance movements. Indeed, two individuals have been documented to disperse long distances, with one traveling 1339 km along the western North Atlantic (Kohler and Turner 2001) and the other ~2200 km in the eastern Pacific (EP) (MigraMar 2011). The majority of the tagging data available for silky sharks, however, indicate far shorter displacement distances (Kohler et al. 1998; Clarke et al. 2011).
The wide distribution and habitat preference of silky sharks exposes them to strong direct and bycatch fisheries pressure in international pelagic fisheries (Beerkircher et al. 2004; Watson et al. 2008). Their fins constitute a major portion of the global fin trade (Clarke et al. 2006). Silky sharks are listed by the International Union for the Conservation of Nature (IUCN) as Vulnerable in the eastern central and southeast Pacific, northwest and west central Atlantic, and Near Threatened in the southwest Atlantic, Indian and western central Pacific oceans (Bonfil et al. 2009). Furthermore, this species’ high vulnerability in ecological risk assessment studies has led the ICCAT (International Commission for the Conservation of Tuna) to mandate that silky sharks captured in the Atlantic by ICCAT contracting nation fisheries be released whether dead or alive and that silky shark body parts not be retained or transshipped (ICCAT 2011).
Despite their highly exploited status and related population viability concerns, there is still little information on genetic diversity and population structure of silky sharks to guide management and conservation of this generally poorly understood species (Bonfil 2008; Watson et al. 2008). A recent study (Galván-Tirado et al. 2013) of silky sharks in the Pacific Ocean based on assessing a 732-base-pair (bp) portion of the mitochondrial DNA (mt DNA) control region revealed a low level of population subdivision at fairly large geographic scales (i.e., eastern versus western Pacific locations). However, tempering these conclusions is that although the latter study included robust sample sizes from the eastern Pacific (EP), there were only two sample sites, both possessing limited sample sizes, assessed from the western Pacific. Our study reports the first genetic assessment of mitochondrial population structure and phylogeography of the silky shark from across major parts of its global range. Although our inferences are based on mitochondrial DNA sequences only, this single marker continues to provide a robust and informative first approach for phylogeographic studies in species of conservation concern (Bowen et al. 2014).
Materials and methods
Samples and DNA sequencing
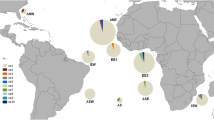
Silky shark fin clips or occasionally muscle sections were obtained from 276 animals caught in artisanal or commercial fisheries from six, globally distributed regions (i.e., from local markets or directly from fishing vessels operating in these regions) (Fig. 1) and stored in 95 % ethanol. Genomic DNA was extracted from ~25 mg of tissue using the QIAGEN DNeasy kit (QIAGEN Inc, Valencia, CA, USA). Polymerase chain reaction (PCR) amplification of the entire mitochondrial control region (mtCR) was performed using external primers CR-F6 (5′-AAGCGTCGACCTTGTAAGTC-3′) and DAS-R2 (5′-GCTGAAACTTGCATGTGTAA-3′) that we designed based on conserved flanking regions from elasmobranch mitochondrial DNA sequences in GenBank. A species-specific, internal forward primer CRint455-F (5′-ACGGTTTGTGGTACATTAC-3′) was designed to sequence through the entire mtCR. The three primers yielded single-stranded coverage of ~60 % and double-stranded coverage of ~40 % of the ~1069 bp mtDNA control region. PCR amplifications (50 μl total volume) consisted of 50 µM of each dNTP, 10 pmol of each primer, 10–25 ng of extracted DNA, 1X PCR buffer and 1 unit of HotStar Taq Plus DNA polymerase (QIAGEN Inc, Valencia, CA, USA). The PCR cycling profile consisted of 15 min (min) at 95 °C, followed by 35 cycles at 94 °C for 1 min, 50 °C for 1 min, 72 °C for 2 min and a final 5-min extension at 72 °C. Amplification specificity and efficiency were determined via visualization on a 1.2 % ethidium bromide-stained agarose gel, and amplicons were purified using the QIAquick PCR purification kit (QIAGEN Inc, Valencia, CA, USA) following the manufacturer’s protocol. Cycle sequencing was performed using the Applied Biosystems BigDye Terminator v3.1 kit (Life Technologies, Carlsbad, CA, USA), following manufacturer’s instructions. Resulting products were sequenced on an Applied Biosystems 3130 Genetic Analyzer and bases called using Applied Biosystems Sequencing Analysis Software version 5.2.

Sequence and genealogical analyses
Sequences were aligned using geneious pro v. 3.5.6 software (Biomatters Ltd, Auckland, NZ) and edited manually. Owing to high sequence quality and minimal base ambiguity, the single-stranded portions of the mtCR were checked by manual examination of each electropherogram and the few base uncertainties seen were resolved by re-amplification and re-sequencing. The number of mtCR haplotypes was identified using FaBox 1.41 (Villesen 2007). Relationships between the haplotypes and concordance with geographic sampling locations were assessed by constructing an unrooted statistical parsimony network at the 95 % confidence interval criterion using the software tcs v. 1.13 (Clement et al. 2000), and a median-joining (MJ) network (Bandelt et al. 1999) using the program PopART (http://popart.otago.ac.nz).
Genetic diversity and population structure
Haplotype (h) and nucleotide (π) diversities, and the number of polymorphic sites (S) were calculated using arlequin 3.1 (Excoffier et al. 2005). An analysis of molecular variance (AMOVA; arlequin 3.1) was performed to estimate the overall extent of genetic subdivision. The extent of genetic differentiation among sharks from the different sampling regions (regional groups) was assessed using two statistical methods that rely on different approaches to obtain a comparative view: (1) pairwise estimates of Φ ST using arlequin 3.1 (Excoffier et al. 2005) with significance determined by 10,000 data permutations and (2) Hudson’s (2000) nearest-neighbor statistic (S NN), a measure of how often the nearest neighbors in sequence space are from the same locality in geographic space. The S NN test estimates genetic differentiation among regional groups based on the number of sequence differences between haplotypes, and not just haplotype frequencies, and can be more powerful when the haplotype diversity is high (Hudson 2000). The S NN statistic was calculated in dnasp v5 (Librado and Rozas 2009), and significance was estimated by 1000 data permutations. Significance values were adjusted using the false discovery rate (FDR) method to accommodate multiple comparisons (Benjamini and Hochberg 1995).
Because our silky shark samples did not extend to the EP, and to obtain a broader geographic perspective on population structure for this species, we also reanalyzed all our shark samples by including all the mtCR sequences obtained by Galván-Tirado et al. (2013) from the EP (N = 242; Region code EPO, Table 1 in their paper). To use their sequences, we trimmed our full-length mtCR sequences to match the sequence length from their study. Variation in sequence length (732 bp in Galván-Tirado et al. (2013) vs. 734 bp in our study) was due to the presence of indels once the datasets were combined. All statistical analyses conducted on this combined, trimmed dataset were as described above.
Results
The full-length mtCR fragment in the 276 analyzed silky sharks was 1063–1069 base pairs, with a GC content of 33.65 %. The sequences resolved 62 total haplotypes defined by 34 polymorphic sites (GenBank accession numbers KM267565–KM267626). The frequency of each haplotype in the various regional groups is shown in Supplementary Material Table S1. Fifty-three percent of the haplotypes were singletons. Global haplotype and nucleotide diversity were relatively high (h = 0.93 ± 0.01; π = 0.61 ± 0.32 %). The average haplotype and nucleotide diversity across samples was computed to compare these metrics to those determined from 15 other shark species for the mtCR (Karl et al. 2011; Whitney et al. 2012). The average silky shark haplotype and nucleotide diversities were 0.86 ± 0.04 and 0.28 ± 0.16 %, respectively. Although nearly equal numbers of haplotypes were observed in the western Atlantic and Indo-Pacific, the nucleotide diversities were significantly lower for silky sharks from the Indo-Pacific (Table 1; two-tailed t test, P = 0.003 with logit-transformed data).
Population structure
Full-length mtCR dataset (without EP samples)
Globally, silky sharks were found (AMOVA) to be markedly structured with a highly significant overall Φ ST (0.49, P < 0.001). Pairwise comparisons among silky shark regional groups (Φ ST; Table 2) showed strong genetic differentiation between the western Atlantic and Indo-Pacific sites. Within the Indo-Pacific, the regional groups were statistically differentiated, with the exception of samples from the Line Islands and Red Sea. None of the three sites in the Atlantic, however, were statistically significantly different (P > 0.05). Hudson’s nearest-neighbor analysis (S NN) mirrored the Φ ST analysis, demonstrating largely strong genetic differentiation among Indo-Pacific regional groups and genetic homogeneity among western Atlantic collections (Table 3).
Trimmed length mtCR dataset (including EP samples)
Analysis of this extended geographic sequence dataset provided generally the same population differentiation results. The AMOVA results again demonstrated silky sharks to be markedly structured globally with a highly significant overall Φ ST (0.59, P < 0.001). Within the Indo-Pacific and among the Indo-Pacific and Atlantic Oceans sites, the regional groups were strongly differentiated (Supplementary Material Table S2). There was no statistical differentiation (P > 0.05) among the three Atlantic sites. The eastern Pacific (EP) samples were significantly differentiated from all other global sampling sites, except the Line Islands. However, in contrast to the results from full-length mtCR sequences, the pairwise Φ ST between the Andaman Sea and Line Island samples was not significant.
Pairwise Hudson’s nearest-neighbor analysis (S NN) again mirrored Φ ST analysis, including significantly differentiating the EP silky sharks from all other global sampling locations except the Line Islands (Supplementary Material Table S3).
Evolutionary relationships
The statistical parsimony network derived from the full-length mtCR silky shark haplotypes revealed two highly distinct lineages separated by seven mutational steps and strong phylogeographic patterning (Fig. 2). Within each lineage, most haplotypes differed from their closest haplotype by a single mutational step. Several individuals sampled in the Atlantic had haplotypes identical or very similar to haplotypes from individuals sampled in the Indo-Pacific. The median-joining network analysis provided the same overall phylogeographic patterning as the statistical parsimony network analysis, with two highly distinct lineages and clustering of some Atlantic individual haplotypes in the Indo-Pacific clade (not shown).

Statistical parsimony network with haplotypes connected at the 95 % confidence interval. Colors represent geographic sampling locations (regional groups) of silky sharks (C. falciformis). Size of each circle is proportional to the frequency of that haplotype. Small solid black circles represent hypothetical haplotypes not sampled in this study. Each connecting line between haplotypes represents one mutational step
The statistical parsimony network derived from the combined, trimmed mtCR silky shark dataset revealed the same two highly distinct lineages separated by five mutational steps. The nine haplotypes observed in the EP samples grouped exclusively with the Indo-Pacific clade (Supplementary Material Fig. S1).
Discussion
Genetic diversity
This work focuses mainly, except as noted, on results from the full-length mitochondrial dataset since it provides more sequence length for assessment. Overall (global-scale) mtCR genetic diversity of the silky sharks was high, with average haplotype diversity (0.86) the third highest relative to 15 other shark species studied to date (Benavides et al. 2011; Karl et al. 2011; Whitney et al. 2012). The only sharks with higher mtCR haplotype diversity (Prionace glauca and Rhincodon typus) are both pelagic species with global distributions. The silky shark average nucleotide diversity was seventh highest compared to values estimated for the other shark species. Interestingly, four of the six species with higher nucleotide diversities also are primarily oceanic in habitat (Karl et al. 2011). The silky shark genetic diversity findings, therefore, are consistent with the conclusion of Karl et al. (2011) that, in general, pelagic sharks that can move long distances may have greater genetic diversity than near-shore species. These findings also are consistent with the general principle that species with broad ecological niches, as is expected to be the case with silky sharks given their global distribution and continental shelf to pelagic habitat utilization, tend to have higher genetic diversities than species with narrow ecological niches (Habel and Schmitt 2012).
The finding of overall high genetic diversity in silky sharks is, however, inconsistent with the substantial exploitation this species encounters in global pelagic fisheries (Clarke et al. 2006, 2012; Cortés et al. 2008; Watson et al. 2008). In general, large population reductions could result in a population bottleneck and concomitant loss of genetic diversity (Nei et al. 1975; Beebee and Rowe 2008). Indeed, reductions in genetic diversity related to overexploitation have been documented in diverse marine fishes (Pinsky and Palumbi 2014). Notably, such a situation has not so far been observed in any elasmobranch, possibly because their particular life histories make them less susceptible to anthropogenic-caused genetic bottlenecks (Chapman et al. 2011). Another possible explanation for the high genetic diversity in silky sharks in the face of reductions in population sizes is that bottlenecks caused by fishery exploitation have occurred relatively recently (over the past 40–50 years) and there has been insufficient time for genetic drift to reduce genetic diversity in a relatively long-lived species (13–20 years; Sánchez-de Ita et al. 2011; Nei et al. 1975). Alternatively, very large, pre-industrial fishing population sizes for silky sharks globally may have helped buffer the loss of genetic diversity from high fishery exploitation. Regardless, the finding of high genetic diversity provides tempered optimism for the mitochondrial genetic health of silky sharks. An assessment of nuclear genetic diversity, however, is prudent to provide a comprehensive view of the overall genetic health of this species and to test for sex-specific dispersal patterns.
Population structure
The semi-pelagic habit of silky sharks makes the findings of substantial matrilineal population structure across much of this shark’s global distribution, intriguing. The fact that silky sharks are routinely captured in both continental shelf and open-ocean fisheries argues for the ability of this species to move long distances and occupy diverse habitats, leading to an expectation of little population structure at least within ocean basins. The absence of population structure across the western North and South Atlantic is consistent with this expectation. We were unable to gain access to silky shark samples from the eastern Atlantic, leaving a conspicuous gap in obtaining a holistic view of matrilineal population structure in the Atlantic. However, the few shark phylogeography studies that have include samples from the eastern Atlantic (scalloped hammerhead sharks, Duncan et al. 2006; blacktip sharks, Keeney and Heist 2006; lemon sharks, Schultz et al. 2008) have all indicated population subdivision across the Atlantic Ocean. Although these inferences were made from analyses of limited sample sizes, they leave open the distinct possibility that silky sharks may also be genetically structured across the Atlantic.
In contrast to the western Atlantic, the silky shark Φ ST and S NN indicated shallow, statistically significant population differentiation among most of the Indo-Pacific sites. The only exceptions both involve samples from the Line Islands, which are not statistically differentiated from the Red Sea and EP silky sharks. The absence of differentiation between the Line Islands and Red Sea is unexpected due to the tremendous distance between these two sites (>20,000 km straight line distance) and the significant differentiation of both of these sites with the intervening Andaman Sea site. It should be noted, however, that the Line Islands represent the smallest sample size and lowest haplotype diversity (Table 1), and the absence of significant differences may be a statistical artifact or reflect a lack of power. This statistical artifact may also explain the absence of differentiation (based on the trimmed sequence dataset) between the Line Islands and EP sites which are separated by the East Pacific Barrier, a soft biogeographic break (Cowman and Bellwood 2013) for many marine species including the scalloped hammerhead and sicklefin lemon sharks (Duncan et al. 2006; Schultz et al. 2008). Although the possibility of long-distance matrilineal connectivity remains (i.e., the lack of silky shark differentiation between the Line Islands and Red Sea reflects the true state of nature), a statistical artifact/lack of statistical power is a more likely explanation. As a precautionary management view, therefore, silky sharks from all the Indo-Pacific sites examined should be considered differentiated from each other and to constitute discrete population sub-segments.
The Line Islands population was likely separated from the Red Sea and Andaman Sea populations during last glacial maximum that exposed the Sunda Shelf approximately 20,000 years ago (Siddall et al. 2003; Bailey 2009). The Red Sea also is a well-documented region with a very high, and likely underestimated, level of endemism (DiBattista et al. 2013) and likely was isolated from the Indian Ocean as well during the last glacial maximum because the shallow (~130 m) strait of the Bab al Mandab is the only connection with the Indian Ocean. Our results are broadly similar to the previous study (Galván-Tirado et al. 2013) with silky sharks showing population genetic differentiation, at least in the Pacific, over broad geographic areas. A caveat to our conclusions, however, is that they are based on a single, maternally inherited locus. Even though there is population subdivision in mtDNA, this does not mean that silky sharks from these locations are isolated from each other. If females exhibit natal site philopatry, there could still be substantial male-mediated gene flow among sites.
That silky sharks in the western Atlantic appear panmictic is somewhat surprising. The major potential geographic barrier to gene flow for silky sharks in the western Atlantic is the soft-barrier, freshwater outfall from the Amazon River separating the Caribbean and Brazilian eco-provinces (Rocha et al. 2005a; Briggs and Bowen 2013). Previous mtDNA studies of elasmobranchs in this region provide contrasting results. For the bull shark, Carcharhinus leucas, (Karl et al. 2011) and the lemon shark, Negaprion brevirostris, (Feldheim et al. 2001; Schultz et al. 2008), there was marked subdivision between the Caribbean and Brazil in mtDNA, but little to none in nuclear microsatellite DNA. In both cases, the pattern was thought to be due to female philopatry on a background of male-mediated gene flow. The strong separation between the Caribbean and Brazil was also seen in mtDNA of the blacktip shark, Carcharhinus limbatus, although no nuclear DNA data are available to differentiate male-mediated gene flow from female philopatry (Sodré et al. 2012). There was, however, clear population subdivision seen in both mtDNA and microsatellite loci in the sand tiger shark, also known as ragged-tooth shark or gray nurse shark (Carcharias taurus, Ahonen et al. 2009). For the relatively sedentary nurse shark, Ginglymostoma cirratum, there was moderate subdivision between the northwestern Atlantic and Brazilian samples at microsatellite loci; however, the samples from Belize and coastal Brazil were not differentiated at mtDNA.
There are a variety of explanations for the lack of a clear pattern across these various species. The most likely explanation is differences in the life histories (Karl et al. 2012b). Whether a species exhibits female (or male) philopatry will have a significant influence on the resulting population structuring and patterns seen in mtDNA versus nuclear loci. Other attributes such as habitat preference may also affect the pattern and degree of population subdivision. The bull and lemon sharks are likely capable migrating between the Caribbean and Brazil; however, female philopatry produces discordant patterns when assessing mitochondrial versus nuclear DNA. That the sand tiger and the nurse shark show population subdivision is likely because they are more strongly coastally associated species. The lack of population subdivision in silky sharks in this region is probably due to their more pelagic existence accompanied by longer distance movements. Without accompanying nuclear DNA data (e.g., microsatellites), our inferences are necessarily limited. We can say, however, that the lack of population subdivision in the western Atlantic is consistent with the lack of female philopatry. If this is the common behavioral mode for silky sharks generally, then the population subdivision seen in the Indo-Pacific Oceans would be due to true population isolation and not philopatry.
Evolutionary relationships and phylogeography
The statistical parsimony network identified two evolutionarily divergent maternal lineages of silky sharks across their sampled global range. This finding supports a relatively long-term, historical separation of female silky sharks from the western Atlantic and Indo-Pacific. Evolutionary, matrilineal separation between these two major ocean basins has also been observed in the congener sharks C. limbatus (blacktip;, Keeney and Heist 2006, Keeney et al. 2005), C. obscurus (dusky, Benavides et al. 2011) and C. plumbeus (sandbar, Portnoy et al. 2010) and the hammerhead shark (Sphyrna lewini, Duncan et al. 2006; Daly-Engel et al. 2012), hinting that this type of evolutionary separation may be a common feature in the globally distributed, large-bodied carcharhiniform sharks.
Of note is that several silky shark individuals sampled from the western Atlantic possessed haplotypes that were shared with or very closely related to individuals sampled in the Indo-Pacific (Fig. 2). Interestingly, clustering of these Atlantic animal haplotypes with the Indo-Pacific lineage was entirely non-reciprocal. Further, the divergence between the Atlantic and Indo-Pacific groups (Fig. 2) is relatively large and suggests that the western Atlantic individuals with Pacific haplotypes are not likely due to incomplete lineage sorting. This suggests relatively recent (i.e., post-ocean basin divergence), unidirectional, secondary contact with dispersal likely from the Indian Ocean into the Atlantic probably around the tip of South Africa. The reason for the unidirectionality is unclear, but may be related to the pattern of currents around the tip of South Africa. The cold Benguela Current flows northwards along the western coast of South Africa, causing upwelling, and is considered a barrier to Atlantic warm water species moving into the Indian Ocean. Countering this cold current is the southwestern flowing, warm Agulhas Current and together they define the Benguela (Atlantic Ocean) and Agulhas (Indian Ocean) provinces (Briggs and Bowen 2013). Easing of the Benguela Current and expansion of the Agulhas Current into the Atlantic has been posited to explain similar Indian to Atlantic Ocean movements of several species such as fish (Bowen et al. 2001, 2006; Rocha et al. 2005b; Reece et al. 2010), sea turtles (Roberts et al. 2004) and rays (Richards et al. 2009).
Conservation implications
Two findings from this study have direct conservation implications. The high overall mitochondrial genetic diversity in silky sharks suggests this species has not experienced a matrilineal genetic bottleneck, despite the heavy exploitation silky sharks encounter in pelagic fisheries worldwide. A related caveat, however, is the unexpected finding that silky sharks from the Indo-Pacific show lower genetic diversity, with nucleotide diversity about half that in the western Atlantic. An even lower level of mtCR genetic diversity in silky sharks from the Indo-Pacific (albeit mainly EP samples) was reported by Galván-Tirado et al. (2013), further highlighting the large diversity difference between ocean basins. A reason for this lower diversity could be much higher exploitation pressure on silky sharks in the Indo-Pacific. Regardless, this lower diversity suggests Indo-Pacific silky sharks, and especially populations from the central and EP, are at a higher risk of genetic diversity loss than western Atlantic sharks in the face of continued high exploitation pressure.
The conservation of genetic diversity is now even more of a priority in the context of possibly large-scale climate changes and associated ocean ecosystem alterations. Our genetic assessment of silky sharks on a worldwide scale shows two clear evolutionary lineages exist between the western Atlantic and Indo-Pacific, indicating that management of this species should occur on, at the very least, on a two mitochondrial-stock basis. The strong divergence between the two lineages suggests that the western Atlantic population will not effectively serve as a source of replenishment by female immigrants should the Indo-Pacific lineage become depleted from disproportionate overfishing. The low but statistically significant genetic differentiation within the Indo-Pacific also raises a cautionary note that connectivity among these regional populations may be low enough to hinder effective replacement if any one of them becomes extirpated. We acknowledge, however, that if the lack of statistical differentiation seen across the vast distance separating the Red Sea and Line Islands is the true state of matrilineal connectivity, at least female immigration from other regions could potentially temper local reductions in population size. Within-ocean basin differentiation was also seen in a previous mitochondrial DNA study that recommended managing silky sharks in the Pacific as two genetically differentiated stocks (Galván-Tirado et al. 2013). With the addition of the present data, at least five mitochondrial DNA populations of silky sharks can be identified globally. Further genetic studies that include more Indo-Pacific sampling sites and nuclear DNA markers are necessary to clarify the extent and magnitude of structure throughout the Indo-Pacific. In the interim, given widespread and growing concerns about their apparent declining trajectory globally, a precautionary strategy that treats and manages silky sharks from the western and EP Ocean, Indian Ocean, Red Sea and the western Atlantic Ocean as individual stocks (management units) is advisable.
References
Ahonen H, Harcourt RG, Stow AJ (2009) Nuclear and mitochondrial DNA reveals isolation of imperiled grey nurse shark populations (Carcharias taurus). Mol Ecol 18:4409–4421
Andrews KR, Karczmarski L, Au WWL, Rickards SH, Vanderlip CA, Bowen BW, Grau EG, Toonen RJ (2010) Rolling stones and stable homes: social structure, habitat diversity and population genetics of the Hawaiian spinner dolphin (Stenella longirostris). Mol Ecol 19:732–748
Aoki M, Wada K (2013) Genetic structure of the wide-ranging fiddler crab Uca crassipes in the west Pacific region. J Mar Biol Assoc UK 93:789–795
Bailey G (2009) The Red Sea, coastal landscapes, and hominin dispersals. In: Petyraglia M, Rose J (eds) The evolution of human populations in Arabia. Springer, Dordrecht
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Beebee T, Rowe G (2008) An introduction to molecular ecology, 2nd edn. Oxford University Press, Oxford
Beerkircher L, Cortes E, Shivji MS (2004) Characteristics of shark bycatch observed on pelagic longlines off the southeastern United States, 1992–2000. Mar Fish Rev 64:40–49
Benavides MT, Horn RL, Feldheim KA, Shivji MS, Clarke SC, Wintner S, Natanson L, Braccini M, Boomer JJ, Gulak SJB, Chapman DD (2011) Global phylogeography of the dusky shark, Carcharhinus obscurus: implications for fisheries management and monitoring the shark fin trade. Endanger Species Res 14:13–22
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57(1):289–300
Bonfil R, Amorim A, Anderson C, Arauz R, Baum J, Clarke SC, Graha, RT, Gonzalez M, Jolón M, Kyne PM, Mancini P, Márquez F, Ruí C, Smith W (2009) Carcharhinus falciformis. Version 2014.3. The IUCN Red List of Threatened Species. http://www.iucnredlist.org/details/39370/0. Accessed 28 Oct 2014
Bonfil R (2008) The biology and ecology of the silky shark, Carcharhinus falciformis. In: Camhi MD, Pikitch EK, Babcock EA (eds) Sharks of the open ocean: biology, fisheries and conservation. Fish and aquatic resources series 13. Blackwell Publishing Ltd., UK, pp 114–127
Bowen BW, Bass AL, Rocha LA, Grant WS, Robertson DR (2001) Phylogeography of the trumpetfish, genus Aulostomus: ring species complex on a planetary scale. Evolution 55:1029–1039
Bowen BW, Muss A, Rocha LA, Grant WS (2006) Shallow mtDNA coalescence in Atlantic pygmy anglefishes (genus Centropyge) indicates a recent invasion from the Indian Ocean. J Hered 97:1–12
Bowen BW, Shanker K, Yasuda N, Malay MCD, von der Heyden S, Paulay G, Rocha LA, Selkoe KA, Barber PH, Williams ST, Lessios HA, Crandall ED, Bernardi G, Meyer CP, Carpenter KE, Toonen RJ (2014) Phylogeography unplugged: comparative surveys in the genomic era. Bull Mar Sci 90:13–46
Briggs JC, Bowen BW (2012) A realignment of marine biogeographic provinces with particular reference to fish distributions. J Biogeogr 39:12–31
Briggs JC, Bowen BW (2013) Marine shelf habitat: biogeography and evolution. J Biogeogr 40:1023–1035
Camhi MD, Valenti SV, Fordham SV, Fowler SL, Gibson C (2009) The conservation status of pelagic sharks and rays: report of the IUCN Shark Specialist Group Pelagic Shark Red List Workshop. IUCN Species Survival Commission Shark Specialist Group. Newbury, UK. x + 78p
Castro ALF, Stewart BS, Wilson SG, Hueter RE, Meekan MG, Motta PJ, Bowen BW, Karl SA (2007) Population genetic structure of Earth’s largest fish, the whale shark (Rhincodon typus). Mol Ecol 16:5183–5192
Chapman DD, Pinhal D, Shivji MS (2009) Tracking the fin trade: genetic stock identification in Western Atlantic scalloped hammerheads sharks (Sphyrna lewini). Endanger Species Res 9:221–228
Chapman DD, Simpfendorfer CA, Wiley TR, Poulakis GR, Curtis C, Tringali M, Carlson JK, Feldheim KA (2011) Genetic diversity despite population collapse in a critically endangered marine fish: the smalltooth sawfish (Pristis pectinata). J Hered 102:643–652
Chow S, Jeffs A, Miyake Y, Konishi K, Okazaki M, Suzuki N, Abdullah MF, Imai H, Wakabayasi T, Sakai M (2011) Genetic isolation between the western and eastern Pacific populations of Pronghorn spiny lobster Panulirus penicillatus. PLoS One 6:e29280. doi:10.1371/journal.pone.0029280
Clarke SC, McAllister MK, Milner-Gulland EJ, Kirkwood GP, Michielsens CGJ, Agnew DJ, Pikitch EK, Nakano H, Shivji MS (2006) Global estimates of shark catches using trade records from commercial markets. Ecol Lett 9:1115–1126
Clarke C, Lea JSE, Ormond RFG (2011) Reef-use and residency patterns of a baited population of silky sharks, Carcharhinus falciformis, in the Red Sea. Mar Freshw Res 62:668–675
Clarke SC, Harley SJ, Hoyle SD, Rice JS (2012) Population trends in Pacific oceanic sharks and the utility of regulations on shark finning. Conserv Biol 27(1):197–209
Clement M, Posada D, Crandall KA (2000) tcs: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1660
Cortés E, Arocha F, Beerkircher L, Carvalho F, Domingo A, Heupel M, Holtzhausen H, Santos MN, Ribera M, Simpfendorfer C (2008) Ecological risk assessment of pelagic sharks caught in Atlantic pelagic longline fisheries. Aquat Living Resour 23:25–34
Cowman PF, Bellwood DR (2013) Vicariance across major marine biogeographic barriers: temporal concordance and the relative intensity of hard versus soft barriers. Proc R Soc B Biol Sci 280:20131541
Daly-Engel TS, Seraphin KD, Holland KN, Coffey JP, Nance HA, Toonen RT, Bowen BW (2012) Global phylogeography with mixed-marker analysis reveals male-mediated dispersal in the endangered scalloped hammerhead shark (Sphyrna lewini). PLoS One 7:e29986. doi:10.1371/journal.pone.0029986
DiBattista JD, Berumen ML, Gaither MR, Rocha LA, Eble JA, Choat JH, Craig MT, Skillings DJ, Bowen BW (2013) After continents divide: comparative phylogeography of reef fishes from the Red Sea and Indian Ocean. J Biogeogr 40:1170–1181
Duncan KM, Martin AP, Bowen BW, de Couet G (2006) Global phylogeography of the scalloped hammerhead shark (Sphyrna lewini). Mol Ecol 15:2239–2251
Eble JA, Toonen RJ, Sorenson L, Basch LB, Papastamatiou YP, Bowen BW (2011) Escaping paradise: larval export form Hawaii in an Indo-Pacific reef fish, the yellow tang Zebrasoma flavescens. Mar Ecol Prog Ser 428:245–258
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Feldheim KA, Gruber SH, Ashley V (2001) Population genetic structure of the lemon shark (Negaprion brevirostris) in the western Atlantic: DNA microsatellite variation. Mol Ecol 10:295–303
Ferretti F, Myers RA, Serena F, Lotze HK (2008) Loss of large predatory sharks from the Mediterranean Sea. Conserv Biol 22:952–964
Galván-Tirado C, Díaz-Jaimes P, García-de León FJ, Galván-Magaña FG, Uribe-Alcocer M (2013) Historical demography and genetic differentiation inferred from the mitochondrial DNA of the silky shark (Carcharhinus falciformis) in the Pacific Ocean. Fish Res 147:36–46
Geraghty PT, Williamson JE, Macbeth WG, Wintner SP, Harry AV, Ovenden JR, Gillings MR (2013) Population expansion and genetic structure in Carcharhinus brevipinna in the Southern Indo-Pacific. PLoS One 8(9):e75169. doi:10.1371/journal.pone.0075169
Geraghty PT, Williamson JE, Macbeth WG, Blower MJ, Johnson G, Ovenden JR, Gillings MR (2014) Genetic structure and diversity of two highly vulnerable carcharhinids in Australian waters. Endanger Species Res 24:45–60
Giles JL, Ovenden JR, Dharmadi AD, Garvilles E, Khampetch K, Manjebrayakath H, Riginos C (2014) Extensive genetic population structure in the Indo-West Pacific spot-tail shark, Carcharhinus sorrah. Bull Mar Sci 90:427–454
Habel JC, Schmitt T (2012) The burden of genetic diversity. Biol Conserv 147:270–274
Hoelzel AR, Shivji MS, Magnussen J, Francis MP (2006) Low worldwide genetic diversity in the basking shark (Cetorhinus maximus). Biol Lett 2:639–642
Hudson RR (2000) A new statistic for detecting genetic differentiation. Genetics 155:2011–2014
ICCAT (2011) Recommendation by ICCAT on the conservation of silky sharks caught in association with ICCAT fisheries, 11-08 BYC. http://www.ccsbt.org/userfiles/file/other_rfmo_measures/iccat/ICCAT_2011-08.pdf. Accessed 28 Oct 2014
Karl SA, Castro ALF, Lopez JA, Charvet P, Burgess GH (2011) Phylogeography and conservation of the bull shark (Carcharhinus leucas) inferred from mitochondrial and microsatellite DNA. Conserv Genet 12:371–382
Karl SA, Castro ALF, Garla RC (2012a) Population genetics of the nurse shark (Ginglymostoma cirratum) in the western Atlantic. Mar Biol 159:489–498
Karl SA, Toonen RJ, Grant WS, Bowen BW (2012b) Common misconceptions in molecular ecology: echoes of the modern synthesis. Mol Ecol 21:4171–4189
Keeney DB, Heist EJ (2006) Worldwide phylogeography of the blacktip shark (Carcharhinus limbatus) inferred from mitochondrial DNA reveals isolation of western Atlantic populations coupled with recent Pacific dispersal. Mol Ecol 15:3669–3679
Keeney DB, Heupel MR, Hueter RE, Heist EJ (2005) Microsatellite and mitochondrial DNA analyses of the genetic structure of blacktip shark (Carcharhinus limbatus) nurseries in the northwestern Atlantic, Gulf of Mexico, and Caribbean Sea. Mol Ecol 14:1911–1923
Kohler NE, Turner PA (2001) Shark tagging: a review of conventional methods and studies. Environ Biol Fishes 60:191–223
Kohler NE, Casey JG, Turner PA (1998) NMFS cooperative shark tagging program, 1962–1993: an atlas of shark tag and recapture data. Mar Fish Rev 60:1–87
Librado P, Rozas J (2009) dnasp v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Marko PB (2002) Fossil calibration of molecular clocks and the divergence times of geminate species pairs separated by the Isthmus of Panama. Mol Biol Evol 19:2005–2021
MigraMar (2011) Silky shark tagged in Galapagos is detected at Clipperton Atoll. http://migramar.org/hi/home/silky-shark-tagged-in-galapagos-is-detected-at-clipperton-atoll/. Accessed 28 Oct 2014
Nance HA, Klimley P, Galván-Magan M-OJ, Marko PB (2011) Demographic processes underlying subtle patterns of population structure in the scalloped hammerhead shark, Sphyrna lewini. PLoS One 6:e21459. doi:10.1371/journal.pone.0021459
Nei M, Maruyama T, Chakraborty R (1975) The bottleneck effect and genetic variability in populations. Evolution 29:1–10
Pérez-Jiménez JC (2014) Historical records reveal potential extirpation of four hammerhead sharks (Sphyrna spp.) in Mexican Pacific waters. Rev Fish Biol Fish 24:671–683
Pinsky ML, Palumbi SR (2014) Meta-analysis reveals lower genetic diversity in overfished populations. Mol Ecol 23:29–39
Portnoy DS, McDowell JR, Heist EJ, Musick JA, Graves JA (2010) World phylogeography and male-mediated gene flow in the sandbar shark, Carcharhinus plumbeus. Mol Ecol 19:1994–2010
Reece JS, Bowen BW, Smith DG, Larson AF (2010) Molecular phylogenetics of moray eels (Muraenidae) demonstrates multiple origins of shell-crushing jaw (Gymnomuraena, Echidna) and multiple colonization of the Atlantic Ocean. Mol Phylogenet Evol 57:829–835
Richards VP, Henning M, Witzell W, Shivji MS (2009) Species delineation and evolutionary history of the globally distributed spotted eagle ray (Aetobatus narinari). J Hered 100:273–283
Roberts MA, Schwartz TS, Karl SA (2004) Global population genetic structure and male-mediated gene flow in the green sea turtle (Chelonia mydas): analysis of microsatellite loci. Genetics 166:1857–1870
Rocha LA, Robertson DR, Roman J, Bowen BW (2005a) Ecological speciation in tropical reef fishes. Proc R Soc B Biol Sci 272:573–579
Rocha LA, Robertson DR, Rocha CR, Van Tassell JL, Craig MT, Bowen BW (2005b) Recent invasion of the tropical Atlantic by an Indo-Pacific coral reef fish. Mol Ecol 14:3921–3928
Sánchez-de Ita JA, Quiñónez-Velázquez C, Galván-Magaña F, Bocanegra-Castillo N, Félix-Uraga R (2011) Age and growth of the silky shark Carcharhinus falciformis from the west coast of Baja California Sur, Mexico. J App Ichthyol 27:20–24
Schmidt JV, Schmidt CL, Ozer F, Ernst RE, Feldheim KA, Ashley M, Levine M (2009) Low genetic differentiation across three major ocean populations of the Whale shark, Rhincodon typus. PLoS One 4:e4988. doi:10.1371/journal.pone.0004988
Schrey AW, Heist EJ (2003) Microsatellite analysis of population structure in the shortfin mako (Isurus oxyrinchus). Can J Fish Aquat Sci 60:670–675
Schultz JK, Feldehim K, Gruber SH, Ashley MV, McGovern TM, Bowen BW (2008) Global phylogeography and seascape genetics of the lemon sharks (genus Negaprion). Mol Ecol 17:5336–5348
Shivji MS (2010) DNA forensic applications in shark management and conservation. In: Carrier JC, Heithaus MR, Musick JA (eds) Sharks and their relatives II: biodiversity, adaptive physiology, and conservation. CRC Press, Boca Raton, pp 593–610
Siddall M, Rohling EJ, Almogi-Labin A, Hemleben C, Meischner D, Schmelzer I, Smeed DA (2003) Sea-level fluctuations during the last glacial cycle. Nature 423:853–858
Sodré D, Rodrigues-Filho LFS, Souza RFC, Rêgo PS, Schneider H, Sampaio I, Vallinoto M (2012) Inclusion of South American samples reveals new population structuring of the blacktip shark (Carcharhinus limbatus) in the western Atlantic. Genet Mol Biol 35:752–760
Villesen P (2007) FaBox: an online toolbox for fasta sequences. Mol Ecol Notes 7(6):965–968
Ward-Paige CA, Mora C, Lotze HK, Pattengill-Semmens C, McClenachan L, Arias-Castro E, Myers RA (2010) Large-scale absence of sharks on reefs in the greater-Caribbean: a footprint of human pressures. PLoS One 5:e11968. doi:10.1371/journal.pone.0011968
Watson JT, Essington TE, Lennert-Cody CE, Hall MA (2008) Trade-offs in the design of fishery closures: management of silky shark bycatch in the eastern Pacific ocean tuna fishery. Conserv Biol 23:626–635
Whitney NM, Robbins WD, Schultz JK, Bowen BW, Holland KN (2012) Oceanic dispersal in a sedentary reef shark (Triaenodon obesus): genetic evidence for extensive connectivity without a pelagic larval stage. J Biogeogr 39:1144–1156
Yokota L, Lessa RP (2006) A nursery area for sharks and rays in northeastern Brazil. Environ Biol Fishes 75:349–360
Acknowledgments
Funding for this study was provided by the Save Our Seas Foundation, Save Our Seas Shark Research Center, Danah Divers, the Guy Harvey Ocean Foundation and Nova Southeastern University. We thank S. Gulak and D. Bethea (NMFS Panama City Laboratory), and D. McCauley and C. Wilson for contributing shark samples. We are grateful to C. Galván-Tirado and colleagues for providing additional silky shark sequences from their paper, which allowed us to extend our shark assessment to the EP. We specially thank the founder of Save Our Seas Foundation for his vision, passion and efforts in supporting shark research to help understand, preserve and protect these dwindling species around the world.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by T. Reusch.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Clarke, C.R., Karl, S.A., Horn, R.L. et al. Global mitochondrial DNA phylogeography and population structure of the silky shark, Carcharhinus falciformis . Mar Biol 162, 945–955 (2015). https://doi.org/10.1007/s00227-015-2636-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00227-015-2636-6