
Abstract
Gymnocanthus is the most widely distributed genus in the family Cottidae, with six species distributed in the high-latitude area of northern hemisphere. To clarify the phylogenetic relationships and to estimate the divergence times of species in the genus, 2,548 bp of the partial sequences of the 12–16S rRNA, cytochrome oxidase subunit I and cytochrome b gene were analyzed. Our results suggest the monophyletic genus, which arose in the Aleutian Archipelago, divided into a shallow-water group and a deep-water group 8.1 million years ago (Ma). G. tricuspis of the shallow-water group firstly migrated from the Pacific to the Arctic Ocean 5.0 Ma when the Bering Strait first opened. A second migration occurred in the late Pliocene to early Pleistocene after which G. pistilliger and G. intermedius diverged 3.9 Ma. Our findings are discussed within an evolutionary and zoogeographic context.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The opening and closing of the Bering Strait have been major events for many biotas (Briggs 2003). When the strait joining Eurasia and North America was closed, it blocked the ocean connection between the North Pacific and the Arctic Ocean, preventing marine organisms from migrating (Briggs 2003). However, when it first opened at 5.5–4.8 million years ago (Ma, Marincovich and Gladenkov 1999) or 3.0 Ma, some species moved northwards, and speciations have been reported in some groups including invertebrates (about 3.0 Ma, Vermeij 1991) and in Myoxocephalus in the later period (about 1.0 Ma, Kontula and Vainola 2003).
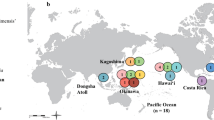
The Cottidae is one of the most diverse fish families found along the North Pacific coastline (Yabe 1985; Mecklenburg et al. 2011), with 275 described species in 70 genera (Nelson 2006). The genera Gymnocanthus, Enophyris, Triglops, and Myoxocephalus occur not only in the North Pacific Ocean, but also in the Atlantic and Arctic oceans. Of these genera, Gymnocanthus is the most widely distributed, inhabiting the high-latitude coastal area in the Pacific, Arctic, and Atlantic Ocean. Gymnocanthus comprises six species: G. intermedius (Temminck and Schlegel), G. pistilliger (Pallas), G. tricuspis (Reinhardt), G. herzensteini Jordan and Starks, G. detrisus Gilbert and Burke, and G. galeatus Bean. G. intermedius and G. herzensteini occur in the northwest Pacific around Japan, G. detrisus occurs in the northwest Pacific and western Bering Sea, G. galeatus occurs in the North Pacific, G. tricuspis occurs in the Bering Sea, Arctic Ocean, and North Atlantic, and G. pistilliger occurs in the North Pacific and North Atlantic, but not in the Arctic Ocean (Fig. 1).

Distributions of Gymnocanthus spp. G. intermedius extends from Iwate and Ishikawa Prefectures to the northern Sea of Japan. G. pistilliger, from the Sea of Japan near South Korea through the Aleutian Archipelago, the Chukchi Peninsula, Southeastern Alaska, and the North Atlantic. G. tricuspis, from the Gulf of St. Lawrence in Canada and Maine, USA, in the Northwest Atlantic to the eastern coasts of Greenland, Iceland, northern coast of Norway, White Sea and throughout the Barents Sea to Spitsbergen and Novaya Zemlya in the Arctic Ocean. G. herzensteini, from the Sea of Japan in northern Japan to the Sea of Okhotsk. G. galeatus, from Hokkaido in Japan to the Aleutian Archipelago, Wales Island, British Columbia in Canada and south of St. Lawrence Island. G. detrisus, from eastern Hokkaido in Japan to the Bering Sea (e.g., Eschmeyer et al. 1983; Masuda et al. 1984; Fedorov 1986; Mecklenburg et al. 2002; Amaoka et al. 2011). Color version is shown in Supplementary material 1. Note: numbers showing sampling sites in Table 2
These six species occur above sandy bottoms and reproduce from winter to early spring (Mecklenburg et al. 2002; fish base) and spawn demersal eggs on the sandy bottom around rocks. Wilson (1973) who clarified these phylogenetic relationships from morphological observations divided into two groups, one included G. herzensteini, G. galeatus, and G. detrisus, and another included G. intermedius, G. pistilliger, and G. tricuspis. The depth distributions also differ among species. Wilson stated G. galeatus is seldom taken at less than 50 m, and G. pistilliger is seldom taken at over 50 m, and G. herzensteini and G. detrisus showed similar depth distributions with G. galeatus, and G. tricuspis showed similar depth distributions with G. pistilliger (Table 1). In this study, therefore, we defined these two groups as a “deep-water group”, which includes the former three species and “shallow-water group”, which includes the latter three species.
The observed ranges in depth preferences may reflect the fluctuations in sea level that occurred during the Pliocene, and that this may have contributed to the speciation observed in the genus. This study was undertaken to clarify the phylogenetic relationships among Gymnocanthus species based on partial mitochondrial DNA (mtDNA) sequences of the 12–16S rRNA, cytochrome b (cytb), and cytochrome oxidase subunit I (COI) genes. We also provide a synthesis of the evolutionary and zoogeographic characteristics of the global cottid fauna.
Materials and methods
Sample collection
A total of 108 individuals representing seven species of marine sculpins (six Gymnocanthus and one Myoxocephalus) were collected in Japan (Usujiri, Kushiro, and Shiretoko), Russia (Vladivostok), the United States (Dutch Harbor, Alaska), the Bering Sea, and the Chukchi Sea during 2006–2010 (Fig. 1). Sampling sites and other details for each species are shown in Table 2. Scorpaenichthys marmoratus, Hemitripterus villosus, and M. stelleri were used as out-groups.
DNA extraction and amplification
Total DNA was extracted from fin clips or muscle tissue preserved in 95 % ethanol and prepared for analysis using the QuickGene Plasmid Kit S II (QuickGene 800, Fujifilm, Japan) following the manufacturer’s instructions. Three mtDNA loci (partial cytb, partial 12–16S rRNA and COI) were amplified by the polymerase chain reaction (PCR). The primers used were Cytb-L 5′-ATGGCAAGCCTACGAAAAA-3′ and Cytb-R 5′-TCCTAAGGCCAAGTTTTCTA-3′ (Kimura et al. 2007) for cytb, 12SA-L 5′-CGGGAACTACGAGAAAAG-3′ and 16SA-H 5′-TCTTTTAGTCTTTCCCTGGGG-3′ (Kimura et al. 2007) for 12-16S rRNA, and FishF1 5′-TCAACCAACCACAAAGACATTGGCAC-3′ and FishR1 5′-TAGACTTCTGGGTGGCCAAAGAATCA-3′ (Ward et al. 2005) for COI. For some specimens of G. herzensteini and G. intermedius, 12SA-L1067 5′-AAACTGGGATTAGATACCCCACTAT-3′ and 16SA-H2492 5′-ATGTTTTTGATAAACAGGCG-3′ were used (Yanagimoto 2003). PCR was performed using 25 μl of EmeraldAmp® PCR Master Mix (Takara, Japan), 0.5 μl (0.1 μg) of each primer, 1 μl (50–100 ng) of template DNA, and 23 μl of sterile distilled water. The thermal cycler conditions consisted of an initial denaturation step at 94 °C for 30 s, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing (cyt b: 58 °C, 12–16S rRNA: 55 °C, and COI 58 °C) for 30 s, and extension at 72 °C for 30 s, with a final extension step at 72 °C for 7 min. After purification by SUPREC filter cartridges (Takara), PCR products were sequenced with an auto-sequencer (3130 Genetic Analyzer, Applied Biosystems, CA) at Macrogen Japan Corporation using the same PCR primers.
Phylogenetic analysis
Sequences were aligned using MEGA5 (Tamura et al. 2011) with default settings and adjusted by eye. Insertions/deletions were included in all phylogenetic analyses. Analyses were performed independently on each gene and on a concatenated matrix in which different set of partitions and no partitions scenarios were explored. Kakusan4 (Tanabe 2011) was used to determine the appropriate model of sequence evolution for each gene. Bayesian Inference was accessed in MrBayes 3.2.1 (Ronquist et al. 2011) setting priors to fit the evolutionary model suggested by Kakusan4 but allowing the parameters to be recalculated during the run. Markov chain Monte Carlo (MCMC) chains were used to sample the probability space in two simultaneous but completely independent runs starting from different random trees (default option); the number of generations fluctuated depending on the convergence of chains, a sample frequency every 100 generations was performed. The two runs were combined, and 25 % of the initial trees and parameters sampled were discarded as the burn-in phase. To evaluate whether the run was long enough to allow good chain mixing and accurately represent the posterior probability distribution of all the parameters, the Effective Sample Size (ESS) statistic was evaluated using Tracer v1.5 software (http://tree.bio.ed.ac.uk/software/tracer/). An ESS greater than 200 suggests that the MCMC chains were run long enough to get a valid estimate of the parameters.
The maximum likelihood (ML) method was also explored. Phylogenetic relationships between each partition were inferred by the ML method using RAxML ver. 7.2.8 (Stamatakis 2006). The robustness of nodes was estimated from 1,000 bootstrap replicates (Felsenstein 1985). Scorpaenichthys marmoratus (GenBank Accession No. AY833368, GU440517), Hemitripterus villosus (GenBank Accession No. AB126382, EU200467), and Myoxocephalus stelleri were used as out-groups in all analyses.
Divergence time estimation
Branch lengths of the phylogenies were reestimated in an ML framework under a generally unconstrained model (no clock) assuming a single substitution rate (clock), which was implemented in PAML4.5 (Yang 2007). In the divergence time estimations, as the 95 % CI of BI framework became wider than ML and showed low responsibility, we used only the ML framework. To test for the presence of a molecular clock, a likelihood ratio test (LRT) was used to compare the performance of both models in each data set, and the molecular clock hypothesis was rejected (no clock: −InL = −3,589.07, clock: −InL = −3,637.60, freedom value = 57). A relaxed molecular clock Bayesian method implemented in MCMC TREE program in PAML was used for dating analysis. The ML estimates of branch lengths were obtained using BASEML (in PAML) programs under the GTR +G substitution models for combined dataset, with the gamma priors set at 0.5. The prior, the overall substitution rate (rgene gamma), and rate-drift parameter (sigma2 gamma) were set at G(1, 330) and G(1, 10) using the strict molecular clock assumption with 5.5–4.8 Ma constraint to the divergence between G. tricuspis and G. intermedius-G. pistilliger. This time constraint is provided with a unit of 10 Million years (Myr) (i.e., 1 = 10 Myr) because some of the model components in the Bayesian analysis are scale variant, and the node ages should fall between 0.01 and 10 (Yang 2007).
Markov chain Monte Carlo (MCMC) approximation with a burn-in period of 1,000,000 cycles was obtained, and every 100 cycles was taken to create a total of 10,000 samples. To diagnose possible failure of the Markov chains to converge to their stationary distribution, at least two replicate MCMC runs were performed with two different random seeds for each analysis. Also, distributions of parameter values from MCMC samples were visualized using Tracer v1.5 to check mixing, choose a suitable burn-in, and look for trends that might suggest problems with convergence. The number of samples (10,000) was large enough to reach effective sample size (>200) for all parameters estimated in this study.
Results
Sequences alignment
Approximately 2,548 bp from 108 individuals belonging to seven cottid species were sequenced. For the six Gymnocanthus species, the lengths of the partial cytb, partial 12–16S rRNA, and partial COI sequences were 1,022 bp including 95 variable sites and 85 parsimony informative sites, 845–846 bp including 52 variable sites and 43 parsimony informative sites, and 679 bp including 71 variable sites and 64 parsimony informative sites, respectively. Only 66 of 108 samples were used to analyze these phylogenetic relationships, because in several samples, we failed to amplify the cytb region. An insertion of A was observed at site 492 in 12–16S rRNA of G. intermedius and G. pistilliger. Although the data were analyzed with and without indels, indels were only included in the final analysis because there was no difference in the obtained results. Cytb and COI sequences were used to estimate a phylogenetic tree because of the lack of sequence information of some outgroup species in 12–16S rRNA, but all regions were used to estimate the divergence time. All sequence data were deposited in GenBank under the following accession numbers (JQ406194-JQ406483).
Phylogenetic analysis
The six Gymnocanthus species formed a monophyletic group with the high bootstrap value (100 %) in all examinations. G. tricuspis was included in G. intermedius and G. pistilliger (lineage A) in ML topology, but included in G. herzensteini, G. galeatus, and G. detrisus (lineage B) in BI topology. Lineage A comprised G. intermedius, G. pistilliger, and G. tricuspis and was supported by bootstrap values of 39 % (ML) and 100 % (BI). Lineage B comprised G. herzensteini, G. detrisus, and G. galeatus and was supported by bootstrap values of 92 and 100 % (Fig. 2). All of the nodes that separated species had high bootstrap values (80–100 %, except G. tricuspis) in each analysis. However, intraspecific nodes within G. intermedius were relatively weakly supported (bootstrap values <60 %), indicating that G. intermedius has several intraspecific variable sites (Table 2).

The molecular phylogenetic tree of the Gymnocanthus maximum likelihood and Bayesian inference tree representing mitochondrial DNA phylogeny among Gymnocanthus species based on cytb and COI dataset (1,700 bp). Numbers above branches indicate bootstrap values (%). Nodes with low levels of statistical support (<50 %) are not shown. The scale bars are expressed as units of expected nucleotide substitutions per site
Haplotype sharing
Some haplotypes were shared between species (Supplementary materials 2). The cyt b haplotypes of two G. herzensteini specimens (h1 and h5) were shared with all G. intermedius specimens. Similarly, one G. tricuspis individual (t6) shared a COI haplotype with G. intermedius (Supplementary material 2). The relationships between these individuals coincided with the morphological characters and the results generated by the remaining two markers.
Divergence time
If G. tricuspis first migrated to the Arctic Ocean when the Bering Strait opened 5.5–3.0 Ma (Marincovich and Gladenkov 1999), then the estimated divergence time of G. intermedius and G. pistilliger was 3.9 Ma (95 % CI 5.3–2.8 Ma), and the deep-water group were estimated to speciate approximately 5.0 Ma [(G. herzensteini: 5.3 Ma (95 % CI 10.6–2.9 Ma), G. detrisus and G. galeatus: 4.3 Ma (95 % CI 8.4–2.7 Ma)].
Discussion
Phylogenetic relationship among Gymnocanthus
The molecular data obtained for the six Gymnocanthus species supported the monophyly of the genus. This supports the results of Wilson (1973), who compared the six species using meristic characters (e.g., overall body shape and development of sexual dimorphism, skeletal characteristics and head depth, interorbital width, and preopercular spine length). The results of that study suggest that certain morphological characters can be used to infer phylogenetic relationships within this genus. Thus, using two different analytical methods, Wilson (1973) and the present study grouped the six species into two major lineages: lineage A, which contained smaller species (G. intermedius, maximum standard lengths (MSL) = 25 cm; G. pistilliger, MSL = 24 cm; and G. tricuspis, MSL = 15 cm) and lineage B, which comprised the larger species (G. herzensteini MSL = 33 cm; G. detrisus, MSL = 30 cm; and G. galeatus, MSL = 30 cm) (Fig. 2). In addition, the clustering of the species into two groups might reflect differences in the habitat preference of the constituent species of the lineages (see “Evolutionary and zoogeographic synthesis of Gymnocanthus” section below).
Estimation of divergence time
Since Beringia acted as a barrier to migration (Briggs 2003), the molecular clock of Gymnocanthus could be calibrated based on the time when marine organisms from the Pacific Ocean invaded the Arctic Ocean after the Bering Strait opened in the Pliocene approximately 5.5–4.8 Ma (Marincovich and Gladenkov 1999). The Pacific and Atlantic populations of G. pistilliger have not been speciated and most developed species, so we did not add 5.5–4.8 Ma as a constraint at the divergence of G. pistilliger. The divergence times estimated from cytb, 12–16S rRNA and COI are plotted on a chronogram in Fig. 3.

Divergence time of six Gymnocanthus species Scale bars above and below the tree indicate the general time reversible model (GTR) of nucleotide substitution with the gamma distribution shape parameter (G) distance between sequences. Lineages refer to those presented in Fig. 2
Evolutionary and zoogeographic synthesis of Gymnocanthus
Numerous cottid species inhabit the North Pacific Ocean, where considerable diversification of the family has occurred (Yabe 1985; Briggs 2000, 2003; Mecklenburg et al. 2011). Bolin (1947) proposed that Gymnocanthus is a primitive genus, because it has numerous ancestral characters, including being slightly compressed, heavy-bodied, large-headed, and having four preopercular spines and that this genus then diverged at an early stage in the morphological phylogeny of the Cottidae (Bolin 1947; Yabe 1985).
This genus is thought to have originated somewhere in the North Pacific Ocean. In a comparative study of the morphology and zoogeography of the six Gymnocanthus species, Wilson (1973) speculated that Gymnocanthus likely originated near the eastern Aleutian archipelago where most Gymnocanthus species occur. Wilson (1973) also proposed that westward coastal currents (e.g., the Alaska Coastal Current, Alaskan Stream, Kamchatka Current, and Oyashio Current) promoted migration and subsequent diversification of members of this genus. Indeed, the results of the molecular phylogenetic approach presented here appear to corroborate those of Wilson (1973). If one considers the divergence time of each species, then the process of diversification of Gymnocanthus species can be summarized as follows.
The ancestral Gymnocanthus species that arose in the Aleutian Archipelago formed two groups approximately 8.1 million years ago (95 % CI 12.1–3.8 Ma): Lineage A became adapted to shallow habitats (0–100 m, but mainly 0–50 m), were smaller, exhibited marked sexual dimorphism, and developed a pair of heavy cleithral spines. Lineage B, became adapted to deeper habitats (50–150 m or more), developed larger bodies, exhibited no marked sexual dimorphism, and developed a pair of relatively smaller cleithral spines.
Deep-water group (lineage B)
The ancestral species of lineage B arose from the ancestor of this genus 8.1 Ma and diverged into three species about 5.0 Ma [G. herzensteini: 5.3 Ma (95 % CI 10.6–2.9 Ma), G. detrisus—G. galeatus: 4.3 Ma (95 % CI 8.4–2.7 Ma)]. The ranges of 95 % CI were wide in these branches, so we could not clarify the exact divergence time because of no constraint. We assumed that G. herzensteini is endemic to Japan, and then added 3.5 Ma when the Sea of Japan began to close (Tada 1994) as a constraint G. herzensteini’s divergence. However, the range of 95 % CI became wider than the estimation without constraint. This might mean that G. herzensteini is a “paleoendemic species”, whereas G. intermedius is a “neoendemic species” around the Sea of Japan. The genetic diversity of G. herzensteini, which has a smaller distribution area, was lower (Table 2). Such a correlation between distribution range and genetic variability has been reported previously in the Zoarcidae (Bothrocara hollandi, Kojima et al. 2007) and Cerithioidea (Cerithidea and Cerithium, Miura et al. 2011).
Shallow-water group (lineage A)
The climate in the early Pliocene (approximately 5.0–3.0 Ma) was warm with global surface temperatures and sea levels that were approximately 0–3 °C and 10–20 m higher than current levels (Raymo et al. 1996; Ravelo and Andreasen 2000). The higher sea levels during this warm period approximately 5.5–4.8 Ma opened the Bering Strait, allowing the ancestral species of lineage A to migrate northward across the Bering Strait and into the Arctic Ocean (Marincovich and Gladenkov 1999). Approximately, 2.8 Ma extensive glaciation occurred in the northern hemisphere (Ravelo et al. 2004), closing the Bering Strait, and promoting the divergence of the lineage A species that migrated to the Arctic Ocean approximately 4.0 Ma (95 % CI 5.5–3.0 Ma) into G. tricuspis (Fig. 3). After the Bering Strait closed, some of these G. tricuspis migrated into the North Atlantic in a clockwise direction with the Beaufort Gyre and Transpolar Drift. This extensive migration may have produced the remarkably high genetic variation in G. tricuspis (Table 2).
Speciation is not likely to have occurred in lineage A in the same way as it did in lineage B. This is because lineage A was mainly distributed at depths of 0–50 m, which meant that it could likely move across shallow straits more easily than species in lineage B; this increased propensity for movement is likely to have limited genetic divergence. When the straits closed during the ice age, a population of the lineage A ancestral species became isolated in the Sea of Japan and developed into G. intermedius, and the remaining populations in lineage A formed G. pistilliger. Most of the speciation events in the genus Gymnocanthus are considered to have arisen through geographic isolation resulting from the ice ages. Thus, the sympatric distribution of shallow and deep-water groups, such as that of G. intermedius and G. herzensteini in northern Japan, is not the result of sympatric speciation, but rather of secondary contact.
G. pistilliger, which ranges from the North Pacific Ocean to the Arctic Ocean and the North Atlantic Ocean, also migrated northward into the Arctic Ocean through the Bering Strait. G. pistilliger diverged in the shallow waters of the Pliocene–Pleistocene approximately 3.3 Ma (95 % CI 5.0–1.8 Ma) when the sea level was 40 m below the current level (e.g., Haq et al. 1987, see Fig. 4). Currently, the deepest areas of the Bering Strait are about 40 m, so G. pistilliger was presumably able to migrate to the Arctic Ocean by moving along the coast when the sea level was above −40 m. During subsequent cool periods, the fish migrated southward to the North Atlantic and the North Pacific. Since relatively little time has passed since these populations diverged, the Atlantic population may not have differentiated markedly from the Pacific population. Future studies should include Atlantic population of G. pistilliger and estimate these divergence times more precisely.

Divergence time as a function of changes in sea level Zero on the vertical axis indicates extant sea level and the horizontal axis shows time (Mya). Allows mean the 95 % confidence interval and the white circles on each allow means the estimated divergence time
References
Allen MJ, Smith GB (1988) Atlas and zoogeography of common fishes in the Bering Sea and northeastern Pacific. NOAA Tech Rep NMFS 66:151
Amaoka K, Nakaya K, Yabe M (2011) The pictorial book of all of the fishes of Hokkaido. The Hokkaido Shimbun Press, Sapporo In Japanese
Bolin RL (1947) The evolution of the marine Cottidae with a discussion of the genus as a systematic category. Stanf Ichtyol Bull 3(3):153–168
Briggs JC (2000) Centrifugal speciation and centres of origin. J Biogeogr 27:1183–1188
Briggs JC (2003) Marine centres of origin as evolutionary engines. J Biogeogr 30:1–18
Eschmeyer WN, Herald ES, Hammann H (1983) A field guide to Pacific coast fishes of North America. Houghton Mifflin, Boston
Fedorov VV (1986) Fishes of the North-eastern Atlantic and the Mediterranean, V.III. UNESCO pp 1243–1260
Fedorov VV, Chereshnev IA, Nazarkin MV, Shestakov AV, Volobuev VV (2003) Catalog of marine and freshwater fishes of the northern part of the Sea of Okhotsk. Dalnauka, Vladivostok, p 204
Felsenstein J (1985) In: Whitehead PJP, Bauchot ML, Hureau JC, Nielsen J, Tortonese E (eds), Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Haq BU, Hardenbol J, Vail PR (1987) Chronology of fluctuating sea levels since the triassic. Science 235:1156–1167
Kimura MR, Yanagimoto T, Munehara H (2007) Maternal identification of hybrid eggs in Hexagrammos spp. By means of multiplex amplified product length polymorphism of mitochondrial DNA. Aquat Biol 1:187–194
Kojima S, Adachi K, Kodama Y (2007) Formation of deep-sea fauna and changes of marine environment in the Japan Sea. Foss Palaeontol Soc Jpn 82:67–71
Kontula T, Vainola R (2003) Relationships of Palearctic and Nearctic ‘glacial relict’ Myoxocephalus sculpins from mitochondrial DNA data. Mol Ecol 12:3179–3184
Marincovich L Jr, Gladenkov AY (1999) Evidence for an early opening of the Bering Strait. Nature 397:149–151
Masuda H, Amaoka K, Araga C, Uyeno T, Yoshino T (eds.) (1984) The fishes of the Japanese Archipelago. Tokai University Press Tokyo
Mecklenburg CW, Mecklenburg TA, Thorsteinson LK (2002) Fishes of Alaska. American Fisheries Society Bethesda, Maryland
Mecklenburg CW, Moller PR, Steinke D (2011) Biodiversity of Arctic marine fishes: taxonomy and zoogeography. Mar Biodiv 41:109–140
Miura O, Torchin ME, Eldredge B, Jacobs DK, Hechinger RF (2011) Flying shells: historical dispersal of marine snails across Central America. Proceedings of the Royal Society B: Biological Science
Nelson JS (2006) Fishes of the World, 4th edn. Wiley, New York
Panchenko VV (2009) Summer distribution of the Purplegray Sculpin Gymnocanthus detrisus (Cottidae) in Waters of Primorsky Krai, Sea of Japan. Rus J Mar Biol 35:15–19
Parin NV, Fedorov VV, Sheiko BA (2002) An annotated catalogue of fish-like vertebrates and fishes of the seas of Russia and adjacent countries: part 2. Order Scorpaeniformes. J Ichthyol 42(Suppl.1):60–135
Ravelo AC, Adreasen DH (2000) Enhanced curculatio during a warm period. Geophys Res Lett 27:1001–1004
Ravelo AC, Dyke HA, Mitchell L, Annette OL, Michael WW (2004) Regional climate shifts caused by gradual global cooling in the Pliocene epoch. Nature 429:263–267
Raymo ME, Grant B, Horowitz M, Rau GH (1996) Mid-Pliocene warmth: stronger greenhouse and stronger conveyer. Mar Micropaleo 27:313–326
Ronquist F, Teskenko M, van der Mark P, Ayres D, Darling A,Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2011) MRBAYES 3.2: efficient Bayesian phylogenetic inference and model selection across a large model space. Systematic biology 61 (in press)
Russian Academy of Sciences (2000) Catalog of vertebrates of Kamchatka and adjacent waters
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690
Tada R (1994) Paleoceanographic evolution of the Japan Sea. Palaeogeogr Paleoclimatol Palaeoecol 108:487–508
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol (Submitted)
Tanabe AS (2011) Kakusan4 and Aminosan: two programs for comparing nonpartitioned, proportional and separate models for combined molecular phylogenetic analyses of multilocus sequence data. Mol Ecol Resour 11:914–921
Vermeij GJ (1991) Anatomy of an invasion: the trans-Arctic interchange. Paleobiology 17:281–307
Ward RD, Zemlak TS, Innes BH, Last PR, Hebert PDN (2005) DNA barcoding Australia’s fish species. Philos Trans Roy Soc B 360:1847–1857
Wilson DE (1973) Revision of the cottid genus Gymnocanthus, with a description of their osteology. Department of zoology, the University of British Columbia
Yabe M (1985) Comparative osteology and myology of the superfamily Cottoidea (Pisces: Scorpaeniformes), and its phylogenetic classification. Hokkaido University, Japan
Yanagimoto T (2003) The identifications for Japanese greenlings: seven Hexagrammidae species, using PCR-RFLP analysis from mitochondrial DNA. Japanese Soc Fish Sci 69(5):726–732 In Japanese
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Acknowledgments
We thank T.W. Pietch, D. Pitruk, M. Yabe, O. Tsuruoka, R. Yokoyama and J. Inoue for their constructive suggestions, and the Alaska Department of Fish and Game, Russian Academy of Sciences, Oshoro maru (a training ship of Hokkaido University), O. Yamamura, Y. Koya, N. Sato, T. Abe and S. Awata for collecting fish samples. We also thank J. Bower for checking English grammar. This study was funded by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan. We also thank three anonymous reviewers who gave us useful advices.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by T. Reusch.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Yamazaki, A., Markevich, A. & Munehara, H. Molecular phylogeny and zoogeography of marine sculpins in the genus Gymnocanthus (Teleostei; Cottidae) based on mitochondrial DNA sequences. Mar Biol 160, 2581–2589 (2013). https://doi.org/10.1007/s00227-013-2250-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00227-013-2250-4