
Abstract
The finless porpoise (Neophocaena phocaenoides) inhabits a wide range of tropical and temperate waters of the Indo-Pacific region. Genetic structure of finless porpoises in Chinese waters in three regions (Yangtze River, Yellow Sea, and South China Sea) was analyzed, including the Yangtze finless porpoise which is widely known because of its highly endangered status and unusual adaptation to freshwater. To assist in conservation and management of this species, ten microsatellite loci were used to genotype 125 individuals from the three regions. Contrary to the low genetic diversity revealed in previous mtDNA control region sequence analyses, relatively high levels of genetic variation in microsatellite profiles (H E = 0.732–0.795) were found. Bayesian clustering analysis suggested that finless porpoises in Chinese waters could be described as three distinct genetic groups, which corresponded well to population “units” (populations, subspecies, or species) delimited in earlier studies, based on morphological variation, distribution, and genetic analyses. Genetic differentiation between regions was significant, with F ST values ranging from 0.07 to 0.137. Immigration rates estimated using a Bayesian method and population ancestry analyses suggested no or very limited gene flow among regional types, even in the area of overlap between types. These results strongly support the classification of porpoises in these regions into distinct evolutionarily significant units, including at least two separate species, and therefore they should be treated as different management units in the design and implementation of conservation programmes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
A long-standing goal in evolutionary and conservation biology is to understand the extent to which current patterns of diversity in separated populations have been influenced by natural selection, genetic drift, gene flow coupled with their interactions (Palstra et al. 2007). Small, isolated populations are assumed to be vulnerable to local extinctions as a result of stochastic factors and more prone than large populations to lose genetic variation through random drift, resulting in a decrease in adaptive fitness (Frankham et al. 2003; Gilpin and Soule 1986; Lynch et al. 1995; Westemeier et al. 1998), which may further exacerbate population decline. As predicted by population genetic theory, due to genetic drift and/or inbreeding, small populations should show lower levels of genetic variability than large populations (Hartl and Clark 1997). However, gene flow can counteract the potentially adverse effects of genetic isolation and the loss of genetic diversity in small populations while simultaneously limiting genetic differentiation between populations (Palstra et al. 2007; Van Rossum et al. 1997). Conversely, speciation may result from more limited gene flow and subsequent differentiation (e.g., Terry et al. 2000). In either case, knowledge of population structure and natural patterns of gene flow can contribute to a better understanding of species’s ecology and evolution, and to the implementation of informed conservation and management measures (Parsons et al. 2006).
The finless porpoise (Neophocaena phocaenoides) inhabits a wide range of tropical and temperate waters in the Indo-Pacific region (Reeves et al. 1997; Kasuya 1999). Obvious variation at morphology and different geographic distribution pattern have been noted for a long time, and individuals from different geographic regions have been identified to be different species (e.g., Pilleri and Chen 1980, Wang et al. 2008) or subspecies within a single species (e.g., Rice 1998). Geographic variation in external morphology and differences in growth and reproduction pattern of finless porpoises in Chinese waters were noted, and three populations (or ecological/morphological forms) were suggested by Gao (1991) and Gao and Zhou (1993): the Yellow Sea population, the South China Sea population, and the freshwater Yangtze River population. Gao and Zhou (1995a, b, c) further conducted a series of careful and comprehensive surveys on the external morphometrics, skull, and postcranial skeleton of these populations. They found that the width of denticulate region, the height of dorsal ridge, the number of rows of dorsal denticles, and stepwise discriminant analysis of skeletal morphology could reliably identify the aforementioned three populations in Chinese waters. The freshwater-adapted Yangtze River population, endemic to the middle and lower reaches of the Yangtze River, is characterized by a narrow tuberculed area on the dorsal ridge and was referred to N. p. asiaeorientalis (Gao and Zhou 1995a). Due to its small population size and rapid decline, the Yangtze finless porpoise has attracted wide attention in recent years, especially when the extinction of another freshwater cetacean sympatric with the finless porpoise in the Yangtze River (the Baiji Lipotes vexillifer) has likely already occurred (Turvey et al. 2007). Surveys conducted in some segments of the Yangtze River found that finless porpoises have decreased annually by approximately 7.3% from 1989 to 1999 (Wei et al. 2002) and only around 1,000 porpoises were estimated to remain in the river system (Wang et al. 2006). The Yellow Sea population, although also characterized by a similar dorsal surface as the Yangtze River porpoise, is distributed in the Yellow/Bohai Sea and the northern East China Sea and was referred to N. p. sunameri, whereas the South China Sea population, which is characterized by a wide area of tubercules on the dorsal surface and distributed in the South China Sea and the southern East China Sea, was referred to N. p. phocaenoides (Gao and Zhou 1995a). Although the Yellow Sea population (or N. p. sunameri) and the South China Sea population (or N. p. phocaenoides) are potentially sympatric in the southern East China Sea and the northern South China Sea (especially in the Taiwan Straits), genetic and morphological evidence suggests little genetic exchange in the overlap area, and likely (although relatively recent) species-level differentiation (Wang et al. 2008). Although the Yangtze River population (or N. p. asiaeorientalis) inhabits a habitat distinct from adjacent marine waters, it is possible that the Yellow Sea population and the Yangtze River population intermix in the river mouth area. However, genetic differentiation between freshwater and oceanic finless porpoises was not addressed in that study as no specimens from the Yangtze River were included (Wang et al. 2008).
To effectively implement conservation measures for the endangered finless porpoise, it is important to identify population structure, rates of gene flow among regions (or ecological/morphological forms), and the degree of demographic isolation from other populations (Caughley 1994) through a systematic population genetic structure analysis. For most cetacean species, direct observation of interchange and interbreeding is almost impossible, due to the nature and scale of the aquatic environment (Parsons et al. 2006). Investigations are further limited by the considerable difficulties involved in long generation times and the dearth of understanding of the barriers to gene flow encountered by these marine animals in a seemingly continuous aquatic habitat (Rosel et al. 1995; Avise 1998). Researchers are increasingly turning to indirect assessment of population structure, such as by analysis of the partitioning of molecular genetic variation. Consequently, modern molecular genetic methods have played an important role in assessing the levels of genetic structuring among many coastal and oceanic cetacean populations (Bérubé et al. 1998; Escorza-Trevino and Dizon 2000; Gaspari et al. 2007; Krützen et al. 2004; Parsons et al. 2006; Fontaine et al. 2007).
Previous genetic studies, mainly based on mitochondrial control region data, have investigated variation mainly across the extant range of the finless porpoises in Chinese and Japanese waters, which was characterized by a high level of among-region genetic structure with low levels of within-region genetic diversity (Yang et al. 2002, 2003, 2008; Yoshida et al. 2001). However, high levels of mtDNA differentiation cannot provide a full accounting of reproductive isolation/genetic interchange because male-mediated gene flow is not measured using maternally inherited mtDNA (Racey et al. 2007). In addition to mitochondrial DNA, evaluation of nuclear microsatellite DNA diversity is applied more and more widely in conservation programs because of the high variability and precision of these markers, rendering them valuable for the study of fine-scale population structure and contemporary population processes (Crandall et al. 2000). In order to have a better knowledge of ecology and population biology relevant to conservation issues, we investigated microsatellite polymorphism in the finless porpoise populations in Chinese waters. The objectives of this study were to: (i) re-examine the pattern of genetic variation of finless porpoises based on biparentally inherited nuclear markers; (ii) highlight possible gene flow between regions which would provide genetic information for the ESUs (evolutionarily significant units) status of finless porpoises in those areas; (iii) provide a broader understanding of the taxonomic status of Neophocaena population units; and (iv) recommend conservation and management priorities for finless porpoises in Chinese waters.
Materials and methods
Sampling and DNA extraction
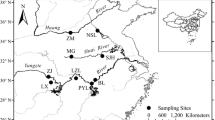
A total of 125 finless porpoises (33 individuals from the Yangtze River, 23 individuals from the Yellow Sea, and 69 individuals from the South China Sea) were collected from 12 locations in coastal China, as well as the middle and lower reaches of the Yangtze River (Fig. 1). All the samples were collected from incidentally captured individuals coastal and Yangtze River fisheries. These samples were a priori assigned to the aforementioned three populations primarily according to their sampling localities as shown in Fig. 1. Voucher specimens are preserved in the College of Life Sciences, Nanjing Normal University, China. Genomic DNA was extracted from blood or muscle using the DNeasy tissue kit (Qiagen) following the manufacturer’s instruction.

Schematic map showing the sampling localities of this study, sampling size for each sampling locality was shown in the parenthesis. Sampling localities abbreviations are as follows: the Yellow Sea population units (filled triangle) DL (Dalian), LS (Lusi), ZS (Zhoushan); HZ (Hangzhou), and YX (Yixing); the Yangtze River population (filled circle): ZJ (Zhenjiang), NJ (Nanjing), TL (Tongling), and HK (Hukou); and the South China Sea population (inverted filled triangle): PT (Pingtan), DS (Dongshan), and BH (Beihai)
Microsatellite genotyping
Seven dinucleotide and three tetranucleotide loci were used for genotyping: EV37Mn and EV104Mn from Megaptera novaeangliae (Valsecchi and Amos 1996); PPHO110, PPHO130 from Phocoena phocoena (Rosel et al. 1999); and Np368, Np391, Np398, Np403, Np409, Np428 from Neophocaena phocaenoides (Chen et al. 2007; Chen and Yang 2008). Each forward primer had a M13 tail at its 5′end (5′-CACGACGTTGTAAAACGAC-3′) to allow for labeling with fluorescent primers. PCR amplifications were conducted in 15-μL volumes containing 1 μL of dilute template, 7.5 μL Ex Taq premix buffer (Takara), 5 pm of each primer, 0.5 pmol of dye-labeled M13 primer [either IRD700 or IRD800 (LI-COR)] and ultrapure water to volume. Polymerase chain reaction (PCR) conditions were as reported in Valsecchi and Amos (1996), Rosel et al. (1999), Chen et al. (2007), and Chen and Yang (2008). Amplified products were genotyped on 6.5% polyacrylamide denaturing gels using fluorescent imaging on a LI-COR 4300 automated DNA sequencer and analyzed using LI-COR SAGAGT software.
Statistical analyses
Genetic variation
Each locus per population unit was tested for deviation from Hardy–Weinberg equilibrium (HWE) and for genetic linkage disequilibrium using GENEPOP version 3.4 (Raymond and Rousset 1995). Exact p-values for both tests were obtained using a Markov chain method with 10,000 dememorizations, 500 batches, and 10,000 iterations per batch. Significance levels were adjusted using the sequential Bonferroni correction for multiple comparisons (Holm 1979). The number of private alleles and the observed (H O) and expected (H E) heterozygosities were all estimated at each locus and for each population unit with FSTAT version 2.9.3.2 (Goudet 2001).
Population structure and differentiation
STRUCTURE (Pritchard et al. 2000), which implements a Bayesian model-based clustering method, was used to determine the optimal number of genetically distinct clusters based on allele frequencies across loci without imposing any preconceived ideas of population information for each individual (e.g., based on morphology or geographical sampling). The method statistically assigns individuals, based on Hardy–Weinberg expectations, to a user-defined number of anonymous genetic clusters (K), thus elucidating genetic structure that may not be apparent otherwise. The most likely value of K is now most commonly assessed by comparing the rate of change in the likelihood of the data for different values of K. Calculations were conducted under the admixture model and the assumption of correlated allele frequencies with a burn-in period of 100,000, followed by 1,000,000 iterations. The number of populations (clusters), K, was set from 1 to 6. Ten independent runs of the Markov chain were performed to check for convergence of the chain and homogeneity among runs for each K.
Genetic differentiation among population units was further quantified by calculating fixation indices based on an infinite alleles model and a stepwise mutation model. F ST values (more precisely θ, Weir and Cockerham 1984) were estimated by using ARLEQUIN version 3.11 (Excoffier et al. 2005) and were tested for significance using 10,000 permutations. For the latter, the program RSTCALC (Goodman 1997) calculates rho ST (ρ) an analogue of Slatkin’s R ST (Slatkin 1995) in which differences in sample size and variance between loci are accounted for. The algorithm weights R ST by allele size, standardizing across the whole data set, to make different loci more directly comparable. A permutation test (10,000 permutations) was used to determine if rho ST estimates were significantly different from zero. SMOGD v 1.2.5 (Crawford 2009) was used to calculate a differentiation statistics, D EST, based on 1,000 permutations.
Migration and gene flow
A non-equilibrium Bayesian method was applied to estimate current immigration rates among population using BAYESASS 1.3 (Wilson and Rannala 2003). BAYESASS 1.3 relies on multilocus genotypes and a Markov chain Monte Carlo (MCMC) algorithm to estimate proportions of non-migrants and the source of migrants for each sampling site (Wilson and Rannala 2003) and does not require the populations to be in migration drift or Hardy–Weinberg equilibrium. The MCMC method used in this program was run for 3 × 106 iterations with sampling every 2,000 iterations, of which the first 106 iterations were discarded as burn-in. As suggested by Wilson and Rannala (2003), more accurate results are obtained from runs when the number of proposed changes for the variables m (migration rate), P (allele frequencies), and F (inbreeding coefficient) are between 40 and 60% of the total chain length. Based on preliminary runs using different delta values ranging from 0.05 to 0.35, our final runs used delta values of 0.10 for the parameters estimated for allele frequency, migration, and inbreeding, respectively, to ensure optimal mixing and maximize log likelihood values.
In order to assess gene flow among three population units, we used the USEPOPINFO option (Pritchard et al. 2000) incorporated in STRUCTURE. This model assumes that most individuals classified having pure ancestry but that a proportion of individuals may also have ancestry from other populations. We set GENBACK = 2, so that showing the probability that each individual is from that population, have a parent, grandparent from alternative populations. In order to ensure a strong statistical support for any inference of mixed ancestry, we further set MIGRPRIOR = 0.001.
Results
Levels of variability
As shown in Table 1, the ten microsatellite loci examined had a total of 136 alleles in the three population units screened. Hardy–Weinberg tests revealed that only three locus-population unit comparisons significantly deviated from Hardy–Weinberg equilibrium after Bonferroni correction. No evidence of linkage disequilibrium between pairs of loci was found after adjustment for multiple comparisons. Heterozygosity ranged from 0.751 in the South China Sea population unit to 0.770 in the Yellow Sea population unit (Table 1). Fifty-nine alleles were found in all three population units, whereas thirty-one alleles were shared by two population units, and 46 alleles were specific to one of the three population units (three in the Yangtze River, nine in the Yellow Sea, and 34 in the South China Sea).
Population structure and genetic differentiation
The highest log-likelihood value was obtained for three clusters (Ln Pr (X/K) = −4,458.5), which indicated that the individuals examined were most likely to be divided into three genetically distinct populations. At K = 3, all individuals from the Yangtze River (Yixing, Hukou, Nanjing, and Tongling) were grouped into a single cluster. The second cluster contained individuals from the Yellow/Bohai Sea (Lusi and Dalian) and the East China Sea (Hangzhou and Zhoushan), whereas the third comprised the South China Sea samples (Dongshan, Pingtan, and Beihai). Zhoushan and Pingtan showed some admixture between the Yellow Sea and South China Sea clusters (Table 2; Fig. 2). Some individuals from Zhoushan and Pingtan showed contrasting patterns of assignment to the original clusters, which was supported by individual assignments using Bayesian model-based STRUCTURE analysis as shown in Table 2. F ST and R ST values computed from composite allele frequencies at ten microsatellite loci confirmed significant genetic differentiation in all pairwise comparisons (Table 3) as revealed by previous mtDNA analyses (Yang et al. 2002, 2003, 2008). Analyses with both F ST (=0.131) and R ST (=0.341) showed high levels of genetic differentiation between the Yangtze River population unit and the South China Sea population unit. Of special note was the lower, but still highly significant, F ST (=0.070) and R ST (=0.067) values for the comparison of the Yangtze River population unit and the Yellow Sea population unit. D values also showed a similar pattern of differentiation to that of F ST and R ST. The D value between the Yangtze River population and the South China Sea population was above 0.3, whereas the values between either the Yangtze River population unit or the South China Sea population unit and the Yellow Sea population unit were lower than 0.3 (Table 3).

Estimated population structure. Each individual is represented by a vertical line, which is partitioned into K colored segments representing the individual’s estimated membership fractions in K clusters. Black lines separate individuals into different population units. Population units are labeled in numbers below the figure. Sample names corresponding to numbers are indicated to the right of the figure. Ten STRUCTURE runs at each K produced nearly identical individual membership coefficients; the figure shown for a given K is based on the highest probability run at that K. 1 Yixing, 2 Zhenjiang, 3 Hukou, 4 Nanjing, 5 Tongling, 6 Lusi, 7 Hangzhou, 8, Zhoushan, 9 Dalian, 10 Pingtan, 11 Dongshan, 12 Beihai
Migration and gene flow among populations
The mean posterior probabilities and 95% confidence intervals for migration rates between sampling sites are shown in Table 4. From Bayesian MCMC simulation analyses, we obtained a 95% confidence interval of approximately (0.675, 0.992) for non-migration rates and of (0.001, 0.261) for migration rates. Confidence intervals recovered from this data set were considerably smaller than those obtained from the scenario mentioned earlier, suggesting that our microsatellite data set contained an appreciable amount of information from which to estimate migration rates and other parameters of interest. Almost all porpoises from the Yangtze River (98%) were identified as non-migrants, whereas the Yellow Sea and the South China Sea population units showed a slightly lower non-migrant proportion (92%). The migration rate from the Yellow Sea to the South China Sea was slightly higher than the reverse, but the large overlap of 95% CI bounds did not allow us to conclude there was asymmetry in migration rates.
Estimates of the amount of finless porpoise ancestry revealed 13 individuals not fitting the original assignments (based on geography only). One individual sampled in the Yangtze River has a very high probability (99.6%) of being from the South China Sea, whereas one individual in Zhoushan has a high probability (99.4%) of being from the South China Sea, and eleven individuals in Pingtan have probabilities >0.9 of being from the Yellow Sea population unit except for samples 60 and 84, which had pure ancestry proportions of Yellow Sea population unit less than 0.9). Of the 125 individuals examined, only four showed signs of genetic admixture between different populations or of ancestry from other populations (sample 60, 71, 84, 121), but all these inferences were supported by relatively low probability values (ranging from 0.093 to 0.270), which suggested that only weak evidence indicated that these individuals are the product of hybridization between different population units.
Disscussion
Population structure and genetic diversity
The ability to identify and define biological “populations” is crucial for making informed decisions concerning conservation and management (Waples and Gaggiotti 2006). Three population units of finless porpoises in Chinese waters have been recognized based on obvious external and skeletal differentiation (Gao and Zhou 1995a) and stepwise discriminate analysis of skeletal morphology (Gao and Zhou 1995 b c). Previous mtDNA analyses (Yang et al. 2002, 2003, 2008) suggested significant genetic structure among these regions, and mtDNA/microsatellite/morphological analyses further suggested relatively recent species-level differences between oceanic finless porpoises in the South China Sea and East China Sea, with an area of sympatry in the Strait of Taiwan (Wang et al. 2008). However, the genetic differentiation between the Yangtze River population unit and the Yellow Sea population unit was previously thought to be relatively small (Yang et al. 2008).
The application of highly polymorphic microsatellite loci and assignment tests has greatly increased the potential for understanding population structure across large and small geographic scales. The Bayesian cluster method implemented in STRUCTURE has been proven a powerful instrument in instances where genetic differentiation is high (Maudet et al. 2002; Pritchard et al. 2000; Randi and Lucchini 2002; Rosenberg et al. 2001) and can also be used when population structure is more complex (Rosenberg et al. 2002). In congruence with morphological classification (Gao and Zhou 1995a) and mtDNA control region analysis (Yang et al. 2002, 2003, 2008), the present Bayesian clustering analysis indicated the presence of three distinct genetic groups in spite of admixture in some locations (e.g., Pingtan) providing strong support for the subdivision of finless porpoises in Chinese waters into three distinct units. Subdivision was further supported by pairwise F ST, R ST, and D values (Table 3) and the existence of around 30% of alleles private to each population. Highly significant differentiation between all populations was found, similar to that revealed by mtDNA (Yang et al. 2008) and mtDNA/microsatellite/morphological analyses (Wang et al. 2008).
Does gene flow occur among population units?
Results from previous studies have not been able to clarify whether or not dispersal and/or gene flow occurs between the Yangtze River and marine population units. Although Zhang et al. (1993) suggested that finless porpoises might migrate from the river to sea or vice versa due to seasonal breeding activities, this hypothesis is not supported by any direct field observations of Yangtze River finless porpoises in the sea or marine porpoises in freshwater. Furthermore, obvious external differences exist among the three population units and no specimens with intermediate morphological characteristics have been found so far.
In general, F ST values below 0.05 are expected with current gene flow, whereas values between 0.05 and 0.1 indicate that populations are semi-isolated, and values above 0.1 suggest that populations are isolated (Wilson et al. 2003). Using this criterion, we inferred significant divergence between the Yangtze River porpoises and South China Sea porpoises from an F ST value of 0.131 and a relatively high R ST value of 0.341, suggesting that these two population units might be genetically isolated with limited or no gene flow. In contrast, smaller values of F ST and R ST between the South China Sea porpoises and Yellow Sea porpoises, and between Yangtze River porpoises and Yellow Sea porpoises, suggests relatively lower levels of differentiation. The pattern of differentiation was further evidenced by D-values calculated between populations. A D-value above 0.3 was estimated for the Yangtze River porpoises and the South China Sea porpoises (D = 0.384), suggesting a moderate to high level of differentiation. D-value estimates for the Yangtze River porpoises and Yellow Sea porpoises and the South China Sea porpoises and Yellow Sea porpoises were both much lower than 0.3 (Table 3), suggesting a relatively low level of differentiation.
Significant levels of genetic differentiation between Yellow Sea porpoises and South China Sea porpoises (F ST = 0.075) also suggested limited or no gene flow between them. BAYESASS analysis showed low levels of migration between these regions (m < 0.1). If the demographic history of species being studied fits the assumptions of the inference model, the Bayesian method implemented in BAYESASS can give fairly accurate estimates of migration rates when the genetic differentiation is not too low (F ST ≥ 0.05) (Faubet et al. 2007). Few studies have addressed the question of how small migration rates (m) should be to insure that subpopulations have independent dynamics (Waples and Gaggiotti 2006), but Hastings (1993) suggested that two populations become demographically independent when m falls below about 0.10. In addition, STRUCTURE analysis (USEPOPINFO = 1, K = 3) also suggested no evidence of genetic exchange between these regions. For example, of the 69 samples from the South China Sea, 11 from Pintan were identified as most likely coming from the Yellow Sea rather than resulting from hybridization. Similarly, one individual sampled in Zhoushan (Yellow Sea) was identified to be a new migrant from the South China Sea with a nearly 100% posterior probability. In other words, although Pingtan and Zhoushan were within the overlapping area of Yellow Sea and South China Sea porpoises, there was little evidence for genetic admixture in the last two generations, strongly suggesting no or very limited gene flow between these two regions, even in the area of potential sympatry between them.
Our analyses also provided several pieces of evidence which suggest that the relatively low level of differentiation between the Yangtze River porpoises and the Yellow Sea porpoises might not be due to contemporary gene flow. First, all individuals from the Yangtze River grouped together with very high probability (Table 2; Fig. 2), which underlines the genetic distinctiveness of the freshwater population. Second, STRUCTURE analysis showed that none of recent migrants was likely from the Yangtze River, and no immigrants from the Yellow Sea were found in the Yangtze River. A plausible explanation for the observed low level of differentiation reflected in F ST and R ST values is limited time since divergence for new microsatellite mutations. Incomplete lineage sorting or retained ancestral polymorphism in offspring populations is a viable alternative to contemporary gene flow as an explanation for such low levels of differentiation (Gibbs et al. 2000). Barriers affected the limitation of gene flow between the freshwater and the marine porpoises might be adaptation to different salinity levels and related environmental factors. If this is true, it would be of great interest to investigate isolation and adaptation mechanisms operating to separate freshwater and marine porpoises that may be correlated with molecular markers involved in adaptive changes.
Taxonomic implications and conservation recommendations
Multiple lines of evidence suggest that finless porpoises in Chinese waters found in the three geographical regions represent distinct population units (Fig. 2; Tables 2, 3, 4). As discussed earlier, Bayesian model-based clustering method divided finless porpoises in Chinese waters into three distinct genetic groupings, corresponding to the three populations/subspecies/species previously identified based upon morphological, genetic and ecological evidence. Levels of genetic differentiation between porpoises in the three regions were highly significant (Table 3). Further, migration and ancestry analyses using BAYESASS and STRUCTURE found that although overlapping areas of distribution were found (especially for Yellow Sea and South China Sea porpoises), the source of most individuals could be reliably be identified with high posterior probabilities. In addition, very few individuals were identified as having evidence of admixture from other populations (with relatively low posterior probabilities), which strongly implied little or no gene flow between these population units even in the area of overlap.
One of the objectives of modern conservation biology is not only to preserve species and habitats, but also their evolutionary potential in terms of maintaining the genetic diversity of the extant species. According to the model proposed by Moritz (1994), ESUs are designated on the basis of reciprocal monophyly at mtDNA haplotypes, while MUs are identified by significant differences in allele frequency distributions and significant divergence in mitochondrial or nuclear loci. Although reciprocal monophyly for mtDNA haplotypes were not found for any populations (Yang et al. 2008) which is not well consistent with ESUs criterion defined by Moritz (1994), significant genetic differentiation and the lack of gene flow revealed by previous mtDNA and present microsatellite markers as well as obvious morphological differentiation strongly suggest that these population units might be significantly isolated from one another. Therefore, it is reasonable to give ESUs status for all three populations in Chinese waters, which support the acceptance of their subspecies status as suggested by Gao and Zhou (1995a, b, c) based upon morphological evidence, or species-level status (for oceanic units) as suggested by Wang et al. (2008). Further, in designing conservation programmes, each of these population units should be treated as different management units (MUs). The Yangtze finless porpoise, the sole freshwater-adapted porpoise population, represents unique genetic diversity among finless porpoise population units. Therefore, the protection of Yangtze finless porpoise has crucial implications for finless porpoise conservation worldwide. The observed heterozygosity in Yangtze finless porpoises was comparable to those of the other two population units and other cetaceans, suggesting that inbreeding and low genetic variation may not be an important threat at present, relative to other factors threatening the freshwater form. However, given the very recent and drastic decline in numbers of animals in the Yangtze River, the currently observed “high” levels of heterozygosity can be alternatively explained as a remnant of what has been retained due to the fairly long generation times of cetaceans. Thus, more attention should be paid to the potential loss of genetic diversity in this population in the future. Recent surveys indicate that this population decreased in size to around 1,000 individuals (Wang et al. 2006). Population viability analysis also suggests that this population will become extinct within 100 years if no effective protection is provided (Zhang and Wang 1999). Therefore, special conservation attention should immediately be allocated to Yangtze River finless porpoises.
References
Avise JC (1998) Conservation genetics in the marine realm. J Hered 89:377–382
Bérubé M, Aguilar A, Dendanto D, Larsen F, Disciara GN, Sears R, Sigurjónsson J, Urban-R J, PalsbØll PJ (1998) Population genetic structure of North Atlantic, Mediterranean Sea and Sea of Cortez fin whales, Balaenoptera physalus (Linnaeus 1758): analysis of mitochondrial and nuclear loci. Mol Ecol 7:585–599
Caughley G (1994) Directions in conservation biology. J Anim Ecol 63:215–244
Chen L, Yang G (2008) Development of tetranucleotide microsatellite loci for the finless porpoise (Neophocaena phocaenoides). Conserv Genet 9:1033–1035
Chen L, Bruford M, Yang G (2007) Isolation and characterization of microsatellite loci in the finless porpoise (Neophocaena phocaenoides). Mol Ecol Notes 7:1129–1131
Crandall KA, Bininda-Emonds ORP, Mace GM, Wayne RK (2000) Considering evolutionary processes in conservation biology. Trends Ecol Evol 15:290–291
Crawford NG (2009) SMOGD: software for the measurement of genetic diversity. Mol Ecol Resour (Accepted for publication)
Escorza-Trevino S, Dizon AE (2000) Phylogeography, intraspecific structure and sex-biased dispersal of Dall’s porpoise, Phocoenoides dalli, revealed by mitochondrial and microsatellite DNA analyses. Mol Ecol 9:1049–1060
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Faubet P, Waples RS, Gaggiotti OE (2007) Evaluating the performance of a multilocus Bayesian method for the estimation of migration rates. Mol Ecol 16:1149–1166
Fontaine MC, Baird SJE, Piry S et al (2007) Rise of oceanographic barriers in continuous populations of a cetacean: the genetic structure of harbour porpoises in Old World waters. BMC Biol 5:30
Frankham R, Ballou JD, Briscoe DA (2003) Introduction to conservation genetics. Cambridge University Press, Cambridge
Gao AL (1991) Morphological differences and generic variation among the populations of Neophocaena phocaenoides. Ph.D. thesis, Nanjing Normal University, Nanjing, 116 pp
Gao AL, Zhou KY (1993) Growth and reproduction of three populations of finless porpoise, Neophocaena phocaenoides, in Chinese waters. Aquat Mamm 19:3–12
Gao AL, Zhou KY (1995a) Geographical variation of external measurements and three subspecies of Neophocaena phocaenoides in Chinese waters. Acta Theriol Sinica 15:81–92
Gao AL, Zhou KY (1995b) Geographical variation of skull among the populations of Neophocaena phocaenoides in Chinese waters. Acta Theriol Sinica 15:161–169
Gao AL, Zhou KY (1995c) Geographical variation of postcranial skeleton among the populations of Neophocaena phocaenoides in Chinese waters. Acta Theriol Sinica 15:246–253
Gaspari S, Azzellino A, Airoldi S, Hoelzel AR (2007) Social kin associations and genetic structuring of striped dolphin populations (Stenella coeruleoalba) in the Mediterranean Sea. Mol Ecol 16:2922–2933
Gibbs HL, Dawson RJG, Hobson KA (2000) Limited differentiation in microsatellite DNA variation among northern populations of the yellow warbler: evidence for male-biased gene flow? Mol Ecol 9:2137–2147
Gilpin ME, Soulé ME (1986) Minimum viable populations: the processes of species extinction. In: Soulé ME (ed) Conservation biology. Sinauer Associates, Sunderland, pp 19–34
Goodman SJ (1997) RSTCALC: a collection of computer programs for calculating estimates of genetic differentiation from microsatellite data and determining their significance. Mol Ecol 6:881–885
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Available from http://www2.unil.ch/popgen/softwares/fstat.htm
Hartl D, Clark AG (1997) Principles of population genetics, 3rd edn. Sinauer Associates, Sunderland
Hastings A (1993) Complex interactions between dispersal and dynamics–lessons from coupled logistic equations. Ecology 74:1362–1372
Holm S (1979) A simple sequentially rejective multiple test procedure. Scand J Stat 6:65–70
Kasuya T (1999) Finless porpoise, Neophocaena phocaenoides (G. Cuvier 1829). In: Ridgway SH, Harrison R (eds) Handbook of marine mammals, vol 6: The second book of dolphins and the porpoises. Academic, San Diego, pp 411–442
Krützen M, Sherwin WB, Berggren P, Gales N (2004) Population structure in an inshore cetacean revealed by microsatellite and mtDNA analysis: bottlenose dolphins (Tursiops sp.) in shark bay, western Australia. Mar Mamm Sci 20:28–47
Lynch M, Conery J, Burger R (1995) Mutation accumulation and the extinction of small populations. Am Nat 146:489–518
Maudet C, Miller C, Bassano B, Breitenrnoser-Würsten C, Gauthier D, Obexer-Ruff G, Michallet J, Luikart G (2002) Microsatellite DNA and recent statistical methods in wildlife conservation management: applications in alpine ibex (Capra ibex [ibex]). Mol Ecol 11:421–436
Moritz C (1994) Defining ‘evolutionary significant units’ for conservation. Trends Ecol Evol 9:373–375
Palstra FP, O’Connell MF, Ruzzante DE (2007) Population structure and gene flow reversals in Atlantic salmon (Salmo salar) over contemporary and long-term temporal scales: effects of population size and life history. Mol Ecol 16:4504–4522
Parsons KM, Durban JW, Claridge DE (2006) Population genetic structure of coastal bottlenose dolphins (Tursiops Truncatus) in the northern Bahamas. Mar Mamm Sci 22:276–298
Pilleri G, Chen P (1980) Neophocaena phocaenoides and Neophocaena asiaeorientalis: taxonomical differences. Invest Cetacea 11:25–32
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Racey PA, Barratt EM, Burland TM, Deaville R, Gotelli D, Jones G, Piertney SB (2007) Microsatellite DNA polymorphism confirms reproductive isolation and reveals differences in population genetic structure of cryptic pipistrelle bat species. Biol J Linn Soc 90:539–550
Randi E, Lucchini V (2002) Detecting rare introgression of domestic dog genes into wild wolf (Canis lupus) populations by Bayesian admixture analyses of microsatellite variation. Conserv Genet 3:29–43
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Reeves RR, Wang JY, Leatherwood S (1997) The finless porpoise, Neophocaena phocaenoides (G. Cuvier 1829): a summary of current knowledge and recommendations for conservation action. Asian Mar Biol 14:111–143
Rice DW (1998) Marine mammals of the world: systematics and distribution. Society for Marine Mammalogy Special Publication 4:1–231
Rosel PE, Dizon AE, Haygood MG (1995) Variability of the mitochondrial control region in populations of the harbour porpoise, Phocoena phocoena, on interoceanic and regional scales. Can J Fish Aquat Sci 52:1210–1219
Rosel PE, France SC, Wang JY, Kocher TD (1999) Genetic structure of harbour porpoise Phocoena phocoena populations in the northwest Atlantic based on mitochondrial and nuclear markers. Mol Ecol 8:41–54
Rosenberg NA, Burke T, Elo K, Feldman MW, Freidlin PJ, Groenen MAM, Hillel J, Maki-Tanila A, Tixier-Boichard M, Vignal A, Wimmers K, Weigend S (2001) Empirical evaluation of genetic clustering methods using multilocus genotypes from 20 chicken breeds. Genetics 159:699–713
Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, Feldman MW (2002) Genetic structure of human populations. Science 298:2981–2985
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Terry A, Bucciarelli G, Bernardi G (2000) Restricted gene flow and incipient speciation in disjunct Pacific Ocean and sea of Cortez populations of a reef fish species, Girella nigricans. Evolution 54:652–659
Turvey ST, Pitman RL, Taylor BL, Barlow J, Akamatsu T, Barrett LA, Zhao XJ, Reeves RR, Stewart BS, Wang KX, Wei Z, Zhang XF, Pusser LT, Richlen M, Brandon JR, Wang D (2007) First human-caused extinction of a cetacean species? Biol Lett 3:537–540
Valsecchi E, Amos W (1996) Microsatellite markers for the study of cetacean populations. Mol Ecol 5:151–156
Van Rossum F, Vekemans X, Meerts P, Gratia E, Lefèbvre C (1997) Allozyme variation in relation to ecotypic differentiation and population size in marginal populations of Silene nutans. Heredity 78:552–560
Wang KX, Wang D, Zhang XF, Pfluger A, Barrett L (2006) Range-wide Yangtze freshwater dolphin expedition: The last chance to see Baiji? Environ Sci Pollut R 13:418–424
Wang JY, Frasier TR, Yang SC, White BN (2008) Detecting recent speciation events: the case of the finless porpoise (genus Neophocaena). Heredity 101:145–155
Waples RS, Gaggiotti OE (2006) What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol 15:1419–1439
Wei Z, Wang D, Zhang XF, Zhao QZ, Wang KX, Kuang XA (2002) Population size, behavior, movement pattern and protection of Yangtze finless porpoise at Balijiang section of the Yangtze River. Resour Environ Yangtze Basin 11:427–432
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Westemeier RL, Brawn JD, Simpson SA, Esker TL, Jansen RW, Walk JW, Kershner EL, Bouzat JL, Paige KN (1998) Tracking the long-term decline and recovery of an isolated population. Science 282:1695–1698
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Wilson PJ, Grewal S, Rodgers A, Rempel R, Saquet J, Hristienko H et al (2003) Genetic variation and population structure of moose (Alces alces) at neutral and functional loci. Can J Zool 81:670–683
Yang G, Ren WH, Zhou KY, Liu S, Ji GQ, Yan J, Wang LM (2002) Population genetic structure of finless porpoises, Neophocaena phocaenoides, in Chinese waters, inferred from mitochondrial control region sequences. Mar Mamm Sci 18:336–347
Yang G, Liu S, Ren WH, Zhou KY, Wei FW (2003) Mitochondrial control region variability of baiji and the Yangtze finless porpoises, two sympatric small cetaceans in the Yangtze River. Acta Theriol 48:469–483
Yang G, Guo L, Bruford M, Wei FW, Zhou KY (2008) Mitochondrial phylogeography and population history of finless porpoises in Sino-Japanese waters. Biol J Linn Soc 95:193–204
Yoshida H, Yoshioka M, Shirakihara M, Chow S (2001) Population structure of finless porpoises (Neophocaena phocaenoides) in coastal waters of Japan based on mitochondrial DNA sequences. J Mammal 82:123–130
Zhang XF, Wang KX (1999) Population variability analysis of the Yangtze finless porpoises. Acta Ecol Sinica 19:529–533
Zhang XF, Liu RJ, Zhao QZ, Zhang GC, Wei Z, Wang XQ, Yang J (1993) The population of finless porpoise in the middle and lower reaches of Yangtze River. Acta Theriol Sin 13:260–270
Acknowledgments
This research was supported by the National Natural Science Foundation of China (NSFC) key project grant no. 30830016 to Yang, NSFC grant no. 30670294 to Yang, the Program for New Century Excellent Talents in University (NCET-07-0445), the Ministry of Education of China to Yang, the Specialized Research Fund for the Doctoral Program of Higher Education (SRFDP 20060319002), the Ministry of Education of China to Yang, and the major project of the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (07KJA18016) to Yang, and a UK Royal Society Incoming Fellowship to Bruford and Yang. All samples examined in this study were collected from already-dead individuals incidentally captured in fisheries bycatch, and no ethics approval is needed in such cases in China. We thank two anonymous referees for their critical comments which greatly improved the submission.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. I. Taylor.
Rights and permissions
About this article
Cite this article
Chen, L., Bruford, M.W., Xu, S. et al. Microsatellite variation and significant population genetic structure of endangered finless porpoises (Neophocaena phocaenoides) in Chinese coastal waters and the Yangtze River. Mar Biol 157, 1453–1462 (2010). https://doi.org/10.1007/s00227-010-1420-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00227-010-1420-x