
Abstract
Squids of the genus Illex are representative of the family Ommastrephidae and account for 65% of the world’s cephalopod captures. Illex is formed by four taxa distributed throughout the Atlantic Ocean (I. argentinus, I. coindetii, I. illecebrosus and I. oxygonius), whose identification and phylogenetic relationships based on morphological characters remain controversial. Thirty-seven enzyme-coding loci were analysed in 230 individuals from seven populations of Illex and ten specimens of Todaropsis eblanae, which were used as the outgroup. Two to four enzyme loci (ALPDH*, IDHP-1*, MEP* and SOD*) were diagnostic among Illex species depending on the species-pair comparison. Individuals morphologically identified as I. oxygonius were also found genetically distinct, which proves the taxonomic validity of this species. No significant intraspecific genetic heterogeneity was detected within Illex argentinus, I. coindetii and I. illecebrosus (Mean G ST= 0.011, 0.003, 0.017, respectively). I. illecebrosus and I. oxygonius were shown as sister species with a close relationship to I. argentinus, whereas I. coindetii formed a different lineage.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cephalopods have come to constitute one of the top invertebrate fisheries with catches of about 2.9 million tons per year (FAO 1998). Among them, the short-finned squids of the genus Illex Steenstrup 1880 (Ommastrephidae, Illicinae) account for about 65% of the world’s commercial cephalopod catches (Caddy 1995). At present, the genus is considered to be constituted by four species: Illex argentinus (Castellanos 1960), I. coindetii (Vérany 1839), I. illecebrosus (Le Sueur 1821) and I. oxygonius Roper, Lu and Mangold 1969, which are distributed throughout the Atlantic Ocean (Roper and Mangold 1998). I. argentinus inhabits the southwest Atlantic, I. coindetii is found along the eastern Atlantic and Mediterranean Sea, as well as in the Gulf of Mexico and Caribbean Sea, while I. illecebrosus lives along the northwestern Atlantic and I. oxygonius off the eastern USA, South to the Mexican Gulf (Lu 1973; Roper et al. 1998) (Fig. 1). Therefore, Illex coindetii, I. illecebrosus and I. oxygonius are sympatric in the northwestern Atlantic Ocean. The morphological similarity of these three species, their overlapping distributions and the disjunctive occurrence of I. coindetii on opposite sides of the Atlantic has led to problems of species identification, which in turn are an impediment for understanding Illex systematics (Roper and Mangold 1998; Roper et al. 1998).

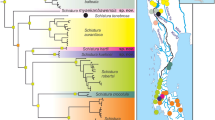
Illex spp. Schematic distribution of Illex species and sampled areas (*) for Illex argentinus (IaF, Ia42), I. illecebrosus (IiN, IiWo), I. coindetii (IcW, IcM), I. oxygonius (IoF), and Todaropsis eblanae (TeW). Sample codes are as in Table 1
Previous morphometric and meristic analyses (e.g. Roper et al. 1998) have not been able to consistently identify all of the Illex species because such characters are influenced by sex, age, growth or environment so that variability within and among taxa is high and overlapping measurements may occur (Lu 1973; Roper and Mangold 1998). Recently, taxonomic identification on I. argentinus, I. coindetii and I. illecebrosus has been assessed based on body and beak structures through stepwise discrimination analysis, with an average of 83% correct designations (Martínez et al. 2002). However, important systematic questions such as the following are still unresolved within the genus Illex: (1) the existence of an I. coindetii complex of morphotypes (Roper and Mangold 1998); (2) the uncertain recognition of I. oxygonius as a valid species sensu stricto due to the difficulty of obtaining wild specimens and its morphological similarity to the other three Illex when mature (Laptikhovsky and Nigmatullin 1993); (3) the existence of I. illecebrosus and I. coindetii morphotypes not attributable to either one taxon or the other in some geographical areas (Lu 1973); and (4) the possible occurrence of subspecies within I. argentinus (Carvalho et al. 1992; Thorpe et al. 1986, cited in Carvalho and Nigmatullin 1998).
Molecular markers such as allozyme polymorphisms are an alternative to morphology and their application to population, taxonomic and phylogenetic studies has been largely proved (Whitmore 1990; Pérez-Losada et al. 1999; Wiens 2000). Allozyme polymorphisms are conservative characters for identifying sibling or morphologically indistinguishable taxa, and for providing an independent estimate of the species phylogeny (Ayala 1983; Wiens 2000). Cephalopods seem to contain lower levels of allozyme variability than other invertebrates (Sanjuan et al. 1996; Carvalho and Nigmatullin 1998; Pérez-Losada et al. 1999), which may make the intraspecific analysis of their populations difficult (Nevo et al. 1984; Ward et al. 1992). However, complex population structures and even cryptic taxa have been inferred using allozyme polymorphisms (e.g. Brierley et al. 1993; Yeatman and Benzie 1994; Pérez-Losada et al. 1999). At present, few genetic studies have been carried out within the genus Illex (Carvalho and Nigmatullin 1998; Jerez et al. 1998; Adcock et al. 1999, Martínez et al. 2005) and none of them look at the four species combined. Hence, Illex species identification and evolutionary relationships as well as their intraspecific population structure remain controversial. Therefore, the main aims of this study were to genetically characterize the four Illex taxa using allozyme polymorphisms, to validate the taxonomic status of the presumptive I. oxygonius, and to infer the phylogenetic relationships within the genus.
Materials and methods
Sampling
Seven Illex populations (N=230 specimens) were collected in 1996–1997 throughout the Atlantic Ocean (Table 1, Fig. 1). Frozen lots from the Falkland Islands and the 42°S international waters fishing areas were collected for I. argentinus (IaF and Ia42, respectively). Fresh specimens of I. coindetii and I. illecebrosus were sampled from fishing ports off Atlantic Northwest (NW; IcW) and Mediterranean East Iberian Peninsula (IcM), and off Newfoundland (Hollyrood Bay, Canada; IiN) and Woods Hole (USA; IiWo). A sample (N=5) of putative I. oxygonius was captured off Florida (USA; IoF) at 200–250 m, within the geographic species range of I. oxygonius, I. coindetii and I. illecebrosus. Identification on morphological traits in I. oxygonius following identification keys from Roper et al. (1998) can only be surely done when individuals are fully mature. Four of the five putative I. oxygonius were mature (individuals 1–4), whereas one was a juvenile (individual 5). Moreover, a sample of ten individuals of Todaropsis eblanae from the NW Iberian waters (TeW) was used for comparison (Table 1). Both fresh individuals and those from commercial lots were frozen after capture and stored at −72°C until required.
Electrophoresis
Horizontal starch-gel electrophoresis was carried out based on the method of Murphy et al. (1996). A section of mantle muscle (11×11 mm) was sliced out for homogenisation with 0.01 M dithiothreitol (DTT) solution. The homogenate was centrifuged at 12,000 g for 10 min at 4°C and the supernatant was embedded in Whatman strips nos. 2 and 3. Hydrolysed-starch gels (13%, Starch-art) were run at constant voltage at 4°C. The enzyme systems routinely examined that showed adequate activity and resolution were: aspartate transaminase (AAT; E.C. 2.6.1.1.), acid phosphatase (ACP; E.C. 3.1.3.2), adenosine deaminase (ADA; E.C. 3.5.4.4), adenylate kinase (AK; E.C. 2.7.4.3), alanopine dehydrogenase (ALPDH; E.C. 1.5.1.17), arginine kinase (ARK; E.C. 2.7.3.3), dihydrolipoamide transaminase (DDH; E.C. 1.8.1.4), carboxylic ester hydrolase (EST; E.C. 3.1.1.-; substrate: α-naphthyl acetate), methylumbelliferyl-acetate deacetylase (ESTD; E.C. 3.1.1.56), glycerol-3-phosphate dehydrogenase (NAD+) (G3PDH; E.C. 1.1.1.8), glucose-6-phosphate dehydrogenase (G6PDH; 1.1.1.49), gliceraldehyde-3-phosphate dehydrogenase, phosphorilating (GAPDH; E.C. 1.2.1.12), glucose-6-phosphate isomerase (GPI; E.C. 5.3.1.9), L-iditol 2-dehydrogenase (IDDH; E.C. 1.1.1.14), isocitrate dehydrogenase (NADP+) (IDHP; E.C. 1.1.1.42), L-leucyl aminopeptidase (LAP; E.C. 3.4.11.1), malate dehydrogenase (MDH; E.C. 1.1.1.37), malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+) (MEP; E.C. 1.1.1.40), mannose-6-phosphate isomerase (MPI; E.C. 5.3.1.8), D-octopine dehydrogenase (OPDH; E.C. 1.5.1.11), cytosol non-specific dipeptidase (PEPA; E.C. 3.4.13.18; substrate: gly-leu), tripeptide aminopeptidase (PEPB; E.C. 3.4.11.4; substrate: leu-gly-gly), X-pro dipeptidase (PEPD; E.C. 3.4.13.9; substrate: phe-pro), peptidase-S (PEPS; E.C. 3.4.11.-; substrate: leucyl-tyrosine), phosphogluconate dehydrogenase (decarboxylating) (PGDH; E.C. 1.1.1.44), phosphoglucomutase (PGM; E.C. 5.4.2.2), pyruvate kinase (PK; E.C. 2.7.1.40), and superoxide dismutase (SOD; E.C. 1.15.1.1). The Tris-citrate pH 8.0 buffer system (gel buffer dilution 1:11) of Ward and Beardmore (1977) was used for most of the enzymes at a voltage of 4.6 V cm−1. The Tris-borate-EDTA pH 8.7 buffer (dilution 1:9) of Boyer et al. (1963) was used for ADA at 3.6 V cm−1 and for G6PDH and GPI (dilution 1:5) at 10 V cm−1. The citrate morpholine pH 7.4 buffer (dilution 1:9) was used for MEP and the locus OPDH-3* at a voltage of 4.6 V cm−1. Enzymes were stained according to recipes in Murphy et al. (1996), with the exception of ACP, DDH, MPI and PK (Harris and Hopkinson 1976), ESTD, LAP, PEPA, PEPB and PEPD (Ahmad et al. 1977) and AAT, IDHP, PGDH and PGM (Shaw and Prasad 1970). The 28 enzymes resolved 37 putative enzyme-coding loci. Banding patterns of the presumptive loci were interpreted according to the current subunit structure of each enzyme. Terminology and notation for allozymes are based on recommendations by Shaklee et al. (1990) and IUBMB (1992). Arabic numerical suffixes for multiple loci (1, 2,...) and for alleles (*100,*105,...) are presented in order of decreasing and increasing anodal mobility, respectively, with *100 corresponding to the most abundant allele in I. coindetii. Cross-comparisons were made among species and gels to ensure scoring accuracy.
Data analysis
Genotype frequencies at polymorphic loci were tested for agreement with Hardy-Weinberg (HW) equilibrium expectations using chi-squared tests, and the probability of the null hypothesis was estimated by the Markov Chain method (Guo and Thompson 1992) as implemented in GENEPOP version 1.2 (Raymond and Rousset 1995). Mean unbiased estimate of expected heterozygosity, mean observed heterozygosity, mean number of alleles and proportion of polymorphic loci (Nei 1987) were calculated for each sample. Variability values were adjusted according to Levene’s (1949) correction for small sample size. BYOSIS-1 (Swofford and Selander 1981) was used to estimate allele frequencies and variability estimates. The genetic structure of each species was analysed by means of F-statistics (G ST, Nei 1987) where G ST significance was calculated by a log-likelihood G-statistic; the probability of the null hypothesis was estimated based on 3,600 permutations (FSTAT version 2.9.1; Goudet 1999). Nei’s (1972) (D N) and modified Cavalli-Sforza & Edwards’ (1967) chord (D cho) genetic distances among samples were used to build UPGMA (Sokal and Michener 1958) and neighbour-joining (NJ; Saitou and Nei 1987) trees (bootstrap procedure, Felsenstein 1985) using PHYLIP version 3.56; Felsenstein 1993. In parallel, the program Dbot (Zaykin and Pudovkin 1993) was used to calculate bootstraps estimates (1,000 replicates) of genetic identities I (Nei 1972) (data not shown). Phylogenetic relationships among the four Illex species (all samples combined) were inferred using four different approaches: NJ, UPGMA, maximum likelihood (ML) and frequency parsimony methods. These methods are the most accurate for analysing allozyme characters (Wiens 2000, and references therein). As previously, NJ and UPGMA were also applied to both D N and D cho genetic distances among species. The UPGMA analysis was carried out to obtain an estimate of the tree under the assumption of a molecular clock (Nei 1987) and for comparison with previous studies. Phylogenetic relationships were also assessed using the allele frequencies under the ML method (Felsenstein 1981) as implemented in PHYLIP. Optimum phylogenetic trees were searched (1,000 replicates) by branch-and-bound (PAUP version 4.0; Swofford 1999). The frequency parsimony approach of Swofford and Berlocher (1987) was applied using FREQPARS version 1.0. Comparison of the defined tree topologies was performed using the usertree option in FREQPARS (Swofford and Berlocher 1987).
Results
Diagnosis and genetic variability
Allele frequencies for the 37 enzyme-coding loci are shown in Table 2. Four loci (ALPDH*, IDHP-1*, MEP* and SOD*) discriminated among the four Illex species. The ALPDH* locus almost completely distinguished I. argentinus and I. illecebrosus (ALPDH 105) from I. coindetii and I. oxygonius (ALPDH 100). The IDHP-1* and SOD* loci discriminated among I. argentinus (IDHP-1 110 and SOD*60), I. coindetii (IDHP-1*100 and SOD*100) and both I. illecebrosus and I. oxygonius (IDHP-1 90 and SOD*90). The MEP* locus was diagnostic for the five individuals labelled as presumptive I. oxygonius (MEP*105) with respect to the other three Illex species (MEP*100). The four mature individuals identified on morphological basis as I. oxygonius showed the allele MEP*105, whereas the juvenile not morphologically identified (individual 5) possessed this same allele.
Polymorphic loci for each population showed no significant deviation from HW expected proportions except for PEPS-2* and PEPS-4* at IcW and for PEPS-4* at IcM, which showed heterozygote deficits (homozygotes for the alleles *90, *85 and *115, respectively (data not shown). Twelve enzyme loci were monomorphic in all the analysed Illex populations (Table 2). Genetic variability estimates (Table 2) indicated low values for all of the seven samples of Illex (e.g. He and Ho ranged between 0.016 and 0.060).
Population structure and phylogenetic analysis
Mean G ST values within the taxa I. argentinus, I. coindetii and I. illecebrosus were relatively low (0.011, 0.003 and 0.017, respectively) indicating intraspecific homogeneity (Table 3). The highest values of G ST per locus were found to be significant (P<0.05) for ALPDH* in I. coindetii due to presence of heterozygotes for the alleles *90 and *105 in IcM, and for IDHP-2* (P<0.01) and PEPD* (P<0.05) in I. illecebrosus, mainly due to four and three heterozygotes for alleles *90 and *80 in IiWo, respectively.
Nei’s (1972) (D N) and modified Cavalli-Sforza & Edwards chord (1967) (D cho) genetic distances using 35 allozyme loci among all samples are presented in Table 4. Both distances showed the lowest values for all pairwise population comparisons within species (D N<0.002; D cho<0.03). I. argentinus and I. oxygonius showed the greatest genetic distances (D N=0.11; D cho=0.196) between species, whereas I. illecebrosus and I. oxygonius presented the lowest values (D N≤0.058; D cho≤0.119), followed by the I. illecebrosus and I. argentinus pair (D N=0.062–0.064; D cho=0.122–0.134). The low genetic distances observed between the latter two pairs is due to the fact that these species share alleles for the diagnostic loci IDHP-1*, SOD* and ALPDH* (Table 2). The NJ tree based on D cho distances among populations for 35 enzyme loci generated the intraspecific clades with the highest bootstrap values (88–96%) (Fig. 2). The NJ tree and similarly the UPGMA analysis (data not shown) depicted I. illecebrosus and I. oxygonius populations as the closest related taxa with a bootstrap support of 62–64%, with this pair showing a sister relationship to I. argentinus (bootstrap support of 70% for the NJ tree and 33% for the UPGMA tree).

Illex spp. Neighbour-joining dendrogram of populations of IaF, Ia42; IiN, IiWo; IcW, IcM; and IoF, and the outgroup TeW based on the modified Cavalli-Sforza & Edwards’ (1967) chord genetic distances among 35 enzyme loci. Estimated branch length connecting TeW to the ingroup is 10 times longer than presented. Bootstrap values (100 replicates) are shown for every clade. Sample codes are as in Table 1
Genetic distances of Nei (1972) (D N) and modified Cavalli-Sforza & Edwards (1967) chord (D cho) were calculated among species considering I. argentinus as constituted by IaF and Ia42, I. coindetii by IcW and IcM, and I. illecebrosus by IiN and IiWo, and using 36 enzyme loci (data not shown). The neighbour-joining and UPGMA trees based on Nei’s (1972) genetic distances showed I. illecebrosus and I. oxygonius as a sister group connected to I. argentinus (Fig. 3a and 3b). The tree topology of the NJ tree based on D cho distances was the same as NJ/Nei’s tree, with bootstrap values slightly higher for the I. illecebrosus and I. oxygonius clade (63%), and 65% for the I. illecebrosus, I. oxygonius and I. argentinus clade. In the UPGMA/D cho tree I. illecebrosus and I. oxygonius formed a sister clade (59% bootstrap support), as in the above trees, but I. argentinus and I. coindetii were grouped together, although with a low support (<50%, data not shown). The ML tree resulted after 189 topologies was identical to the NJ/Nei, UPGMA/Nei and NJ/CSE trees, with higher bootstrap values (Fig. 3c). The modified Wagner’s parsimony analysis (MANAD) grouped I. argentinus and I. illecebrosus species (Fig. 3d). Shortest MANAD tree after MANOB branch-and-bound search obtained the most parsimonious tree after 1,000 replicates. FREQPARS estimation of tree lengths for the main topologies resulted in very similar L values, with L=76.125 for the parsimony analysis and L=77.919 for the ML, NJ/Nei and UPGMA/Nei analyses. Thus, the I. illecebrosus and I. oxygonius clade was supported by ML and distance-based methods (Fig. 3a–c), whereas I. argentinus and I. illecebrosus species were clustered together in the parsimony analysis (Fig. 3d). All these topologies showed I. coindetii as forming an independent lineage (Fig. 3). The clade formed by the Western Atlantic species I. illecebrosus, I. oxygonius and I. argentinus was supported by bootstrap values of 61–67% in the NJ/Nei, NJ/D cho, ML and parsimony analyses, and with less support in the UPGMA/Nei and UPGMA/D cho trees (<50%, tree not shown). A lower clade support obtained in the UPGMA analyses may be due to the performance of this procedure for estimating branch lengths (Wiens and Servedio 1998).

Illex spp. Phylogenetic relationships among Illex species based on 36 enzyme loci and using TeW as the outgroup. a Neighbour-joining tree based on Nei’s (1972) genetic distance (DN) (estimated TeW branch length is 10 times-folded); b UPGMA tree using DN (the cophenetic correlation coefficient was 0.998); c Maximum likelihood tree after 189 topologies, where branch length indicates expected accumulated variance as rate of character evolution (estimated TeW branch length is 2 times-folded); and d Shortest MANAD tree after MANOB branch-and-bound search obtaining most parsimonious tree (after 1,000 replicates). Bootstrap values (≥50) are shown above branches for distance and likelihood methods
Discussion
Diagnosis of the genus Illex: I. oxygonius validation
Four enzyme-coding loci (ALPDH*, IDHP-1*, MEP*, SOD*) were found diagnostic among Illex species (Tables 2 and 5). An enzyme-coding locus is defined as diagnostic if an individual can be correctly assigned to one of two species with a probability of 99% or higher (Ayala and Powell 1972). Each of the four Illex species could be genetically recognised with 2–4 enzyme loci depending on each pairwise comparison (Table 5). A high percentage (83%) of correct designations among I. argentinus, I. coindetii and I. illecebrosus was also achieved using morphometric beak-character analyses (Martínez et al. 2002). In the present work, I. argentinus could be distinguished from I. coindetii, I. illecebrosus and I. oxygonius by 3, 2 and 4 allozyme loci, respectively, I. coindetii from the other Illex species by 3 loci, and I. illecebrosus from I. oxygonius by 2 loci. The one juvenile and four mature specimens of the putative I. oxygonius showed the diagnostic allele MEP*105, although only the former four specimens have been preliminary considered I. oxygonius based on morphological evidence. The most geographically restricted species I. oxygonius occurs in the overlapping distributions between I. coindetii and I. illecebrosus (Fig. 1), being taxonomically the most controversial. Some authors have postulated that I. oxygonius is a hybrid between I. coindetii and I. illecebrosus (Roper et al. 1998); however, no heterozygotes for the diagnostic loci IDHP-1* and SOD* between both species were found in the I. oxygonius sample. Moreover, each recognised Illex species could be genetically differentiated and clearly distinguished from the putative I. oxygonius by 2–4 loci (Table 5). Genetic identities (Nei 1972) among Illex taxa (I=0.8–0.9) fell within the range of conspecific populations and congeneric species (Thorpe 1983). However, considering the very low levels of allozyme variability shown by all the populations, it would be difficult to explain the observed abrupt changes in allele frequencies for several loci if we assume that all of the Illex populations belong to the same species. Moreover, congruence between morphological and allozyme characters argues for the consideration of the four Illex taxa as four different species. Nevertheless, a more extensive sampling, mainly from Florida waters (off southern USA), where some Illex species are sympatric, would be desirable to complement this study.
General low allozyme variability levels were found for all the Illex taxa (Table 2), for example, Ho=0.02–0.06, in agreement with previous studies on I. argentinus (Ho= 0.038) and other cephalopods (Ho=0.018–0.083) (Carvalho et al. 1992; Carvalho and Nigmatullin 1998; Pérez-Losada et al. 1999). Low genetic variability estimates have been associated with generalist-habitat species that migrate long distances and inhabit wide bathymetric ranges, as occurs in Illex taxa when compared to other less mobile cephalopods (see Carvalho and Nigmatullin 1998 for a review). Another potential explanation for these low levels of allozyme variation could be intensive fishing pressure, which may involve population size depletion and so the erosion of the genetic pool. Thus, in general, population dynamics of the short-lived cephalopods may be heavily influenced by low diversity balancing the risks of mortality factors and causing periodic local extinctions (Boyle and Boletzky 1996).
Population structure of I. argentinus, I. coindetii and I. illecebrosus
Illex argentinus samples from off North Falkland Islands and the 42°S latitude were found to be genetically homogeneous (Table 3). In contrast with our results, Carvalho et al. (1992) found genetic heterogeneity between northern and southern Illex populations of the 42°S latitude at the Patagonic slope supported by the diagnosis of the loci ADA* and MDH-1*. It may be possible that the 42°S sample used in this work did not belong to the same population analysed by Carvalho et al. (1992), as marked time-related differences in population structure have been found by previous life cycle studies on Illex species (Hatanaka 1988; Nigmatullin 1989; Arkhipkin 1997; Carvalho and Nigmatullin 1998). Moreover, as it has been shown for other invertebrates, differences in reproductive timing may maintain different genetic pools in sympatry (McFadden 1999). However, in spite of the high genetic variability recently found at microsatellite loci within I. argentinus populations, no marked population differentiation was found between northern and southern samples of the 42°S latitude (Adcock et al. 1999a, b), which agrees with the present data. To clarify the population structure of I. argentinus and the other Illex species a more detailed structural analysis including samples captured at different bathymetric and seasonal ranges should be carried out.
Samples of I. coindetii and I. illecebrosus showed no significant overall genetic differences (mean G ST=0.003 and 0.017, respectively; Table 3). Thus, I. coindetii from the northeastern Atlantic (IcW) and Mediterranean (IcM) seem to belong to the same genetic pool, as previously indicated by discriminant analysis of body and beak characters (Martínez et al. 2002) and by a recent molecular study (Martínez et al. 2005). The general lack of intraspecific allozyme differentiation may be due to the long migrations effected by these oceanic species (e.g. 1,260 miles for I. illecebrosus, Dawe et al. 1981), or the discriminatory limitations of the electrophoretic technique. Previous morphological studies on I. coindetii from the Mediterranean and East and West Atlantic, however, have shown possible heterogeneity (Zecchini et al. 1996; Arkhipkin 1997; Hernández-García and Castro 1998). Future genetic studies including more samples and using other molecular markers would help to clarify the population dynamics of Illex across the Atlantic.
Phylogenetic relationships among Illex species
Allozyme data can provide an accurate estimate of the species phylogeny and can be used for reconstructing phylogenies among closely related species (Wiens 2000, and references therein). Phylogenetic relationships shown by distance-based (NJ/DN, NJ/Dchord and UPGMA/DN trees) and ML approaches were in congruence (Fig. 3a, b and c) supporting phylogenetic accuracy (see Wiens 2000). But these relationships were slightly different from those obtained in the maximum parsimony analysis (Fig. 3d): I. illecebrosus is sister to I. oxygonius in the NJ, UPGMA and ML analyses, but sister to I. argentinus in the parsimony analysis. The length difference between both topologies was L=1.79 steps. Although the parsimony search found a slightly shorter tree, distance and likelihood methods are considered more accurate than parsimony methods under most conditions, as independent of spurious allelic frequency variations (Wiens 2000). Thus, the MANAD parsimony analysis is dependent on allelic frequencies (Swofford and Berlocher 1987). Moreover, a close evolutionary relationship between I. illecebrosus and I. oxygonius is supported by their overlapping geographic distributions (Fig. 1).
The monophyletic clade formed by I. argentinus, I. illecebrosus and I. oxygonius species was supported by all phylogenetic analyses (Fig. 3). A previous phylogenetic study on ommmastrephids including Illex species (but not I. oxygonius) was conducted by Yokawa (1994), where an UPGMA/Nei distance tree using 23 enzyme loci showed I. coindetii and I. illecebrosus as sister species separated from I. argentinus. Yet, similar genetic distance values were shown for I. coindetii with respect to the other two Illex species (Yokawa 1994). However, only two individuals per species were included in this analysis, which according to Archie et al. (1989) does not guarantee the inference of accurate phylogenetic relationships. Moreover, several authors (e.g. Graybeal 1998; Poe 1998) have reported that increasing taxon sampling is important to assess evolutionary relationships, so the inclusion of I. oxygonius in the present study should generate more reliable inferences of the Illex phylogeny.
References
Adcock GJ, Carvalho GR, Rodhouse PG, Shaw PW (1999a) Highly polymorphic microsatellite loci of the heavily fished squid genus Illex (Ommastrephidae). Mol Ecol 8:157–168
Adcock GJ, Shaw PW, Rodhouse PG, Carvalho GR (1999b) Microsatellite analysis of genetic diversity in the squid Illex argentinus during a period of intensive fishing. Mar Ecol Prog Ser 187:171–178
Ahmad M, Skibinski DOF, Beardmore JA (1977) An estimate of the amount of genetic variation in the common mussel Mytilus edulis. Biochem. Genetics 15:833–846
Archie JW, Simon C, Martin A (1989). Small sample size does decrease the stability of dendograms calculated from allozyme-frequency data. Evolution 43(3):678–683
Arkhipkin AI (1997) Geographical variation in growth and maturation of the squid Illex coindetii (Oegopsida, Ommastrephidae) off the north-west African coast. J Mar Biol Ass UK 76:1091–1106
Ayala FJ (1983) Enzymes as taxonomic characters. In: Oxford GS, Rollinson D (eds) Systematics Association Special, vol 24. protein polymorphism: adaptive and taxonomic significance. Academic, London
Ayala FJ, Powell JR (1972) Allozymes as diagnostic characters of sibling species of Drosophila. Proc Nat Sci USA 69:1094–1096
Boyer SH, Fainer DC, Naughton MA (1963) Myoglobin: inherited structural variation in man. Science, NY 140:1228–1231
Boyle PR, Boletzky Sv (1996) Cephalopod populations: definition and dynamics. Phil Trans R Soc Lond 351(B):985–1002
Brierley AS, Rodhouse GG, Thorpe JP, Clarke MR (1993) Genetic evidence of population heterogeneity and cryptic speciation in the ommastrephid squid Martialia hyadesi from the Patagonian Shelf and Antarctic Polar Frontal Zone. Mar Biol 116:593–602
Caddy JF (1995) Cephalopods and demersal finfish stocks: some statistical trends and biological interactions. In: Squid 94 Venice, the 3rd international Cephalopod trade conference proceedings, Agra Europe, London
Carvalho GR, Nigmatullin Ch M (1998) Stock structure analysis and species identification. In: PG Rodhouse, EG Dawe, RK O’Dor (eds) Squid Recruitment Dynamics. The genus Illex as a model, the commercial Illex species and influences on variability. FAO Fisheries Tech. Paper 376:199–232
Carvalho GR, Thompson A, Stoner AL (1992) Genetic diversity and population differentiation of the shortfinned squid Illex argentinus in the south-west Atlantic. J Exp Mar Biol Ecol 158:105–121
Cavalli-Sforza LL, Edwards AWF (1967) Phylogenetic analysis: models and estimation procedures. Evolution 32:550–570
Dawe EG, Beck PC, Drew HJ, Winters GH (1981) Long-distance migration of a short-finned squid, Illex illecebrosus. J North Atl Fish Sci 2:75–76
FAO Food and Agriculture Organization Yearbook (1998). Fishery statistics. Capture production, vol 86/1. FAO, Rome
Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17:368–376
Felsenstein J (1985) Confidence limits on phylogenies with a molecular clock. Syst Zool 34:152–161
Felsenstein J (1993) PHYLIP. Phylogeny Inference Package, ver.3.56c, Seattle
Goudet J (1999) FSTAT a program to estimate and test gene diversities and fixation indices (ver. 2.9). Available from http://www.unil.ch/izea/softwares/fstat.html.
Graybeal A (1998) Is it better to add taxa or characters to a difficult phylogenetic problem? Syst Biol 47(1):9–17
Guo SW, Thompson EA (1992) Performing the exact test of Hardy-Weinberg proportions for multiple alleles. Biometrics 48:361–372
Harris H, Hopkinson DA (1976) Handbook of enzyme electrophoresis in human genetics, North-Holland, Amsterdam
Hatanaka H (1988) Feeding migration of short-finned squid Illex argentinus in the waters off Argentina. Nip Suis Gak 54(8):1343–1349
Hernández-García V, Castro JJ (1998) Morphological variability in Illex coindetii (Cephalopoda: Ommastrephidae) along the North-west coast of Africa. J Mar Biol Ass UK 78:1259–1268
IUBMB (1992) International Union of Biochemistry and Molecular Biology (IUBMB). In: Webb EC (ed) Enzyme nomenclature 1992: recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the nomenclature and classification of enzymes. IUMB Press, London
Jerez B, Roldán MI, Pla C (1998) Biochemical genetics in the Argentinean squid, Illex argentinus. Sci Mar 62(1–2):141–149
Laptikhovsky VV, Nigmatullin ChM (1993) Egg size, fecundity and spawning in females of the genus Illex (Cephalopoda: Ommastrephidae). ICES J Mar Sci 50:393–403
Levene H (1949) On a matching problem arising in genetics. Ann Math Stat 20:91–94
Lu CC (1973) Systematics and zoogeography of the squid genus Illex (Oegopsida: Cephalopoda), PhD Thesis, Memorial University of Newfoundland, Canada
Martínez P, Sanjuan A, Guerra A (2005) Allozyme analysis of geographical and seasonal variation of Illex coindetii (Cephalopoda: Ommastrephidae) from Central Mediterranean and Iberian Atlantic. J Mar Biol Ass UK 85:177–184
Martínez P, Sanjuan A, Guerra A (2002) Identification of Illex coindetii, I. illecebrosus and I. argentinus (Cephalopoda: Ommastrephidae) throughout the Atlantic Ocean; by body and beak characters. Mar Biol 141:131–143
McFadden CS (1999) Genetic and taxonomic relationships among north-eastern Atlantic and Mediterranean populations of the soft coral Alcyonium coralloides. Mar Biol 133: 171–184
Murphy RW, Sites CW, JR Buth DG, Haufler CH (1996) Proteins: isozyme electroforesis In: DM Hillis, C Moritz (eds) Molecular Systematics. Sinauer Associates, Sunderland, MA
Nei M (1972) Genetic distance within populations. Amer Naturalist 106:283–292
Nei M (1987) Molecular Evolutionary Genetics. Columbia University Press, New York
Nevo E, Beiles A, Ben-Shlomo R (1984) The evolutionary of genetic diversity: ecological, demographic and life history correlates. Lec Notes Biomath 53:13–213
Nigmatullin ChM (1989) Las especies de calamar más abundantes del Atlántico suroeste y sinopsis sobre la ecología del calamar Illex argentinus. Frente Mar 5(A):39–59
Pérez-Losada M, Guerra A, Sanjuan A (1999) Allozyme differentiation in the cuttlefish Sepia officinalis (Mollusca: Cephalopoda) from the NE Atlantic and Mediterranean. J Heredity 83:280–289
Poe S (1998) Sensitivity of phylogeny estimation to taxonomic sampling. Syst Biol 47 (1):18–31
Raymond M, Rousset F (1995) GENEPOP (ver. 1.2): population genetics software for exact tests and ecumenicism. J Heredity 86:248–249
Roper CFE, Mangold K (1998) Systematic and distributional relationships of I. coindetii to the genus Illex (Cephalopoda: Ommastrephidae). In: Voss N, Vecchione M , Toll RB, Sweeney MJ (eds) Systematics and Biogeography of cephalopods. Smithsonian Contributions to Zoology 586:13–26
Roper CFE, Lu CC, Mangold K (1969) A new species of Illex from the Western Atlantic and distributional aspects of other Illex species (Cephalopoda: Oegopsida). Proc Biol Soc Washington 82:295–322
Roper CFE, Lu CC, Vecchione M (1998) A revision of the systematics and distribution of Illex species (Cephalopoda: Ommastrephidae). In: Voss N, Vecchione M, Toll RB, Sweeney MJ (eds) Systematics and Biogeography of cephalopods. Smithsonian Contributions to Zoology 586:405–423
Saitou N, Nei M. (1987) The neighbour-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sanjuan A, Pérez-Losada M, Guerra A (1996) Genetic differentiation by allozyme electrophoresis in three Sepia spp. (Mollusca: Cephalopoda) from Galizian waters (North-West Iberian Peninsula). Mar Biol 126:253–259
Shaklee JB, Allendorf FW, Morizot DC and Whitt GS (1990) Gene nomenclature for protein-coding loci in fish. Trans Am Fish Soc 119:2–15
Shaw CR, Prasad R (1970) Starch gel electrophoresis of enzymes. A compilation of recipes. Bioch Genet 4:297–320
Sokal RR, Michener CD (1958) A statistical method for evaluating systematic relationships. Univ Kansas Sci Bull 28:1409–1438
Swofford DL (1999) PAUP: beta version of PAUP 4.0. Sinauer, Sunderland
Swofford DL, Berlocher SH (1987) Inferring evolutionary trees from gene frequency data under the principle of maximum parsimony. Syst Zool 36(3):293–325
Swofford DL, Selander RB (1981) BIOSYS-1, FORTRAN program for the comprehensive analysis of electrophoretic data in population genetics and systematics. J Heredity 72:281–283
Thorpe JP (1983) Enzyme variation, genetic distance and evolutionary divergence in relation to levels of taxonomic separation. In: Oxford GS, Rollinson D (eds) Protein polymorphism: adaptive and taxonomic significance. Academic, London
Ward RD, Beardmore JA (1977) Protein variation in the plaice Pleuronectes platessa. Genet Research 30:45–62
Ward RD, Skibinski DOF, Woodwark M (1992) Protein heterozygosity, protein structure and taxonomic differentiation. Evol Biol 26:73–159
Whitmore DH (1990) Electrophoretic and isoelectric focusing techniques in fisheries management. CRC Press, Boca Raton
Wiens JJ (2000) Reconstructing phylogenies from allozyme data: comparing method performance with congruence. Biol J Lin Soc 70:613–632
Wiens JJ, Servedio MR (1998) Phylogenetic analysis and intraspecific variation: performance of parsimony, likelihood, and distance methods. Syst Biol 47(2):228–253
Yeatman J, Benzie JAH (1994) Genetic structure and distribution of Photololigo spp. in Australia. Mar Biol 118:79–87
Yokawa K (1994) Allozyme differentiation of sixteen species of ommastrephid squid (Mollusca, Cephalopoda). Antartic Sci 6(2):201–204
Zaykin DV, Pudovkin AI (1993) Two programs to estimate significance of χ2 values using pseudoprobability tests. J Heredity 84:152
Zecchini F, Vecchione M, Roper CFE (1996) A quantitative comparison of hectocotylus morphology between Mediterranean and Western Atlantic populations of the squid Illex coindetii (Mollusca: Cephalopoda: Ommastrephidae). Proc Biol Soc Washington 109(3):591–599
Acknowledgements
We are grateful to Dr. Earl Dawe from the Department of Fisheries and Oceans, St. John’s Newfoundland, Canada, to Dr. José Luis Sánchez Lizaso from the University of Alicante, Spain, and to Dr. Scott Herke from the School of Biological Sciences, Loussiana State University, USA, for their helpful sample collections. This research was supported by the projects 64102C028 (Universidade de Vigo, Spain) and REN2000-1193 MAR (Ministerio de Ciencia y Tecnología, Spain) to A.S. Portions of this paper fulfilled Ph.D. requirements for P.M., who received a scholarship from University of Vigo. The experimental procedure complied with current national laws.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by O. Kinne, Oldendorf/Luhe
Rights and permissions
About this article
Cite this article
Martínez, P., Pérez-Losada, M., Guerra, A. et al. First genetic validation and diagnosis of the short-finned squid species of the genus Illex (Cephalopoda: Ommastrephidae). Marine Biology 148, 97–108 (2005). https://doi.org/10.1007/s00227-005-0057-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00227-005-0057-7