
Abstract
The depletion of shallow-water fish stocks through overexploitation has led to increasing fishing pressure on deep-sea species. Poor knowledge of the biology of commercially valuable deep-water fish has led to the serial depletion of stocks of several species across the world. Data regarding the genetic structure of deep-sea fish populations is important in determining the impact of overfishing on the overall genetic variability of species and can be used to estimate the likelihood of recolonisation of damaged populations through immigration of individuals from distant localities. Here the genetic structure of the commercially fished deep-water species the blackspot sea bream, Pagellus bogaraveo is investigated in the northeastern Atlantic using partial DNA sequencing of mitochondrial cytochrome b (cyt-b) and D-loop regions and genotyping of microsatellite loci. An absence of variation in cyt-b and low genetic variation in D-loop sequences potentially indicate that P. bogaraveo may have undergone a severe bottleneck in the past. Similar bottlenecks have been detected in other Atlantic species of fish and have possibly originated from the last glaciation. P. bogaraveo may have been particularly vulnerable to the effects of low temperature and a fall in sea level because stages of its life history occur in shallow water and coastal sites. However, there are other explanations of low genetic variability in populations of P. bogaraveo, such as a low population size and the impacts of fishing on population structure. Analysis of population structure using both D-loop and microsatellite analysis indicates low to moderate, but significant, genetic differentiation between populations at a regional level. This study supports studies on other deep-sea fish species that indicate that hydrographic or topographic barriers prevent dispersal of adults and/or larvae between populations at regional and oceanographic scales. The implications for the management and conservation of deep-sea fish populations are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The depletion of fish stocks associated with the continental seas of the world has led to increasing fishing efforts directed at deep-water species (e.g. Haedrich 1995; Koslow et al. 2000). These fisheries have focused on seamounts, continental slopes, and the slopes of oceanic islands. In the deep sea, it is in these habitats that concentrations of heavy-bodied, bentho-pelagic fish species, suitable for commercial exploitation, are found. Technological requirements for deep-sea fishing have meant that the majority of these fisheries have commenced recently, since the late 1960s, starting with the exploitation of species such as slender armourhead (Pseudopentaceros wheeleri Hardy, 1983), in the Pacific, by the Soviet fishing fleet (Sasaki 1986). Since this time, fishing operations in deep waters have expanded and vessels from developed nations now target fish species such as orange roughy (Hoplostethus atlanticus Collett, 1889), oreo (Allocytus niger James, Inada, Nakamura, 1988); A. verrucosus (Gilchrist, 1906), Pseudocyttus maculates Gilchrist, 1906), and grenadiers (Coryphaenoides spp.), down to depths of up to 2,000 m all over the world (Rogers 1994; Gianni 2004).
Several major problems have arisen as a result of deep-sea fisheries. The first of these is the rapid depletion of deep-sea fish populations that are subject to fishing pressure. Fisheries such as the slender armourhead in the North Pacific, orange roughy in the South Pacific, and roundnose grenadier (Coryphaenoides rupestris Gunnarus, 1765) in the Atlantic have all declined or become depleted often within 5–10 years of commencing (Koslow et al. 2000). In most cases, individual stocks have not recovered and catches have been maintained only by the fishing fleets exploiting new stocks in different geographic areas. Rapid depletion of seamount and other deep-sea fisheries, even within managed Exclusive Economic Zones, has taken place because of a lack of understanding of the biology of target species and the intense fishing pressure that can be placed on stocks using modern technology (Rogers 1994). For example, it has been found that species such as orange roughy mature after a long period of time (30 years) and have an extremely high longevity (>100 years; Fenton et al. 1991), and populations show sporadic recruitment (Mace et al. 1990). It has now been estimated that sustainable annual harvests of this species may be only 1–2% of the virgin biomass (Clark 1995), much lower than the annual quotas set for orange roughy in the early years of exploitation. On the high seas, outside of zones of national jurisdiction, such fishing activities are unregulated. Additional problems associated with deep-sea trawling include by-catch of non-target species and destruction of benthic epifaunal communities associated with seamounts and continental slopes (Rogers 1999).
The Azores is the site of one of the few deep-water long-line fisheries that can be regarded as artisanal in origin. Along with Madeira, this archipelago is the site of a traditional open-boat drop-line fishery for species such as scabbard fish [Aphanopus carbo Lowe, 1839; Lepidopus caudatus (Euphrasen, 1788)] that has been practised for several hundred years. However, over the last three decades, traditional fishing methods have been replaced by the use of modern fishing vessels using large arrays of long lines. During the period from 1980 to 1990, the number of fishing vessels in this region halved but the catches landed in the Azores doubled. There has therefore been increasing concern with regards to the sustainability of the deep-water Azorean long-line fisheries. This stimulated a multidisciplinary study of the biology of the main target species of this fishery that commenced in 1997, with an aim of improving the scientific basis of the management strategy for long-line fishing in the Azores. This study included aspects of the population biology, genetics, and reproduction of a number of deep-sea species in this region. The results presented here pertain to the genetics of one of the main target species of the Azores long-line fishery, the black-spot sea bream, Pagellus bogaraveo.
P. bogaraveo is found in the northeast Atlantic, from southern Norway to Cap Blanc in Mauritania, including Madeira, the Azores, and Canary Islands. It is a protandrous hermaphrodite with a peak spawning period of March to April (late winter/spring) in the Azores islands. The larva of P. bogaraveo is planktonic (Karrer 1984); juveniles occur in coastal waters and adults are found on the continental slope down to 700 m depth in the Azores. The maximum age of P. bogaraveo detected in this study was approximately 16 years (Menezes et al. 2001). Despite this species being hermaphroditic, the sex ratio of males to females in the Azores region during the period of study was not significantly different from 1:1 (n=1,806; Menezes et al. 2001; but see Discussion).
Given the potentially dispersive life history of P. bogaraveo, it was expected that populations would be genetically homogenous across the Azores archipelago and even regionally within the northeast Atlantic. Genetic studies on other seamount- and oceanic island–associated species have demonstrated a lack of genetic structure on regional and even on oceanic scales, for example, slender armourhead, alfonsino (Beryx splendens Lowe, 1834), and wreckfish [Polyprion americanus (Bloch and Schneider, 1801)] (Martin et al. 1992; Sedberry et al. 1996; Hoarau and Borsa 2000). However, other studies on deep-sea fish have been contradictory (e.g. orange roughy—reviewed in Creasey and Rogers 1999) or have been reported to show evidence of genetic structure in populations on regional and/or oceanic scales, including: Greenland halibut, Reinhardtius hippoglossoides (Walbaum, 1792); roundnose grenadier, Coryphaenoides rupestris; walleye pollack, Theragra chalcogramma (Pallas, 1814); rockfish, Sebastolobus alascanus Bean, 1890, S. altivelis Gilbert, 1896 and Dover sole, Microstomus pacificus (Lockington, 1879); ling, Genypterus blacodes (Forster, 1801); hoki, Macruronus novaezealandiae (Hector, 1871); black oreo, Allocytus niger (Smith and Francis 1982; Milton and Shaklee 1987; Duschenko 1988; D’yakov 1991; Mulligan et al. 1992; Baker et al. 1995; Bailey et al. 1997; Vis et al. 1997; Ward et al. 1998; Stepien 1999). Many of these species have pelagic larvae but these studies have suggested that topographic and hydrographic barriers to dispersal prevent panmixia at regional and oceanic scales.
This study aimed to test the hypothesis of wide dispersal and homogeneity of the genetic population structure of P. bogaraveo within the Azores archipelago and on a regional basis in the northeastern Atlantic. Populations were sampled from the three island groups of the Azores, including a seamount, the Princess Alice Bank, the island of Madeira, and from the continental slope of Europe, off the coast of Portugal. The genetic composition of individuals within and between different samples was analysed using partial DNA sequencing of the mitochondrial cytochrome b and D-loop regions and microsatellite DNA analysis.
Materials and methods
Sampling of populations
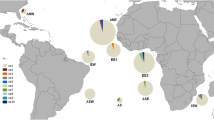
A stratified sampling design was used, focusing on comparing populations in the three island groups of the Azores (east, west, and central), and the Princess Alice Bank. To provide a regional comparison, two additional samples were collected, one by sampling a commercial catch from off Peniche, on the Portuguese continental slope, and one from Madeira, (Fig. 1). Samples in the Azores and Princess Alice Bank were collected using a commercial stone-buoy type of long-line gear deployed from the “R.V. Arquipélago”, the University of the Azores’ research vessel (Menezes et al. 2001).

Pagellus bogaraveo. Map showing relative positions of sampling locations in the northeast Atlantic. AZW Azores west; PAB Princess Alice Bank; AZC Azores central; AZE Azores east
Samples from the Azores were taken on three cruises in 1999 in spring, summer, and autumn, respectively, with long lines being deployed from approximately 500 m to 1,200 m, maximum depth. Line setting commenced 1 h before sunrise and line retrieval occurred 1.5 h after sunset and lasted on average about 3.5 h. Samples from the Portuguese continental slope were collected in February 2000 and those from Madeira were collected in 1997 (locations are given in Table 1).
DNA extraction
Tissue samples of liver, gill, and muscle were taken from freshly caught fish on board the research vessel or sampled from fresh caught fish landed in port (Portuguese slope samples only). Samples were preserved in 96% ethanol (Hayman Ltd., U.K.). In the laboratory excess ethanol was blotted from the tissue and total genomic DNA was then extracted from small (1–3 mg) sections of tissue following a phenol\chloroform protocol based on Sambrook et al. (1989) or extractions were carried out using DNeasy kits (Qiagen, Crawley, West Sussex, U.K.) following the manufacturer’s instructions for animal tissue. The extracted DNA was resuspended in elution buffer and then quantified using a Uvidoc 008-XD documentation system, with Uvisoft version 98 gel quantification software, by comparison with a known standard (1 Kb DNA ladder, Gibco BRL, Rockville, Md, USA) on a 1% agarose gel, following electrophoresis. DNA was subsequently stored at −20°C until used for analysis.
DNA sequencing
Genetic variation in P. bogaraveo samples was examined by partial sequencing of the cytochrome b oxidase gene (cyt-b) and mitochondrial control region (D-loop). All amplifications of these regions were carried out in an MWG Primus 96 Plus PCR (polymerase chain reaction) machine. An approximately 410-bp fragment of the cyt-b mitochondrial gene was amplified from six Eastern Azorian individuals, seven Seamount individuals, and six individuals from Peniche by PCR using universal primers designed by Kocher et al. (1989) and described in Palumbi et al. (1991). These were:
Light strand primer CYB-GLU-L-CP (based upon a carp sequence);
5′-TGA CTT GAA GAA CCA CCG TTG-3′
and primer CYB-CB2-H (based upon a stingray sequence);
5′-CCC TCA GAA TGA TAT TTG TCC TCA-3′
The PCR conditions for cyt-b amplifications were as follows: amplification in a 20-μl volume containing approximately 30 ng of genomic DNA extract, 0.2 μl (1 U) Amplitaq DNA Polymerase [Perkin Elmer (PE)], 1 μl (10 pmol) of each primer, 2 μl of 10× PE buffer (containing 10 mM Tris-HCl, pH 8.3, at 25°C, and 20 mM KCl), 2.5 μl 25 mM MgCl2, and 0.4 μl (0.2 mM) of each dNTP (PE). PCR cycle conditions were as follows: an initial denaturation step of 94°C for 4 min, followed by 30 cycles of 94°C for 50 s, 56°C for 30 s, and 72°C for 50 s. After these cycles, a final extension step of 72°C for 5 min ended the PCR. The resulting amplified DNA fragments were stored at 4°C until cycle sequencing reactions were carried out. No further samples were sequenced because of the lack of variation found for this region.
A 306-bp fragment of the D-loop mitochondrial region was amplified from a total of 140 P. bogaraveo, consisting of: 97 Azorian (26 Azores west, 20 Azores east, 25 Azores central islands, 26 Azores central seamounts) 20 Madeiran, and 23 Peniche specimens (locations given in Table 1 and Fig. 1). Sample numbers for the Azores were regarded as sufficient for comparison with other sites. However, the sample size from Madeira was limited. Given the results of data analysis (see below) it was felt that additional sampling of specimens from Madeira and sequencing of specimens from the Peniche site was not necessary.
PCR was performed using the primers described by Ostellari et al. (1996). These were:
L-pro1 (5′-ACT CTC ACC CCT AGC TCC CAA AG-3′) and
H-DL1 (5′-CCT GAA GTA GGA ACC AGA TGC CAG-3′)
The conditions for these D-loop amplifications were as follows: amplification in a 20-μl volume containing 50 ng of genomic DNA extract, 0.2 μl (1 U) Amplitaq DNA polymerase (PE), 1 μl (10 pmol) of each primer, 2 μl of 10× PE buffer (containing 10 mM Tris-HCl, pH 8.3, at 25°C, and 20 mM KCl), 2.5 μl 25 mM MgCl2, and 0.2 mM of each dNTP (PE). PCR cycle conditions were 30 cycles of 94°C for 50 s, 53°C for 30 s, and 72°C for 50 s, with a final extension phase of 72°C for 5 min. The resulting amplified DNA fragments were stored at 4°C until they were sequenced.
After PCR, the mitochondrial cyt-b and D-loop amplicons were cleaned using Qiaquick PCR purification kits (Qiagen). The resulting cleaned PCR product was quantified, as for genomic DNA, and samples diluted to concentrations suitable for cycle sequencing. Cycle sequencing was carried out using the Bigdye Terminator Ready Reaction Mix [Applied Biosystems (ABI) Ref. 4306158, Warrington, Cheshire, U.K.] according to the manufacturer’s instructions. The resulting products from the cycle-sequencing reactions were cleaned using Dye-X spin-kits (Qiagen), according to the manufacturer’s instructions. The labelled DNA fragments were then visualised by electrophoresis on a 6% polyacrylamide gel on an ABI 377 automatic DNA sequencing machine. Cyt-b and D-loop were sequenced in both directions and sequences checked and aligned by eye using Chromas version 1.45 (McCarthy 1998).
Microsatellites
P. bogaraveo samples from the Azores and Peniche only were used for microsatellite analysis because of the low sample size from Madeira. The numbers of individuals used per site were as follows: Peniche 62, Azores east 72, Azores west 72, Azores central seamounts 72, Azores central islands, 72. Genomic DNA used for microsatellite amplification was obtained by the use of DNeasy genomic DNA extraction kits (Qiagen), because the resultant DNA was of a quality that maximised the reliability of microsatellite amplifications. The resulting genomic DNA samples were stored in 10 mM Tris solution at −20°C until used.
Isolation and initial assessment of variability of P. bogaraveo microsatellite loci are detailed in Stockley et al. (2000). The ten microsatellite loci were amplified using the conditions detailed in Stockley et al. (2000). To reduce the number of PCR reactions performed, multiplex PCR (the amplification of multiple loci in a single reaction) was used. The quantities of all other reagents (including template DNA) were kept constant as described in Stockley et al. (2000) but primers were added in full (10 pmol) quantities. The combinations of loci were as follows [primer pairs x/x, followed by the annealing temperature used (in parentheses)]: PbMS18/PbMS19 (56°C), PbMS2/PbMS17 (56° C), PbMS16/PbMS20 (56°C), PbMS1/PbMS4/PbMS6 (62°C), and PbMS15 (56°C).
The assessment of the variability of these loci was carried out using the Genescan application (manual 4303188, analysis guide 4306603; PE Applied Biosystems, available at http://www.appliedbiosystems.com/techsupp/) on an ABI 377 automatic sequencer. Analysis was performed on a Macintosh computer running both the Genescan and the Genotyper applications (PE Biosystems). Because the fluorescent dyes, attached to the one primer from each pair, fluoresced at different wavelengths, up to three loci were scored on the same gel. Analysis differed for locus PbMS19 compared to the other loci. This locus had no stutter bands, because it was a pure triplet repeat (CAG)10 making it an ideal candidate for automatic scoring using the Genotyper application. This application scored the gel with a high level of accuracy, the results of which were checked for accuracy by eye using Genescan. The other loci (mostly di-nucleotide repeats) possessed stutter bands, and variable levels of amplification. Consequently when the Genotyper application was used on these loci, erroneous genotype assessments were made, false homozygotes were called, or more than two alleles per loci were detected. Therefore, it was decided to score the gels by eye, using empirical binning. This method was applied where possible to all loci, including PbMS19, where the empirical binning method was found to confirm the Genotyper application method (PE Biosystems). Those locus-individual combinations that failed to produce bands of adequate quality for scoring were repeated, as single PCR reactions (i.e. not multiplex) and run again. This process was repeated until no loci had more than 5% missing data at any site.
RST analysis requires that the size difference between microsatellites alleles be known. This was done by sequencing amplicons that were scored as homozygotes by genotyping. The primers used were those appropriate to the locus being sized (Stockley et al. 2000). A DGtp sequencing kit (PE Biosystems) was used to sequence these amplicons. This kit, whilst less accurate for normal sequencing applications, is capable of sequencing through microsatellite repeats (the Big Dye Terminator kits are unreliable for sequencing repeat regions). The resulting sequence outputs were used to correlate size of the alleles to the number of repeats, thus allowing data analysis based upon number of microsatellite repeat units.
The microsatellite data was placed on an Excel spread sheet and converted into ‘microsatellite’ format (data containing number of repeats), and ‘standard’ format (data not containing number of repeats) using the macro ‘Microsatellite toolkit 2.2’ (available from http://acer.gen.tcd.ie/~sdepark/MSmacros.htm). These formats are readable by Arlequin 2.000 (Schneider et al. 2000). This program was used to analyse both spatial and temporal variation of P. bogaraveo in the Azores.
Data analysis
Sequence data
All sequences were aligned using CLUSTAL X (Thompson et al. 1997) using default settings. Overall genetic diversity was estimated using gene diversity (h)—the probability that two randomly chosen haplotypes are different (Nei 1987), and nucleotide diversity (π)—the probability that two randomly chosen homologous nucleotides are different (Tajima 1983; Nei 1987).
Genetic structure of P. bogaraveo in the northeast Atlantic was analysed using pairwise FST estimates to detect short-term genetic distance between populations. In this process, a transformation of the data is used to linearise the genetic distance with the divergence time (Slatkin 1995) and therefore make this F-statistic proportional to divergence time. This approach was used to produce an analogue of FST that can be used to estimate effective migration rates. Slatkin’s (1995) approach calculates FST by finding the average probability of identity in state of two alleles in each population, and the average probability of identity in state of two alleles chosen at random from all the populations together (Slatkin 1991). This is a coalescent approach, and therefore FST is defined in terms of “the ratio of average coalescence times of different pairs of genes”. The significance of these genetic distances is assessed by means of a permutation test. The permutation of haplotypes is between populations. Such changes of the data set results in a null distribution of pairwise FST values under the hypothesis of no differentiation between the populations. The P-value derived from this test is the proportion of permutations leading to an FST value larger than or equal to the observed one. Migration estimates were not derived from FST values because such measures of interpopulation exchange are unlikely to be realistic (Bossart and Prowell 1998; Whitlock and McCauley 1999).
Hierarchical genetic differentiation and the significance of group and population structure were tested using analysis of molecular variance (AMOVA; Excoffier et al. 1992). For this purpose populations were divided into two regional groups based on the results from pairwise FST analyses (all Azores vs Peniche/Madeira) and into populations within groups (Azores east, Azores central, Azores west, Princess Alice Bank, Peniche, Madeira). All population analyses were performed using Arlequin version 2.0 (Schneider et al. 2000).
To make an assessment of the geographic partition of haplotypes it was necessary to carry out phylogeographic analysis. Because phylogenetic analysis makes assumptions that are invalid at the population level (i.e. that ancestral haplotypes are extinct) data were analysed using minimum-spanning network techniques as implemented by the software package TCS version 1.13 (Clement et al. 2000).
Microsatellite data
Observed and expected heterozygosity under the Hardy–Weinberg equilibrium (HWE; Nei 1987) were estimated and the significance of deviation from HWE was assessed using Guo and Thompson’s (1992) analogue of Fisher’s exact test. Hierarchical genetic differentiation and the significance of group and population structure were tested using AMOVA (Excoffier et al. 1992) and pairwise F-statistics (Wright 1951), respectively, as for mitochondrial DNA sequence data. In addition, data were analysed using RST (Slatkin 1995), which is based on a stepwise mutation model (SMM) instead of the infinite alleles model (IAM) used for FST. As well as comparing spatially separated samples of P. bogaraveo, data for samples from the Azores were pooled and different age classes were compared using FST and RST. All analyses were carried out using Arlequin version 2.0 (Schneider et al. 2000).
Results
DNA sequence data
The 306 bp of the left domain of the D-loop in 140 P. bogaraveo produced a perfect alignment with no insertions or deletions. This sequence (described from 5′ to 3′) begins immediately after the tRNApro gene (light strand) and extends into the D-loop (Ostellari et al. 1996). This region is the most variable in the D-loop of other species within the Sparidae (Jean et al. 1995) and flanks the more conserved central segment and 3′ end of this region of mitochondrial DNA. Twenty-five haplotypes were identified with 19 polymorphic nucleotide sites. Two haplotypes dominated the haplotype frequencies (Fig. 2) and accounted for over 77% of the haplotypes from all the sample sites. There were 17 sites showing transitions, and 2 sites showing transversions. The following mean nucleotide frequencies were obtained: A=31.65%, C=12.75%, G=17.37%, T=38.23%. The overall gene (haplotype) diversity (h) was 0.5919 (±0.0459). The overall nucleotide diversity (π) was 0.002111 (±0.001827). The commonest haplotype is deposited in the European Molecular Biology Laboratory (EMBL) sequence database, accession number AJ630508.

P. bogaraveo. Minimum-spanning network for 306 bp of the left domain of the mitochondrial D-loop. The haplotype with the highest outgroup probability is displayed as a square, other haplotypes as circles. The size of the square or circle is proportional to the frequency of the haplotype (n given in parentheses).
The approximately 410-bp fragment of the cyt-b mitochondrial gene sequenced in 19 P. bogaraveo was trimmed to 379 bp and found to contain no variation in its sequence between individuals from the Azores, Peniche, and Madeira. This result demonstrated that this gene was not a useful marker in determining levels of genetic differentiation amongst the samples used in this study. Therefore no further individuals from any of the sample sites were sequenced for this gene. This haplotype is deposited in EMBL, accession number AJ630507, and differs from P. bogaraveo sequences from the Mediterranean by a single nucleotide change (see Desdevises et al. 2000; EMBL/Genbank acc. no. AJ276880/PB0276880).
Pairwise FST estimates are given in Table 2. The highest FST values, and thus the highest differentiation between populations, were found between Peniche, Madeira, and the western island group of the Azores. These are the most geographically (by distance) and oceanographically (by means of currents, and separation by deep sea) separated populations. The lowest FST values were found between Peniche and Madeira, and also between Azores east, Azores central islands, and Azores central seamounts. FST values in pairwise comparisons between Peniche and all Azores populations were significant but comparisons between Peniche and Madeira were not significant. FST values for comparisons between Madeira and Azores populations were significant except for Azores east (which is geographically the closest to Madeira). Comparisons of the Azores west population and Madeira, Peniche, and the Princess Alice Bank were significant. All other comparisons within the Azores were not significant.
AMOVA of the D-loop partial sequence data showed that 87.9% of all variance in haplotype frequencies occurred within populations. Only 2.2% of variance was partitioned between populations within groups, and 9.9% of variance was explained by differences between the two geographical groups defined (i.e. Azorian, and Peniche/Madeira). The variance among populations within groups was insignificant but the partition of variance among the two designated groups was significant (P<0.00000; see Table 3). These results were therefore consistent with those obtained for FST analysis in that the Azores populations showed significant genetic differentiation from the European continental shelf and Madeira.
The parsimonious network of mtDNA haplotypes derived from TCS analysis of the D-loop of P. bogaraveo is shown in Fig. 2. The maximum number of mutational events separating any two haplotypes is six. The most common branch length between two haplotypes is 1. The network is dominated by a single ubiquitous haplotype shared by 87 individuals. The second most common haplotype (21 individuals) is restricted to the Azores area only but is found in all the Azores island groups and the Princess Alice Bank. There are also several haplotypes found both in Madeira and Peniche, with frequencies varying between 2 and 6 individuals per haplotype. There are numerous haplotypes found only in single regions, represented by a single individual. The general pattern of the network is star shaped, rooted in the central, most common haplotype. There is also a second star-shaped portion of the cladogram, which is restricted to the Azores, with the second most common haplotype as its centre.
Microsatellite data
Loci PbMS4,17, and 18 could not be binned according to the empirical binning method, or the automatic Genotyper (PE Biosystems) method. This was because although all three loci were dinucleotide repeats, amplification produced bands that formed a continuum of sizes that did not form discrete two-or-more base-pair jumps. This may be because some amplification products contained an additional ′A base attached by the Taq polymerase (PE Biosystems Genescan manual). Alternatively there may have been a mutation in some alleles where a single base pair had been inserted or deleted (although no such mutation was detected during the sequencing of the calibration band—see Materials and methods). Because of the difficulty of positive identification of a single base-pair size difference on an ABI 377 (B. Stockley, personal observation; PE Biosystems, personal communication), and the possibility of either a mutational or an artifactual cause for the 1-bp difference, it was decided to exclude these loci from the study.
All remaining loci (except PbMS16) showed deviations from the HWE at P=0.05 level at some sampling sites (Table 4). However two loci showed consistent significant heterozygote deficiencies at all localities (P<0.00000): these are loci 1 and 2. Uniform heterozygote deficiency across all geographic localities for these sites could be interpreted as evidence of null alleles (Schlötterer 1998), and these loci were therefore excluded from further data analysis.
The pairwise FST values based on microsatellite loci showed a lack of detectable differentiation (P>0.05) between all sites in the Azores (Table 2). However they detected significant genetic differentiation (P<0.0005) between all sites in the Azores compared to Peniche. The RST statistics failed to detect differentiation between any sample sites, except between Azores west and Azores central islands (P<0.05; Table 5).
The AMOVA for microsatellite loci, based on the FST distance method to calculate covariance partitioning, revealed significant partitioning of variance components (Table 3) between the population groups (P<0.0005). Of the variance in genotype frequencies, 98.07% occurred within populations and 0.13% was found among populations within groups. The AMOVA, based on the RST distance method to calculate covariance partitioning, failed to find significant partitioning of variance components at any level of the hierarchical analysis (Table 3).
Discussion
The absence of genetic diversity in cyt-b for P. bogaraveo is unusual and comparable to species that are known to have undergone extreme population bottlenecks, such as the Coos River striped bass (Morone saxatilis; Waldman et al. 1998), the northern elephant seal (Mirounga angustirostris; Weber et al. 2000), and the whooping crane (Grus americana; Glenn et al. 1999). Partial sequences of the mitochondrial D-loop did reveal variation in P. bogaraveo populations but nucleotide diversity was extremely low (π=0.002) for mitochondrial DNA in fish and haplotype diversities (h=0.5919) were also low to moderate (see Grant and Bowen 1998). Nucleotide diversity was an order of magnitude below that observed for the deep-water fish B. splendens in the same region (B. Stockley, personal observation).
A possible explanation of these data is that P. bogaraveo has undergone a bottleneck during the past, in the northeast Atlantic, during which effective population size must have been reduced to very low numbers. This is supported by the star-shaped haplotype network in P. bogaraveo. In this, two common haplotypes represent the majority of individuals and other low-frequency haplotypes are separated from the common haplotypes by one or two mutations (Fig. 2). A similar pattern was detected previously in a comparison of D-loop haplotypes from P. bogaraveo populations from the Mediterranean and the Azores (Bargelloni et al. 2003). In addition, this study also showed that the number of haplotypes detected for P. bogaraveo was lower than for four other sparid species in the same geographic region.
In populations of marine fish, reduced genetic diversity is commonly ascribed to historical bottlenecks (Bernatchez et al. 1989; Richardson and Gold 1997; Waldman et al. 1998). A severe population bottleneck (i.e. near extinction event) can lead to a drastic reduction of the number of mtDNA haplotypes (Glenn et al. 1999; Weber et al. 2000). The likely origin of the bottleneck in P. bogaraveo populations, in the northeast Atlantic, was the severe environmental effects of the last glacial maximum (approximately 20,000 years ago; Mix et al. 2001), and particularly the last Heinrich event (approximately 17,000 years ago; Adams et al. 1999). During the most recent glacial event (Wisconsin), sea ice reached 50°N and populations of marine animals in the northeast Atlantic, especially of warm water species, are likely to have been severely impacted (Ruddiman and McIntyre 1981; Muss et al. 2001). However, there is evidence that the signature of glacial cycles previous to the last one may also influence the genetic population structure of marine organisms (e.g. Stammatis et al. 2004).
A recent study on the red-lip blenny, Ophioblennius atlanticus analysed variation in the mitochondrial cyt-b region in populations from the tropical and sub-tropical Atlantic (Muss et al. 2001). Samples from the Azores region also showed extremely low nucleotide and haplotype diversities compared to elsewhere in the species range (further south). Studies on Atlantic cod also suggest a recent bottleneck followed by expansion of populations and species range (Carr et al. 1995; Pogson et al. 1995). It is likely that all of these bottlenecks occurred as a result of climate change associated with glaciations. However, lack of any observed variation in the cyt-b region and extremely low diversity in the D-loop, which has a mutation rate more than three times higher than the rest of the mitochondrial genome (Donaldson and Wilson 1999), indicate that this event may have been particularly severe for P. bogaraveo. This is supported by low haplotype diversity and the shallow population structure detected for P. bogaraveo compared to other sparids from the northeast Atlantic region (Bargelloni et al. 2003; see below). Such a bottleneck may have been caused by the combined effects of both low temperature and the decrease in sea-level associated with the last glaciation. The larvae of P. bogaraveo occur in shallow water and juvenile fish are coastal, often inhabiting bays and inlets (Menezes et al. 2001). The availability of juvenile habitats may have been severely impacted by the fall in sea level (see also Fauvelot et al. 2003) and shallow waters will have also been subject to the most extreme effects of low temperatures.
However, there are other explanations for the low genetic variation observed in mitochondrial DNA sequences for P. bogaraveo. Small census population size or effective population size can result in low genetic diversity in populations. This is because genetic drift fixes alleles more rapidly than in large populations and may also negate the influence of weak selection (Wright 1931; Frankham et al. 2002). P. bogaraveo has been subject to heavy fishing pressure that may explain observations of changes in the population structure in the Azores over a 10-year period (Krug 1998). Observations indicated a marked increase in the occurrence of intersexual fish during this period whilst the numbers of male fish decreased. The explanation for this is thought to be that fishing has impacted the larger size classes of P. bogaraveo. Because the species is a protandrous hermaphrodite, this has had a large impact on the female fish in the population (Krug 1998). The population has responded to this loss by changing from male to female at a smaller size. It is notable that during this study, of the 1,806 specimens sampled by long line, 369 were females, 407 were males (not significantly different from a 1:1 ratio), 649 were intersexuals, and the sex of 381 individuals was indeterminate (Menezes et al. 2001). If fishing has led to major mortality amongst female fish then the maternal population of this species may indeed have a reduced population size leading to low diversity in mitochondrial genes. Historical bottlenecks and recent decreases in population size may not be mutually exclusive explanations of the data.
An alternative explanation of low diversity is that of genetic hitchhiking induced genetic sweeps (Barton 2000). Selective sweeps are most frequent in areas of recombination and areas near rapidly evolving functional genes (Nurminsky 2001) and are therefore unlikely to be influential in mtDNA dynamics. It should also be noted that mutation rates do vary considerably within groups of organisms. It has been suggested that differences in animal size, generation time, and metabolic rate can all influence the rate of evolution and therefore the diversity of mitochondrial genes (e.g. Martin and Palumbi 1993; Spradling et al. 2001). A low level of genetic variation in the genes studied could arise from other factors.
The star-shaped network for P. bogaraveo for D-loop, associated with low/moderate haplotype diversity and the variation of microsatellite loci, indicates that this species has undergone population expansion. Both D-loop sequences and microsatellites show evidence of genetic structure in northeast Atlantic populations of P. bogaraveo. The signal for structure is relatively weak, probably because of the recent extreme-bottleneck and population-expansion events, as suggested for D-loop data by Bargelloni et al. (2003).
AMOVA indicated that there was a significant partitioning of haplotype variation between the groups of populations analysed (Azores populations vs Peniche/Madeira). Pairwise FST values between Peniche and the Azores populations and Madeira and the Azores populations were significant (P<0.05) with the greatest FST values found in comparisons with the western Azores locality. The comparison between the western Azores and the Princess Alice Bank was also significant (P<0.05; but insignificant with Bonferroni correction). This supports a restriction of gene flow between the populations located on the Azores and those on the European continental slope and Madeira. It is also suggestive of some restriction of gene flow within the Azores island group between the western islands and the rest of the Archipelago, though evidence for this is weak. Restriction of gene flow between the Azores and continental slope is strongly supported by the haplotype network showing that the second most common haplotype, and many of the less common haplotypes, are restricted to the Azores region only.
For microsatellites, AMOVAs based on FSTs show a significant partition of the variance in genotype frequencies at the level of groups of populations (Azores vs Peniche). Analyses of pairwise FST values are an order of magnitude higher for all comparisons between the Azores and Peniche than for comparisons within the Azores. All pairwise FST values between the Azores and Peniche are significant (P<0.00000). However, AMOVAs based on RST (Table 3), or pairwise comparisons of RST, (Table 5) do not show the same pattern of genetic differentiation between populations on the Azores and those on the European continental shelf.
In the present study the overall variance components for AMOVA from RST are two orders of magnitude higher than the overall variance components in AMOVA based on FST (Table 3). In cases where the variance of the RST estimate is high this test can have a low reliability (O’Connell et al. 1998). It is also notable that the allele frequencies of the microsatellites of P. bogaraveo show multiple peaks across several sizes of repeat. This is not compatible with the stepwise mutation model on which RST is based, in which repeats are gained or lost from a single ancestral allele. It suggests that there are occasional jumps in repeat size or that an extreme bottleneck event in P. bogaraveo has eliminated or reduced the frequency of many ancestral alleles. That this pattern may have arisen from a recent bottleneck may also be responsible for the findings of previous workers, for example, Garcia de Leon et al. (1997), where cod microsatellite loci showed greater concordance with the IAM than the SMM. It may also explain why analyses based on the SMM often seem less reliable for fish microsatellites, as they often exhibit evidence of recent bottleneck events (e.g. Grant and Bowen 1998). The Peniche sample is also of a different size from the Azores samples and this may further influence RST estimates. This is because RST may be more sensitive to unequal sample size than FST (Ruzzante 1998).
Overall the observations on haplotype frequencies and microsatellite loci indicate that P. bogaraveo populations in the northeast Atlantic are structured at a regional level. Populations in the Azores are genetically distinct from those on the European continental slope, off Portugal and off Madeira. Whilst this structure may be strongly influenced by historical factors, current levels of gene flow have been insufficient to homogenise populations. This study adds to the growing evidence that demersal or bentho-pelagic deep-water fish and squid species tend to show limited dispersal between geographic areas on a regional scale (reviewed in Rogers 2003). This has implications for management of deep-water fisheries at regional levels, in that migration between populations separated by major hydrographic features, such as ocean basins, is limited. It is therefore unlikely that migration between such populations will be sufficient to compensate for impacts arising from excessive fishing pressure, in terms of both biomass and genetic variation of the population.
References
Adams J, Maslin M, Thomas E (1999) Sudden climate transitions during the Quaternary. Prog Phys Geogr 23:1–36
Bailey KM, Stabeno PJ, Powers DA (1997) The role of larval retention and transport features in the mortality and potential gene flow of the walleye pollack. J Fish Biol 51 (Suppl A):135–154
Baker CS, Perry A, Chambers GK, Smith PJ (1995) Population variation in the mitochondrial cytochrome b gene of the orange roughy Hoplostethus atlanticus and the hoki Macruronus novaezelandiae. Mar Biol 122:503–509
Bargelloni L, Alarcon JA, Alvarez MC, Penzo E, Margoulas A, Reis C, Patarnello T (2003) Discord in the family Sparidae (Teleostei): divergent phylogeographical patterns across the Atlantic–Mediterranean divide. J Evol Biol 16:1149–1158
Barton NH (2000) Genetic hitchhiking. Philos Trans R Soc Lond B Biol Sci 355:1553–1562
Bernatchez L, Dodson JJ, Bolvin S (1989) Population bottlenecks—influence on mitochondrial-DNA diversity and its effect on coregonine stock discrimination. J Fish Biol 35 (Suppl A):233–244
Bossart JL, Prowell DP (1998) Genetic estimates of population structure and gene flow: limitations, lessons and new directions. Trends Ecol Evol 13:202–206
Carr SM, Snellen AJ, Howse KA, Wroblewski JS (1995) Mitochondrial DNA sequence variation and genetic stock structure of Atlantic cod (Gadus morhua) from bay and offshore locations on the Newfoundland continental shelf. Mol Ecol 4:79–88
Carvalho GR, Hauser L (1995) Molecular genetics and the stock concept in fisheries. In: Carvalho GR, Pitcher TJ (eds) Molecular genetics in fisheries. Chapman & Hall, London, pp 55–79
Clark MR (1995) Experience with the management of orange roughy (Hoplostethus atlanticus) in New Zealand and the effects of commercial fishing on stocks over the period 1980–1993. In: Hopper AG (ed) Deep-water fisheries of the North Atlantic oceanic slope. Kluwer, Dordrecht, pp 251–266
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Creasey S, Rogers AD (1999) Population genetics of bathyal and abyssal organisms. Adv Mar Biol 35:1–151
Desdevises Y, Jovelin R, Jousson O, Morand S (2000) Comparison of ribosomal DNA sequences of Lamellodiscus spp. (Monogenea, Diplectanidae) parasitising Pagellus (Sparidae, Teleostei) in the north Mediterranean Sea: species divergence and coevolutionary interactions. Int J Parasitol 30:741–746
Donaldson KA, Wilson RR (1999) Amphi-panamic geminates of snook (Percoidei: Centropomidae) provide a calibration of the divergence rate in the mitochondrial DNA control region of fishes. Mol Phylogen Evol 13:208–213
Duschenko VV (1988) The formation of the commercial stock of the north Atlantic grenadier. Can Transl Fish Aquat Sci 5340:1–21
D’yakov YP (1991) Population structure of Pacific black halibut, Reinhardtius hippoglossoides. J Icthyol 31:16–28
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplogroups: applications to human mitochondrial DNA restriction data. Genetics 131:479–491
Fauvelot C, Bernardi G, Planes S (2003) Reductions in the mitochondrial DNA diversity of coral reef fish provide evidence of population bottlenecks resulting from Holocene sea-level change. Evolution 57:1571–1583
Fenton GE, Short SA, Ritz DA (1991) Age determination of orange roughy, Hoplostethus atlanticus (Pisces: Trachichthyidae) using 210Pb:226Ra disequilibria. Mar Biol 109:197–202
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Garcia de Leon F, Chikhi JL, Bonhomme F (1997) Microsatellite polymorphism and population subdivision in natural populations of European sea bass Dicentrachus labrax (Linnaeus, 1758) Mol Ecol 6:51–62
Gianni M (2004) High seas bottom fisheries and their impact on the biodiversity of vulnerable deep-sea ecosystems: summary findings. Report to the World Conservation Union, World Wildlife Fund and Natural Resources Defence Council. IUCN, Gland, Switzerland, pp 1–83
Glenn TC, Stephan W, Braun MJ (1999) Effects of a population bottleneck on whooping crane mitochondrial DNA variation. Conserv Biol 13:1097–1107
Grant WS, Bowen BW (1998) Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J Hered 89:415–428
Guo S, Thompson E (1992) Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48:361–372
Haedrich RL (1995) Structure over time of an exploited deepwater fish assemblage. In: Hopper AG (ed) Deep-water fisheries of the North Atlantic oceanic slope. Kluwer, Dordrecht, pp 70–96
Hoarau G, Borsa P (2000) Extensive gene flow within sibling species in the deep-sea fish Beryx splendens. C R Acad Sci Sci Vie 323:315–325
Jean C, Hui C, Lee S, Chen C (1995) Variation in the mitochondrial DNA and phylogenetic relationships of fish of the subfamily Sparinae (Perciformes: Sparidae) in the coastal waters of Taiwan. Zool Stud 34:270–280
Karrer C (1984) Beschreibung von Jungenstadien dreier Spariden-Arten aus dem nordwest-afrikanischen Auftriebsgebeit (Teleostei, Perciformes). Arch Fisch Wissenschaft 35:53–90
Kocher TD, Thomas WK, Meyer A, Edwards SV, Paabo S, Wilson AC (1989) Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc Natl Acad Sci U S A 86:6169–6200
Koslow JA, Boehlert GW, Gordon JDM, Haedrich RL, Lorance P, Parin N (2000) Continental slope and deep-sea fisheries: implications for a fragile ecosystem. ICES J Mar Sci 57:548–557
Krug H (1998) Variation in the reproductive cycle of the black spot seabream, Pagellus bogaraveo (Brünnich, 1768) in the Azores. Arquipelago Bull Univ Azores Life Mar Sci 16A:37–47
Mace PM, Fenaughty JM, Coburn RP, Doonan IJ (1990) Growth and productivity of orange roughy (Hoplostethus atlanticus) on the north Chatham Rise. N Z J Mar Freshw Res 24:105–119
Martin AP, Humphreys R, Palumbi SR (1992) Population genetic structure of the armourhead, Pseudopentaceros wheeleri, in the North Pacific Ocean: application of the polymerase chain reaction to fisheries populations. Can J Fish Aquat Sci 49:2368–2391
Martin AP, Palumbi SR (1993) Body size, metabolic rate, generation time, and the molecular clock. Proc Natl Acad Sci U S A 90:4087–4091
McCarthy C (1998) Chromas, ver 1.45. Available at: http://www.technelysium.com.au/chromas14x.html
Menezes G, Rogers A, Krug H, Mendonça A, Stockley BM, Isidro E, Pinho MR, Fernandes A (2001) Seasonal changes in biological and ecological traits of demersal and deep-water fish species in the Azores. Final Report European Commission DGXIV/C/1 Contract 97/081. University of the Azores, Faial, Azores, Portugal, pp 1–162 plus appendices
Milton DA, Shaklee JB (1987) Biochemical genetics and population structure of blue grenadier, Macruronus novaezelandiae (Hector) (Pisces: Merluccidae), from Australian waters. Aust J Mar Freshw Res 38:727–742
Mix AC, Bard E, Schneider R (2001) Environmental processes of the ice age: land, oceans, glaciers (EPILOG). Quatern Sci Rev 20:627–657
Mulligan TJ, Chapman RW, Brown BL (1992) Mitochondrial DNA analysis of walleye Pollack, Theragra chalcogramma, from the eastern Bering Sea and Shelikof Strait, Gulf of Alaska. Can J Fish Aquat Sci 49:319–326
Muss A, Robertson DR, Stepien CA, Wirtz P, Bowen BW (2001) Phylogeography of Ophioblennius: the role of ocean currents and geography in reef fish evolution. Evolution 55:561–572
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nurminsky DI (2001) Genes in sweeping competition. Cell Mol Life Sci 58:125–134
O’Connell M, Dillon MC, Wright JM, Bentzen P, Merkouris S, Seeb J (1998) Genetic structure among Alaskan Pacific herring populations identified using microsatellite variation. J Fish Biol 53:150–163
Ostellari L, Bargelloni L, Penzo E, Patarnello P, Patarnello T (1996) Optimization of single-strand conformation polymorphism and sequence analysis of the mitochondrial control region in Pagellus bogaraveo (Sparidae, Teleostei): rationalized tools in fish population biology. Anim Genet 27:423–427
Palumbi SR, Martin A, Romano S, McMillan WO, Stice L, Grabowski G (1991) The simple fools guide to PCR, ver 2.0. Kewalo Marine Laboratory, University of Hawaii, Honolulu
Pogson GH, Mesa KA, Boutillier RG (1995) Genetic population structure and gene flow in the Atlantic cod Gadus morhua: a comparison of allozyme and nuclear RFLP loci. Genetics 139:375–385
Richardson LR, Gold JR (1997) Mitochondrial DNA diversity in and population structure of the red grouper, Epinephelus morio, from the Gulf of Mexico. Fish Bull 95:174–179
Rogers AD (1994) The biology of seamounts. Adv Mar Biol 30:305–350
Rogers AD (1999) The biology of Lophelia pertusa (Linnaeus 1758) and other deep-water reef-forming corals and impacts from human activities. Int Rev Hydrobiol 84:315–406
Rogers AD (2003) Molecular ecology and evolution of slope species. In: Wefer G, Billett D, Hebbeln D, Jørgensen B, Schlüter M, Weering T van (eds) Ocean margin systems. Springer, Berlin Heidelberg New York, pp 323–337
Ruddiman WF, McIntyre A (1981) The north Atlantic Ocean during the last glaciation. Palaeogeogr Palaeoclimatol Palaeoecol 35:145–214
Ruzzante DE (1998) A comparison of several measures of genetic distance and population structure with microsatellite data: bias and sampling variance. Can J Fish Aquat Sci 55:1–14
Sambrook J, Fritscher EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Sasaki T (1986) Development and present status of Japanese trawl fisheries in the vicinity of seamounts. In: Uchida RN, Hayasi S, Boehlert GW (eds) Proceedings of the workshop on the environment and resources of seamounts in the North Pacific. U.S. Department of Commerce, NOAA Tech Rep NMFS 43:31–35
Schlötterer C (1998) Microsatellites. In: Hoelzel AR (ed) Molecular genetic analysis of populations: a practical approach, 2nd edn. IRL Press/Oxford University Press, Oxford, pp 237–261
Schneider S, Roessli D, Excoffier L (2000) Arlequin: a software for population genetics data analysis, ver 2.000. Genetics and Biometry Lab, Dept. of Anthropology, University of Geneva
Sedberry GR, Carlin JL, Chapman RW, Eleby B (1996) Population structure in the pan-oceanic wreckfish Polyprion americanus (Teleostei: Polyprionidae), as indicated by mtDNA variation. J Fish Biol 49 (Suppl A):318–329
Slatkin M (1991) Inbreeding coefficients and coalescence times. Genet Res 58:167–175
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Smith PJ, Francis RICC (1982) A glucosephosphate isomerase polymorphism in New Zealand ling Genypterus blacodes. Comp Biochem Physiol 73B:451–455
Spradling TA, Hafner MS, Demastes JW (2001) Differences in the rate of cytochrome-b evolution among species of rodents. J Mammal 82:65–80
Stammatis C, Triantafyllidis A, Moutou A, Mamuris Z (2004) Mitochondrial DNA variation in northeast Atlantic and Mediterranean populations of Norway lobster, Nephrops norvegicus. Mol Ecol 13:1377–1390
Stepien CA (1999) Phylogeographic structure of the Dover sole Microstomus pacificus: the larval retention hypothesis and genetic divergence along the deep continental slope of the northeastern Pacific Ocean. Mol Ecol 8:923–939
Stockley BM, Rogers AD, Iyengar A, Menezes G, Santos R, Long A (2000) Ten microsatellite loci isolated and developed for the blackspot seabream, Pagellus bogaraveo (Brünnich 1768). Mol Ecol 9:999–1000
Tajima F (1983) Evolutionary relationship of DNA sequences in finite populations. Genetics 105:437–460
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Vis ML, Carr SM, Bowering WR, Davidson WS (1997) Greenland halibut (Reinhardtius hippoglossoides) in the North Atlantic are genetically homogenous. Can J Fish Aquat Sci 54:1813–1821
Waldman JR, Bender RE, Wirgin II (1998) Multiple population bottlenecks and DNA diversity in populations of wild striped bass, Morone saxatilis. Fish Bull 96:614–620
Ward RD, Elliot NG, Grewe PM, Last PR, Lowry PS, Innes BH, Yearsley GK (1998) Allozyme and mitochondrial DNA variation in three species of oreos (Teleostei: Oreosomatidae) from Australian waters. N Z J Mar Freshw Res 32:233–245
Weber DS, Stewart BS, Garza JC, Lehman N (2000) An empirical genetic assessment of the severity of the northern elephant seal population bottleneck. Curr Biol 10:1287–1290
Whitlock M, McCauley D (1999) Indirect measures of gene flow and migration: FST doesn’t equal 1/(4Nm+1). Heredity 82:117–125
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Wright S (1951) The genetical structure of populations. Ann Eugen 15:323–354
Acknowledgements
This study was funded by the European Commission DGXIV/C/1 as study contract 97/081 “Seasonal changes in biological and ecological traits of demersal and deep-water fish species in the Azores”. The authors wish to thank the crew of the “R.V. Arquipélago” for invaluable help and commitment during the sampling for this investigation and Maria Aboim for assistance with sampling on the Portuguese mainland. We also acknowledge provision of facilities for this work by Prof. Ricardo Santos, Departamento Oceanografica e Pescas, University of the Azores, Faial Island; Prof. Patrick Holligan at the School of Ocean & Earth Science, University of Southampton, Southampton Oceanography Centre; Professor Christopher Rapley, British Antarctic Survey. Special thanks for technical assistance and help with laboratory work are due to Mark Dixon, School of Biological Science, University of Southampton, and members of the Molecular Ecology Research Group Ex. School of Ocean & Earth Science, University of Southampton.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J.P. Thorpe, Port Erin
Rights and permissions
About this article
Cite this article
Stockley, B., Menezes, G., Pinho, M.R. et al. Genetic population structure in the black-spot sea bream (Pagellus bogaraveo Brünnich, 1768) from the NE Atlantic. Marine Biology 146, 793–804 (2005). https://doi.org/10.1007/s00227-004-1479-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00227-004-1479-3