
Abstract
The transition characteristics of Quercus rubra wood fiber cell walls during carbonization were investigated by means of scanning thermal microscopy (SThM). Wood samples were carbonized at temperatures from 200 to 600 °C. SThM investigations indicated that the secondary wall (S2) and compound middle lamella (CML) of the wooden fiber cell wall (FCW) could easily be distinguished in the cross section because of the difference in thermal conductivity between them, but when the carbonization temperature was increased, the difference decreased and disappeared at carbonization temperatures above 325 °C. Confocal Raman microscopy studies indicated that the concentrations of cellulose and lignin differed significantly in the S2 and CML, and when carbonized at temperatures higher than 325 °C, the chemical component of the FCW became homogenous. The changes of the dimensions of FCW were also investigated. The FCW became noticeably thinner during carbonization. Compared to native wood, the thickness decreased by 62% when carbonized at 600 °C and the loss of thickness occurred simultaneously with weight loss. The key temperature was 300 °C, with most shrinkage and weight loss occurring before 300 °C.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
When heating wood under oxygen free conditions, the structures and compositions of wood will change to those of charcoal. The transitional characteristics from wood to charcoal warrant investigation (Kwon et al. 2009). Intensive investigations will help to understand the performances of wood carbon materials and optimize the process of wood carbonization. These investigations usually focus on the changes of the structure and physical and chemical properties from native wood to charcoal under different carbonization conditions. The mass loss, charcoal yield, density changes and dimensional shrinkage in the longitudinal, radial and tangential directions have been the most often analyzed parameters (McGinnes et al. 1971; Moore et al. 1974; Slocum et al. 1978). Besides that, many details of structure and property changes at microscale have been revealed. The arrangement of microfibrils in the cell walls of charcoal became smooth and amorphous compared to original wood cell walls (McGinnes et al. 1971). The dynamic mechanical modulus of wood decreased significantly if the carbonization temperature approached 673 K (Moore et al. 1974). Although a vast amount of knowledge has been gained from earlier studies, research continues to progress because new methods have been introduced. X-ray scattering, Raman spectroscopy and X-ray diffraction have been used to investigate changes of the mesoscopic and chemical structures of carbonization wood (Kalliat et al. 1983; Kwon et al. 2009; Paris et al. 2005), nanoindentation has been employed to study the micro-mechanical properties of pyrolyzed wood cell walls (Brandt et al. 2010; Zickler et al. 2006), and transmission electron microscopy (TEM) has been used to investigate changes of the ultrastructure of wood cell walls during carbonization (Brandt et al. 2010; Zollfrank and Fromm 2009). The application of new methods in this field of research will help to reveal more details about the changes of the characteristics from wood to carbonized materials.
Scanning thermal microscopy (SThM) was developed from scanning tunneling microscopy. SThM can image the scanning surface at the microscale based on the thermal conductivity (ThC) properties of materials (Pool 1990). It is well known that differences in ThC can correspond to differences in the structure and components between materials; thus, SThM can be used to investigate changes in materials. In recent years, SThM has been applied to research in the field of wood and wood-based composites. Xu et al. (2016) and Konnerth et al. (2008) employed SThM to study how adhesive penetrate into wood cells. Lee et al. (2009) visualized the interfacial zones between cellulose fibers and polypropylene by SThM. Vay et al. (2013) used SThM to investigate the ThC of wood cell walls, and they analyzed the difference in ThC between different layers of cell walls. These studies indicate that SThM is an efficient technique for the study of wood-based materials at the microscale and can provide details of the microstructures and micro-properties of materials according to the images acquired based on materials’ ThC difference.
Combined with confocal Raman microscopy (CRM), this study used SThM to study the carbonization of wood. The objective was to investigate how the structure and components of wood cell walls transformed with increases in the carbonization temperature.
Materials and methods
Sample wood
Wood samples were obtained from a plate of a 45-year-old red oak (Quercus rubra, Tennessee) tree. The samples were prepared as wood slices with dimensions of 15 mm × 3 mm × 1.5 mm (longitudinal × radial × tangential direction) from a location between the 34th and 35th ring and wood fiber cell were chosen from latewood.
Carbonization treatment
A mini-mite tube furnace (The Lab Depot, Inc., Dawsonville, GA) was used for carbonization. Wood samples were carbonized in a nitrogen atmosphere, and the carbonization temperatures ranged from 200 to 600 °C, with 25 °C intervals before 350 °C and 50 °C intervals from 350 to 600 °C. The heating rate was 10 °C/min, and the holding time at the fixed carbonization temperature was 30 min. After carbonization, specimens were kept in a drying oven to cool to room temperature prior to weight testing for mass loss analysis.
Preparation of specimens
The surface of each sample prepared for CRM and SThM scanning should be very smooth. Scanning errors in the SThM will appear if the local roughness of the surface exceeds 30 nm (McConney et al. 2010). To smooth the scanning area of the wood samples, a Leica Ultracut R ultramicrotome was used to prepare the samples. Original wood can be smoothed by the ultramicrotome directly, but after carbonization, wood becomes very crisp and it is very difficult to fix sample in the hold of the ultramicrotome. To solve this problem, Spurr’s resin (which can be easily prepared and is commonly utilized in biomaterials micro-characteristics research) was used to embed the carbonization wood samples (Meng et al. 2013). After embedding in the epoxy resin, the samples were treated in a vacuum-pressure oven for 8 h and the temperature was fixed at 70 °C. Thus, in this process, air was removed from the embedding substance and the resin was cured. Then, the cured samples were fixed on the holder for microtoming. During the microtoming process, a glass knife was used to pretreat the sample surface, and each time, a 500-nm section was cut from the surface. Furthermore, a diamond knife was used to carry out the final microtoming, and each time, a 100-nm section was cut from the surface of the samples. The progression of the sample preparation is shown in Fig. 1.

Scheme of sample preparation
Confocal Raman microscopy (CRM)
An XploRA™ PLUS confocal Raman microscope (Horiba Ltd., Kyoto, Japan) was used to detect changes in the compositions of carbonized wood cell walls. Wood samples were placed on a piezoelectric x, y micro-motional stage, and a polarized laser λ = 785 nm (2 mW) was used as the excitation source. To obtain an image of the wooden fiber cell wall (FCW), a region of the FCW was selected for scanning by optical microscopy; then, the micro-motional stage was moved by 0.5 μm at each step, and the CRM scanned the points of the selected region with each movement of the stage at an acquisition time of 10 s. The confocal aperture and slit width were set to 300 and 100 μm, respectively. Raman spectra were obtained over specific band ranges (200–2000 cm−1). LabSpec 6 software was used to set the scanning parameters, process data and generate Raman images of the scanning regions.
Scanning thermal microscopy (SThM)
SThM was used to investigate the ThC properties of materials at the microscale. The microscopy has two operation modes, ThC contrast microscopy (CCM) and temperature contrast microscopy (TCM). When scanning in CCM mode, the probe maintains a constant temperature, and if the probe comes into contact with a high ThC area, the current flowing through the probe is increased to maintain a constant temperature. Meanwhile, when the probe comes into contact with a low ThC area, the current is decreased. Thus, CCM mode can image the scanning area based on the changes in probe current. When using TCM mode, a constant current is applied to the probe and the resistance of the probe is measured. The ThC of the scanning area can affect the temperature of the probe, and the temperature of the probe corresponds to its resistance. Thus, in TCM mode, SThM can image the scanning area based on changes in the probe’s temperature. This study used CCM mode to image the cross sections of the wooden fiber cell walls of carbonization wood. In this mode, probe current images can be obtained and the current represents the local ThC of the scanning location in the image. SThM scanning was performed using an XE100 atomic force microscope (XE-100, PSIA Corp., Sang-Daewon-dong, Korea). The scanning parameters of SThM were set as 0.35 Hz scanning rate and 0.5 mA current servo.
Results and discussion
Compositional changes of wooden fiber cell walls (FCWs) by CRM
CRM was used to investigate the chemical compositions of FCWs and to determine how the compositions changed at different carbonization temperatures. Figure 2 shows images of FCWs obtained through CRM. The area in the white box is the scanned area in Fig. 2a. From previous works (Agarwal 2006; Lewis et al. 1994; Gierlinger and Schwanninger 2006; Zhang et al. 2012), Raman bands at near 1600 and 1650 cm−1 can be assigned to lignin; thus, the band region from 1520 to 1680 cm−1 was used to generate images of the scanned area, as shown in Fig. 2b. In Fig. 2b, the brighter regions represent higher lignin concentrations, such as in CML and cell corners of FCWs, while the darker region was mainly the S2 of the FCWs. When using the band region from 1050 to 1180 cm−1 (which was mostly assigned to cellulose and hemicellulose) to generate the image, as shown in Fig. 2c, the cell corners and CML regions appear darker than S2 because S2 has higher cellulose and hemicellulose concentrations (Agarwal 2006; Gierlinger and Schwanninger 2006). Using LabSpec 6, Raman spectra of regions 1, 2 and 3, in small boxes in Fig. 2a, which corresponded to CML, S2 and CC, respectively, were acquired as shown in Fig. 2d. Raman spectra indicated that the intensities of the S2 bands at 1600 and 1650 cm−1 were higher than those of the CC and CML bands in the region from 1050 to 1180 cm−1. Both Raman images and spectra showed a clear difference in compositions between S2, CML and CC of the FCWs, compared with S2, CML and CC had higher concentrations of lignin and lower concentrations of cellulose and hemicellulose.

Oak wooden fiber cell wall imaged by CRM: a selected scanning area; b intensity of the lignin band (1520–1680 cm−1); c intensity of cellulose and hemicellulose band (1050–1180 cm−1); d Raman spectra of the area in small boxes 1 (CML), 2 (S2), 3 (CC) of the scanning area
This study attempted to use CRM to investigate how the compositions of FCW changed when wood samples were carbonized at temperatures of 200, 225, 250, 275 and 300 °C. However, due to high noise, Raman spectrum signals could not be detected for wood samples carbonized at these temperatures. It was considered that the noise was caused by fluorescence background due to lignin compositions of wood (Albinsson et al. 1999; Donaldson 2013), because at these temperatures, cellulose and hemicellulose pyrolyzed first, increasing the relative concentration of lignin. When investigating the sample pyrolyzed at 325 °C, Raman signals could be detected as shown in Fig. 3. The square region in the white box with brighter color was the area scanned by CRM in Fig. 3a. The Raman spectrum in Fig. 3c shows the compositions of FCWs in the small box in the scanning area. The spectrum shows that compared with native wood, after carbonization at 325 °C, the Raman bands assigned to cellulose, hemicellulose and lignin could not be identified, and at bands near 1360 and 1580 cm−1 the Raman peaks seem to have appeared. Bands near 1360 and 1580 cm−1 were D-band and G-band assigned to graphitic carbon. Figure 3b is the image generated by using band 1580 cm−1. In this image, the S2, CC and CML of FCWs could not be distinguished, and the compositions of the whole FCW became homogenous. Samples carbonized at 350 and 400 °C were also investigated. Figure 4a, c shows the scanned area with bright color in the white box that was generated by using band 1580 cm−1. In Fig. 4a, c, the brightness shows differences in areas 1 and 2 in the small black boxes. For both the 350 and 400 °C samples, Raman spectra of areas 1 and 2 were acquired, as shown in Fig. 4b, d. The spectra indicated that the Raman bands of the two areas were almost the same except that the intensity was different. It was assumed that the difference in intensity may be caused by excessive roughness in the microtome section of the samples (Agarwal 2006). Figure 4b, d also indicates that with temperature increases, the D-band and G-band become more visible. From the CRM study, it could be concluded that when the carbonization temperature surpassed 325 °C, the compositions difference between different layers of wood cell walls became invisible and graphitic carbon began to appear in carbonized wood.

CRM investigation of oak wooden fiber cell walls pyrolyzed at 325 °C: a selected scanning area; b intensity of the band at 1580 cm−1; c Raman spectra of the area in the small box of the scanning area

CRM investigation of oak wooden fiber cell wall pyrolyzed at 350 and 400 °C: a the selected scanning area of cell wall pyrolyzed at 350 °C is shown by integrating the 1580 cm−1 band; b Raman spectra of region 1, 2 of the cell wall pyrolyzed at 350 °C; c selected scanning area of the cell wall pyrolyzed at 400 °C, as shown by integrating the 1580 cm−1 band; d Raman spectra of region 1, 2 of the cell wall pyrolyzed at 400 °C
Examination of the carbonization process of wooden fiber cell walls by SThM
Figure 5a shows a SThM probe current image of the embedded native wood fiber cell. Contrasting with the scale bar on the left of the probe current image, the brighter color corresponds to a higher current and represents a higher ThC than that of the darker areas. Figure 5a shows that the cell lumens of the embedded wood cell were full of a lower ThC substance. This was the embedding resin that entered the wood structure during treatment in the vacuum-pressure oven. Figure 5b shows the wood cell without embedding resin. When focusing on the cell wall, Fig. 5a, b indicates that the ThC of S2 was higher than that of CML, and the difference in current between the two layers was close to 1 μA, as shown by the line profile. Vay et al. (2013) investigated why S2 has higher ThC than CML, and they assumed that the orientation of cellulose in the S2 layer was nearly parallel to the axial direction, so when the SThM probe heated a cross section of S2, heat transferred along the cellulose fibers. In CML, the content of lignin was higher than in S2 and components were arranged in a disordered fashion, so S2 has an increased thermal conductivity compared to CML.

SThM probe current image (20 μm × 20 μm); a wooden fiber cell with epoxy resin embedding and b wooden fiber cell without epoxy resin embedding
Figure 6 shows the probe current images of wood samples carbonized at different temperatures. As the Figure shows, wood cells kept their original characteristics at lower carbonization temperatures. S2 and CML could still be differentiated before 300 °C in the probe current image by their difference in ThC. When the carbonization temperature was higher than 300 °C, the ThC of the wood cell wall became homogeneous and the ThC difference between S2 and CML disappeared. It is well known that cellulose, hemicellulose and lignin are the three main constituents of wood and that the wood thermal degradation process is mainly composed of the thermal degradation of these three constituents. In the thermal degradation of wood, hemicellulose degrades first, and the temperature interval is from approximately 180 to 240 °C. Then, cellulose degrades from approximately 240 to 310 °C and lignin degrades from approximately 310 to 400 °C (Zeriouh and Belkbir 1995). In previous studies, Paris et al. (2005) used wide-angle X-ray scattering, small-angle X-ray scattering and Raman spectroscopy to study the transition characteristics of a wood biopolymer during the carbonization process. They noted that for the thermal degradation of the crystal structure, there was no difference between cellulose microfibrils and polyoses/lignin in the temperature interval between 580 and 620 K. TEM studies demonstrated that the orientation of cellulose microfibrils could be observed before 250 °C, while the CML continued to fade up to 300 °C. Above 300 °C, the whole cell wall began to appear to be isotropic (Brandt et al. 2010; Zollfrank and Fromm 2009). The change in composition and structure of cell wall during pyrolysis led to the physical properties change. Nanoindentation testing indicated that the modulus of wood cell walls in the axial, radial, tangential directions had obvious differences, but when the carbonization temperature increased to 300 °C, the moduli of the three directions tended to be the same (Brandt et al. 2010). Vay et al. (2013) determined the ThC differences between S2 and CML were caused by differences in the orientation of cellulose. The current CRM studies showed that the compositions of cell walls become homogenized, and it was hard to detect the Raman band assigned to cellulose when the carbonization temperatures reached 325 °C. Carbonized at 325 °C, the decomposition of cellulose caused the orientational characteristics in S2 to vanish. Thus, the difference in ThC between different layers of the cell wall disappeared.

Probe current image of carbonized wood samples at different carbonization temperatures
Dimensional shrinkage of wooden fiber cell walls of carbonized wood
Carbonization does not change the anatomical structure and texture of wood (Ehrburger et al. 1982; Vafaeenezhad et al. 2013). As shown in Fig. 6, the whole structure of the wood cell maintained its integrity even at carbonization temperatures up to 600 °C, but the tissues of wood shrank during carbonization (McGinnes et al. 1971; Ehrburger et al. 1982; Kwon et al. 2009). Many previous studies focused on the shrinkage of carbonization wood. Most studies were performed at the macroscale; usually, wood cubes were prepared as samples and dimensional changes of tangential, radial and longitudinal directions of the cubes from wood to charcoal were measured. Dimensional shrinkage of wood is a complex process. To reveal more details about wood shrinkage during carbonization, other researchers performed investigations at the microscale. Earlier studies performed by McGinnes et al. (1971), by means of SEM, showed that early wood vessels shrank by 25.6 and 38.9% in the tangential direction when samples were carbonized at 400 and 800 °C, respectively. The shrinkage values were greater than the values measured for macrocubes (McGinnes et al. 1971). The difference between the values was caused by different cells and tissues shrinking differently; usually, broad rays shrink less (Slocum et al. 1978). Ehrburger et al. (1982) noted that the cell walls of vessels, rays and fibers were all thinned and the anisotropic shrinkage deformed the shapes of cell lumens. Kwon et al. (2009) also investigated how vessels of Quercus variabilis shrunk during carbonization, and they found that when carbonization temperatures were above 350 °C, vessels shrunk noticeably.
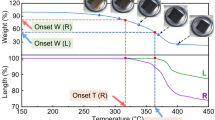
The present study attempted to quantitatively analyze the shrinkage of wooden fiber cell walls during the carbonization process. Figure 7 shows that two adjacent wood cells were selected and a straight line was drawn from one cell lumen to the other one by XEI. The straight line was maintained perpendicular to the CCM as much as possible. Then, a pair of cursors was used to measure the length of the selected line. As shown in Fig. 7, the cursors were marked as two arrows in the line, and the length between the two arrows could be obtained from the line profile. For example, the length between the two arrows in the red line was 8.281 μm. This study assumed that this length was the thickness of the cell walls of the two selected wood cells. Using this method, the thickness of wood cell walls at different carbonization temperatures was measured. For each sample, more than ten areas were scanned, and 50 thickness values were obtained. The thickness of the native wooden fiber cell wall was 8.23 μm, with a standard deviation of 1.71 μm, and when carbonized at 600 °C, the thickness decreased to 3.14 ± 0.65 μm. The thickness decreased by more than half after carbonization. The shrinkage process of the wooden fiber cell walls from native to carbonized at 600 °C is shown in Fig. 8. During carbonization, the thickness shrank noticeably below 300 °C. The thickness at 300 °C was 4.07 ± 0.99 μm, a decrease of approximately 50%. After 300 °C, the cell wall continues to shrink, but the decrease tended to occur more slowly. From 300 to 450 °C, the thickness decreased by approximately 12%, and after 450 °C, the cell wall stopped shrinking until reaching 600 °C. The mass loss of the samples in the carbonization process was also measured. Figure 8 shows that the process of cell wall shrinkage and mass loss was quite similar. The characteristic temperature of mass loss was also 300 °C, and approximately 65% of weight was lost at 300 °C. After that, the mass loss slowed, and between 300 and 600 °C, only approximately 15% mass loss occurred. Combined with ThC analysis, the above results indicate that during carbonization, degradation causes changes in the composition and structure of cell walls, which in turn leads to physical property changes.

SThM image indicating the measurement of the thickness of a wood cell wall

Change of the thickness of wood cell walls and weight of wood samples at different carbonization temperatures
Conclusion
By means of SThM investigations, this study concluded that when the carbonization temperature reached 325 °C, the microstructure and properties of wood cell walls changed dramatically. The ThC of CML and S2 of native wood cell walls were different, but when the carbonization temperature reached 325 °C, the difference vanished. This was caused by changes in the composition and microstructure of the cell walls during carbonization. CRM investigations revealed that when wood sample carbonized at 325 °C, Raman bands assigned to cellulose, hemicellulose and lignin disappeared and the composition of wood cell walls became homogenous in S2, CML and CC. Carbonization decreased the thickness of wood cell walls. From native wood to 600 °C carbonized wood, the thickness decreased by 62%. The key temperature was 300 °C because before 300 °C, the thickness shrank by 50%. Meanwhile, the ThC changes and cell wall thickness decreases occurred simultaneously with mass loss.
References
Agarwal UP (2006) Raman imaging to investigate ultrastructure and composition of plant cell walls: distribution of lignin and cellulose in black spruce wood (Picea mariana). Planta 224:1141–1153
Albinsson B, Li S, Lundquist K, Stomberg R (1999) The origin of lignin fluorescence. J Mol Struct 508:19–27
Brandt B, Zollfrank C, Franke O, Fromm J, Göken M, Durst K (2010) Micromechanics and ultrastructure of pyrolysed softwood cell walls. Acta Biomater 6:4345–4351
Donaldson L (2013) Softwood and hardwood lignin fluorescence spectra of wood cell walls in different mounting media. Iawa J 34:3–19
Ehrburger P, Lahaye J, Wozniak E (1982) Effect of carbonization on the porosity of beechwood. Carbon 20:433–439
Gierlinger N, Schwanninger M (2006) Chemical imaging of poplar wood cell walls by confocal Raman microscopy. Plant Physiol 140:1246–1254
Kalliat M, Kwak CY, Schmidt PW, Cutter BE, McGinnes EA (1983) Small angle X-ray scattering measurement of porosity in wood following pyrolysis. Wood Sci Technol 17:241–257
Konnerth J, Harper D, Lee S-H, Rials TG, Gindl W (2008) Adhesive penetration of wood cell walls investigated by scanning thermal microscopy (SThM). Holzforschung 62:91–98
Kwon SM, Kim NH, Cha DS (2009) An investigation on the transition characteristics of the wood cell walls during carbonization. Wood Sci Technol 43:487–498
Lee SH, Wang S, Endo T, Kim NH (2009) Visualization of interfacial zones in lyocell fiber-reinforced polypropylene composite by AFM contrast imaging based on phase and thermal conductivity measurements. Holzforschung 63:240–247
Lewis IR, Daniel NW Jr, Chaffin NC, Griffiths PR (1994) Raman spectrometry and neural networks for the classification of wood types—1. Spectrochim Acta A Mol Spectrosc 50:1943–1958
McConney ME, Singamaneni S, Tsukruk VV (2010) Probing soft matter with the atomic force microscopies: imaging and force spectroscopy. Poly Rev 50:235–286
McGinnes EA Jr, Kandeel SA, Szopa PS (1971) Some structural changes observed in the transformation of wood into charcoal. Wood Fiber Sci 3:77–83
Meng Y, Wang S, Cai Z, Young TM, Du G, Li Y (2013) A novel sample preparation method to avoid influence of embedding medium during nano-indentation. Appl Phys A 110:361–369
Moore GR, Blankenhorn PR, Beall FC, Kline DE (1974) Some physical properties of birch carbonized in a nitrogen atmosphere. Wood Fiber Sci 6:193–199
Paris O, Zollfrank C, Zickler GA (2005) Decomposition and carbonisation of wood biopolymers—a microstructural study of softwood pyrolysis. Carbon 43:53–66
Pool R (1990) The Children of the STM: the Nobel Prize-winning scanning tunneling microscope has inspired a whole generation of imaging devices that use everything from magnetic forces to sound waves to examine samples. Science 247:634
Slocum DH, McGinnes EA Jr, Beall FC (1978) Charcoal yield, shrinkage, and density changes during carbonization of oak and hickory woods. Wood Sci 11:42–47
Vafaeenezhad H, Zebarjad SM, Khaki JV (2013) Intelligent modeling using fuzzy rule-based technique for evaluating wood carbonization process parameters. Int J Adv Manuf Technol 68:1471–1478
Vay O, Obersriebnig M, Müller U, Konnerth J, Gindl-Altmutter W (2013) Studying thermal conductivity of wood at cell wall level by scanning thermal microscopy (SThM). Holzforschung 67:155–159
Xu D, Zhang Y, Zhou H, Meng Y, Wang S (2016) Characterization of adhesive penetration in wood bond by means of scanning thermal microscopy (SThM). Holzforschung 70:323–330
Zeriouh A, Belkbir L (1995) Thermal decomposition of a Moroccan wood under a nitrogen atmosphere. Thermochim Acta 258:243–248
Zhang ZH, Ma JF, Ji Z, Xu F (2012) Comparison of anatomy and composition distribution between normal and compression wood of Pinus bungeana Zucc. revealed by microscopic imaging techniques. Microsc Microanal 18:1–8
Zickler GA, Schöberl T, Paris O (2006) Mechanical properties of pyrolysed wood: a nanoindentation study. Philos Mag 86:1373–1386
Zollfrank C, Fromm J (2009) Ultrastructural development of the softwood cell wall during pyrolysis. Holzforschung 63:248–253
Acknowledgments
The authors thank the Youth Innovation Fund of Nanjing Forestry University (Grant No. CX2016014), National Natural Science Foundation of China (Grant Nos. 31570552, 31470590) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xu, D., Ding, T., Li, Y. et al. Transition characteristics of a carbonized wood cell wall investigated by scanning thermal microscopy (SThM). Wood Sci Technol 51, 831–843 (2017). https://doi.org/10.1007/s00226-017-0919-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00226-017-0919-4