
Abstract
A fast and reproducible method for estimating the sulfur ester content of cellulose nanocrystals (CNCs) prepared by a pilot plant (Alberta Innovates Technology Futures—Alberta, Canada) is discussed. CNCs from sulfuric acid hydrolysis form electrostatically stabilized suspensions because of the introduction of sulfate ester groups onto the surface of the crystallites during the chemical production. The surface charge of the crystals and indirectly the sulfur ester content has implications when considering several applications. In the past, tedious and time-consuming methods such as X-ray photoelectron spectroscopy, X-ray diffraction, and elemental analysis have been used to estimate the sulfur content of CNCs. As of late, conductometric titrations have surpassed these techniques for estimating the sulfur content of CNC. In this communication, a technique is explored that is specific, fast, and reproducible across a number of samples for determining the quantity of C–O–S linkages in CNC samples. The method is based on integrating key peaks characteristic of sulfur functionalities obtained via Fourier transform infrared spectroscopy. The samples were analyzed as obtained, and no time was needed for extra sample cleanup prior to analysis. Finally, good correlation was obtained between the results obtained for the different samples and comparable to the results obtained from the widely used conductometric method.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cellulose nanocrystals (CNCs) are predominantly produced by a controlled acid hydrolysis process which removes the accessible non-uniform sections of cellulose to produce highly crystalline, nm-sized particles that are rod shaped (Habibi et al. 2010). The natural abundance of the feedstock required for producing CNCs and the green footprint of the many possible products has ignited much attention in this research space. In fact, the preparation of CNCs from treating wood with sulfuric acid was first reported by Ranby (1949), but the research area has seen the publication of close to 160,000 papers pertaining to the CNCs in the last decade (Google Scholar).
CNCs produced via sulfuric acid hydrolysis are electrostatically stabilized in aqueous suspension by the anionic sulfate ester surface groups that are introduced during the reaction (Beck et al. 2012). The polydispersity and average sizes of the particles are influenced by the starting material and reaction parameters employed. In this case, the conditions of acid concentration, reaction time, agitation, and feedstock loading were optimized and controlled by the operators of the pilot plant. This was done to prepare CNCs with the correct particle range and surface properties. The procedure employed currently mirrors the conditions reported by the Gray laboratory (temperature of 45 °C, 64 wt% sulfuric acid, 25–45 min reaction time and pulp consistency of 10%) (Abitbol et al. 2013).
The sulfur ester content has been reported to have implications in dispersibility, which indirectly will have implications in properly storing and transporting the material (Jiang et al. 2010; Kloser and Gray 2010). Additionally, recent interest (Habibi et al. 2010; Kiziltas et al. 2016; de Castro et al. 2016; Qiao et al. 2016) and rapid commercialization of CNCs across Canada (pilot plants in Alberta, Saskatchewan, and Quebec) warrant a holistic characterization study detailing the properties of this material, and what factors contribute to variation in these properties. Hence, accurate determination of sulfur content is important for reproducibility across research labs, comparison across the literature, and to fully understand the behavior of CNCs, not limited to self-assembly and phase separation (Abitbol et al. 2013).
According to Beck-Candanedo et al. (2005), the sulfur ester content of acid hydrolyzed samples is used to compare hydrolysis conditions, to assess or determine the colloidal stability, and charge content of the material. To date, conductometric titration is one of the most commonly used methods for determining the sulfate ester content of CNC samples. This method, with prior sample cleanup via a centrifugation, extensive dialysis, and finally tangential flow filtration, can produce more reproducible results than X-ray photoelectron spectroscopy and elemental analysis (via atomic absorption spectroscopy). To that end, the titrimetric method does not require the elaborate calibration and expense associated with the above-listed instruments (Abitbol et al. 2013). Conductometric titrations work on the principle of quantifying all protonated species in the suspension. These include the R–COOH and R–O–SO3H groups. The method relies on a few key assumptions: (1) There is no free acid in the systems, (2) the acid groups are sulfate esters grafted onto the surfaces of the CNCs, (3) all the sulfate ester groups are in the acidic form, and (4) all ester groups are accessible to the titrant (Abitbol et al. 2013). As a result, as reported by Abitbol et al. (2013) prior to any conductometric titration, there needs to be extensive sample cleanup. Hence, a fast and reproducible method that targets just the sulfur functionalities needs to be developed.
As a result, an infrared method is proposed for quantifying the specific sulfur ester content of the CNC samples obtained from a commercial pilot plant. The results were compared to those obtained using X-ray photoelectron spectroscopy, elemental analysis, and conductometric titration. To aid better resolution for the infrared method, dextran sulfate (DS) was added to the CNC samples at different ratios. Different ratios of CNC with or without DS were studied using the above methods. Specifically, for the infrared method, signatures characteristic of the –C–O–S and total –C–H was studied and reported. A correlation was found between these fractions and the % C–O–S. The results for all the samples studied were within the range of the values obtained via the conductometric titrations.
Materials and methods
Materials
KBr (Mol. wt 119 g/mol, >99% purity) used for all IR analysis was obtained from Sigma (Minnesota, USA). Sodium hydroxide (Mol. wt 40 g/mol, >98% purity), sulfuric acid (Mol. wt 98.08 g/mol−1, >99.99%), sodium chloride (Mol. wt 58.44 g/mol, >99.0% purity), and dextran sulfate (Mol. wt ≈ 40,000, with a sulfur content of 13–15%) were bought from Sigma (Minnesota, USA). A Dowex Marathon C-10 cation exchange resin was sourced from Dow Chemical Company (Alberta, Canada).
CNC preparation
Samples of cellulose nanocrystals (CNC) were obtained from a pilot-scale facility located at Alberta Innovates Technology Futures, Edmonton, Canada. The different samples were stored at room temperature until characterization. The CNC samples (freeze dried, spray dried) were obtained via hydrolysis of dissolving pulp. Reaction parameters of 120 min, 45 °C, stirred at 200 rpm, and a sulfuric acid concentration of 64% were used to produce the samples. An acid:pulp consistency of 12:1 was used for all experiments.
A standard CNC (CNC_Std) sample obtained from sulfuric acid hydrolysis of softwood pulp followed by spray drying was kindly supplied by National Research Council of Canada (via Alberta Innovates Technology Futures). Prior to characterization, no sample cleanup was done, to aid in developing a method that was robust to any sort of contaminations that may be present in the samples.
Determination of sulfur content
As previously mentioned, CNC samples were characterized using X-ray photoelectron spectroscopy, conductometric titrations, elemental analysis, and infrared spectroscopy. Prior to these analyses, CNC samples were mixed with dextran sulfate (DS), at varying ratios. The ratios were chosen based on the approximate sulfur groups in the DS ratio and the amount of CNC that can be added without affecting the dry mixing of these two chemicals. The ratios used are provided below in Table 1.
The different samples evaluated included CNC suspensions dried using freeze-drying (CNC_FD), spray drying (CNC_SD), and a standard CNC (CNC_Std).
X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy measurements were performed with a Kratos Ultra 165 X-ray Photoelectron Spectrometer with energy spectra ranging from 0 to 1000 eV. Samples were scanned for C1s (284 eV) and O1s (530 eV). The low-resolution survey scans were taken with a 50-eV step and 160 eV analyzer pass energy. For each sample, the CNC samples were wrapped with aluminum foil and compressed between nickel plates. The entire sample assembly (4 total samples) was then placed in the sample chamber for subsequent analysis. In these specific instances, an eight-channel multi-detector was employed for analysis.
All raw data obtained were processed using CasaXPS software (Casa Software Ltd, UK), provided by the home laboratory (nanoFab–University of Alberta). C1s spectra were analyzed with a Gaussian product function, by maintaining the full width half maximums (FWHMs) of all components to within the range of 1.200–1.600 (George et al. 2014). The XPS data provided an estimate of the S content.
Conductometric titration
The protonated groups of the different samples were evaluated using conductometric titration (Orion conductivity cell 018010, K = 0.987 cm−1). The conductometric method used was similar to that reported by Abitbol et al. (2013). It should be noted the authors’ team did not subject their samples to the cleanup used by those researchers. The reason for this step was because the authors were interested in determining the concentration of the sulfate half ester content of the material purified by the pilot plant and not subjected to further cleanup. This will enable the timely determination of the sulfate ester content while eliminating the cost and manipulation associated with further cleanup. In addition, one clear distinction was that the cited team produced their own CNC, and hence, had to subject the samples to cleanup. The samples analyzed in this communication were produced at a large-scale facility and subjected to sample cleanup (as detailed previously).
Approximately 0.1 g sample (CNC and DS in different ratios) was dispersed in 3 mL of distilled water. The next step involved adding NaCl (130 g of a 1-mM solution) and a solution of pre-calibrated NaOH (2.5 mM, 20 mL total volume added in increments). The conductivity values (μS/cm) were then corrected for the different dilution effects and plotted against the added volume of NaOH. The titration curves were characterized with two linear sections; the first depicting an initial decrease in conductivity with added NaOH as the protons associated with the sulfate ester groups are consumed, and secondly, an increase in conductivity with added NaOH which occurs when all the charged groups have been fully neutralized. The equation provided below was used for all calculations.
In Eq. 1, the concentration of NaOH (C NaOH), volume of NaOH at the inflection point (VolNaOH), mass of the suspension (m susp), concentration of suspension (C susp), and the molecular weight of sulfur (\( {\mathcal{M}}w \)) were used to calculate the % S for each sample. The inflection point was determined for each set of samples using line fitting. The intersection of the two linear sections gave an estimate of the volume of NaOH required to fully neutralize the sulfur ester for each sample.
In cases where there were slope variations, especially in samples with low DS content, increasing the amount of sample and/or reducing the dilution factor resulted in more data points from which the equivalence point can be estimated. The values of % S were reported as average ± SD.
Elemental analysis
The CHNS content of the different samples was determined using a Thermo Flash 2000 CHNS–O. An autosampler was used to place approximately 10 mg (contained in a tin cup) into a vertical quartz tube called the combustion reaction (heated up to 1000 °C). The autosampler was continuously swept with helium gas. The upper portion of the combustion reactor was filled with an oxidation catalyst (tungsten oxide on alumina), and the lower portion was filled with pure reduced copper wires. Once the sample entered the reactor, a fixed volume of pure oxygen was added to the helium carrier gas. The added oxygen created a highly oxidizing atmosphere, which completely burnt the sample and the tin cup container. The C, H, N, and S present in the sample were converted to CO2, H2O, NO x , and SO2, respectively. The carrier gas then swept the combustion products onto a chromatographic column (Porapak QS, 4 mm ID, 2 m long), where the gases were separated. The detection of the individual gases was achieved with a thermal conductivity detector.
The Eager Xperience software program was used to convert the output signal from the thermal conductivity detector into a chromatogram that displayed the CO2, H2O, NO x , and SO2 peaks. The software then generated the area count data used to calculate the percentage of N, C, H, and C present in the sample.
The instrument was calibrated using at least three samples of an organic analytical standard (OAS) at the start of each analysis day. The standard was accurately weighed directly into tin cups using a Mettler XP2U microgram balance, which is readable to 0.1 µg. The standards span the range from ~0.5 to ~3 mg. Once the standard runs were completed, linear regression analysis was performed on the peak areas for each of the elements (C, H, N, and S) versus the milligrams of each element contained in the standards. The amount of each element was calculated by multiplying the known proportion of each element in the standards by the amount of sample. The equation of the regression line that fits these points was used to calculate the amount of C, H, N, and S in the samples. An acceptable regression line for each element has an R 2 value of at least 0.999. For R 2 values less than these, another set of standards was run and a new calibration curve was obtained. To monitor the accuracy of the instrument, a sample of the OAS was periodically tested (this was done every time; 4 experimental samples were tested). The calibration check was treated as an unknown. The check samples were weighed such that they are within the calibration range of the standards used. The measured values for these standards should produce results that were within ±0.3% of the theoretical values. If they did not agree within ±0.3%, the system was inspected for possible malfunction and if necessary repairs were performed on the instrument and the samples re-analyzed.
Triplicate runs were done for each of the CNC–DS samples. The average and SD of these were reported.
Infrared spectroscopy
Fourier transform infrared spectroscopy (Nicolet 8700 FTIR Spectrometer) was used to investigate the sulfur signatures of the different CNC samples. Samples were conditioned for 4 h at 50 °C and mixed with KBr to minimize the effects of moisture in the samples. FTIR spectra were measured from 4000 to 400 cm−1 at a resolution of 4 cm−1. Samples of each ratio were mixed with KBr to a ratio of 1:30 and pressed in a pellet holder. Each sample was pressed for exactly 3 min at approximately 7000 psi. The samples were then subsequently subjected to infrared radiation. Nitrogen gas was pumped continuously during the reaction to provide an inert atmosphere. Triplicate runs were made on each sample, with each sample scanned for 32 times.
Further sample purification/cleanup
To determine whether the samples obtained from AITF and NRC are laced with minute contaminants that potentially affected the % S values, it was decided to subject the samples to ion exchange treatment. A procedure reported by Abitbol et al. (2013) was closely followed. A Dowex Marathon C-10 strong acid cation exchange resin was used. Briefly, before use, the resin was rinsed with ethanol until the filtrate was no longer pink and then dried. Approximately 0.125–0.150 g resin/g suspension for 24 h was used for all purifications.
In each case, the resin was added to vials of suspensions with no agitation (to prevent fragmentation of resin bead—Abitbol et al. 2013). Each sample of CNC was analyzed in batches of three, and the values reported are the mean ± SD.
Sample desulfation and % S quantification
After understanding the factors impacting the results of each method, it was thought it was imperative to examine samples with varying % S and see how each technique responds. Acid-catalyzed desulfation was chosen because it is fast and reproducible. In all cases, 1 g of lyophilized CNC was treated with 200 mL of 0.025 N HCl at 80 °C for 30, 60, 90, 120, and 150 min. The samples were centrifuged, dialyzed, and sonicated. The method used was adopted from Gu et al. (2013).
Statistical analysis
Statistical analyses were done as outlined by George et al. (2014). Treatments were done in triplicates and results reported as mean ± SD. The statistical analyses of the data were conducted using the statistical software package SAS version 9.4. The Kruskal–Wallis test was applied to the data populations involved, with a 95% confidence level (P < 0.05).
Results and discussion
X-ray photoelectron spectroscopy estimation of sulfur content
All % S for the different samples analyzed will be presented as mass percentage. XPS was used to determine the abundance of S for the different sample sets outlined in Table 1. Survey scans for spray dried CNC samples and DS are presented in Fig. 1. In addition, the S2p peaks are presented in Fig. 2. The S2p peaks were within the concentration limit in most instances. C1s (284 eV) and O1s (543 eV) peaks were very distinct in the survey scans for all the CNC, DS, or CNC + DS samples. On the other hand, the S2p (163 eV) peak was smaller in all the scans and decreased as the amount of CNC increased. It should be noted S2p was detected in all samples analyzed, unlike the results reported by Gu et al. (2013), when they quantified the sulfate esterification content of cellulose nanowhiskers. A few plausible reasons that the S content was higher in this communication (Gu et al. 2013 reported S % values <0.23%) were the longer reaction time (longer exposure to the sulfuric acid) and more rapid agitation (better reaction), different starting material (they used cotton), and a lower acid: starting material ratio used in their study.


Survey scans for the different ratios of CNC to DS for CNC_SD: a 0% CNC, b 25% CNC, c 50% CNC, d 75% CNC, and e 100% CNC. Peaks of interest were S2p of sulfur (163 eV), C1s of carbon (284 eV), and O1s of oxygen (543 eV)


S2p scans for the different ratios of CNC to DS for CNC_SD: a 0% CNC, b 25% CNC, c 50% CNC, d 75% CNC, and e 100% CNC
The S2p peaks presented in Fig. 2 were characterized with similar shape, which allowed for accurate curve fitting, hence better quantification.
The results for the % S for the different samples are presented in Fig. 3. CNC_SD, CNC_FD, and CNC_Std were characterized with S contents (%) of 0.74 ± 0.02, 0.85 ± 0.04, and 0.58 ± 0.05, respectively. The values reported here are higher (except for CNC_Std) than the original samples reported by Jiang et al. (2010), when they studied the production of acid-catalyzed and solvolytic desulfation of H2SO4-produced CNCs. When compared, the only difference in conditions was the longer reaction time employed by the pilot plant at Alberta Innovates Technology Futures. The R 2 values were within acceptable ranges for the determination of the S2p content of the different samples.

% S for the different samples of CNC to DS as determined by XPS: a CNC_SD, b CNC_FD, and c CNC_Std
In the end, XPS gave a total S content of the samples and not specific sulfur ester quantities. Hence, there is a continuous search for a method better suited to determine the specific % sulfate ester groups of CNC samples.
In the end, the values for the systems decreased because the CNC decreased in that order. The DS has 10–15% S per packaging details, and as the CNC is increased to DS, the % S will decrease significantly because the CNC samples have less sulfur than DS.
Conductometric titration
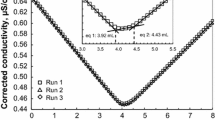
Conductometric titrations were used to estimate the concentration of S of the different CNC samples. The different samples comprising varying ratios of CNC:DS were dispersed in distilled water, and then, a fixed amount of NaCl was added (as detailed in the experimental section). As mentioned before, the authors are interested in assessing the properties of the samples as received. In another section, the effect of ion exchange purification (full protonation using a strong acid resin) on the % S values was investigated. Each plot for the samples was characterized with two distinct linear portions, and the point of inflection was taken as the volume of NaOH required to completely neutralize the sulfur ester groups. A depiction of this is presented in Fig. 4.

Plot of corrected conductivity against volume (mL) of NaOH added. Note, for the sample run 5b (broken line with triangle), lines are extended from the linear portion to give a point of inflection (arrow), which gives the volume of NaOH required for complete neutralization
It should be noted Fig. 4 is being used just for explanation purposes and is not used for calculation of any values. Graphs used for calculations consisted of >120 data points and can be cumbersome when trying to explain a concept or trend.
The above experiment was done for each combination of CNC:DS, and a plot was made of the percentage sulfur (% S) calculated based on the equation given in section “Conductometric titration.” Figure 5 illustrates the plot for the different CNC sample types (CNC_SD, CNC_FD, and CNC_Std) and the corresponding % S at varying abundance. The corresponding equations for the best fitted straight lines and the R 2 value are also provided. Based on the calculations done, CNC_SD, CNC_FD, and CNC_Std were characterized with % S of 0.74 ± 0.08, 0.80 ± 0.07, and 0.81 ± 0.06, respectively. The dextran sulfate was also characterized with an average % S of <15%. The values calculated here are higher than those reported by Abitbol et al. (2013). In fact, they reported % S within the range of 0.54–0.62%. One plausible reason for these differences may be the differing sample preparation parameters (such as higher acid concentration and longer reaction time), and they used a number of different cleanup techniques before running their (their sample was laboratory made, hence the cleanup) titrations while here the samples were used as obtained from the pilot plant.

Plots of the varying % S estimated using conductometric titration against the different percentages of CNC to DS for a CNC_SD, b CNC_FD, and c CNC_Std
The % S of the DS used is between 10 and 15%, but because of its polymeric nature, the majority of the ester groups are not free to the titration. This explains the % S for samples with no CNC added. In addition, the variation of DS % S alone is within 2 SD of the averages. Nevertheless, as will be presented in the next section, when assessed with elemental analysis, it is clear the DS alone samples were characterized within limits of the packaging material.
Elemental analysis
The CHNS concentrations of the different samples were evaluated to determine the % S in the different CNC samples. Based on the result presented in Fig. 6, the % S was approximately 1.10 ± 0.07, 1.10 ± 0.02, and 0.80 ± 0.04 for the CNC_SD, CNC_FD, and CNC_Std, respectively. These values are significantly higher than the ones previously reported with XPS and conductometric titration. Plausible reasons for this may be the presence of trace acid remnants which are not sensitive to the XPS (because XPS is a surface technique and would not pick up impurities in suspension) or the acid groups were not fully protonated when considering the titration. Nevertheless, the main idea of this communication is to develop a sense of strength and weakness of each method, and based on preliminary results, a suitable method to determine the sulfur ester content can be determined.

Plots of the varying % S estimated using elemental analysis against the different percentages of CNC to DS for a CNC_SD, b CNC_FD, and c CNC_Std
It should be noted the % S in these samples is comparably higher than previously reported by Gu et al. (2013), when they investigated the different sulfate esterification levels of cellulose nanowhiskers. In that paper, the authors hydrolyzed cotton cellulose with both sulfuric and hydrochloric acid, to produce nanowhiskers with varying degrees of sulfur esterification. The sulfuric acid-prepared nanowhiskers were characterized with lower sulfur content (0.35 ± 0.01) determined by combustion gas analysis (elemental analysis) when compared to results obtained in this communication. One plausible reason for the higher sulfur content in this present study when compared to Gu et al. (2013) may have been the longer reaction time (120 vs. 90 min) and higher stirring rate (200 rpm) employed by the pilot plant at Alberta Innovates Technology Futures. These conditions allow longer acid exposure and more agitation (hence, higher reaction rate between acid and pulp), respectively, which facilitated higher sulfur content.
Similarly, the % S values reported by Abitbol et al. (2013) are significantly lower when compared to what was estimated for this study. Again, the main reason for this may have been the longer reaction time (120 min) employed by the pilot plant when compared to the 45 min used by Abitbol et al. (2013). In the end, the values estimated using elemental analysis are more a measure of the total sulfur content and not the best indicator of the surface sulfur ester levels of CNCs. In addition, given that the method centers around combustion of the targeted species, all impurities present will be quantified and this makes the need for further sample cleanup paramount.
In a more applied study, Flauzino Neto et al. (2013) extracted CNCs from agro-industrial soy hull residues. The authors used a caustic treatment, then sodium chlorite to remove the hemicellulosic, and lignin fraction of the soy hulls. The cellulose concentrated material was then digested with 64% (w/w) sulfuric acid for 30 and 40 min. Interestingly, the authors choose to report the concentration of SO4 − kg−1 of cellulose, instead of total crude sulfur content. In the authors’ opinion, based on the method used (EA 1110–CHNS/O), this measurement is not possible, instead it should be reported as % S. Nevertheless, if back calculated, 118 and 111 mmol SO4 − kg−1 correspond to approximately 1.13 and 1.06% SO4 −, respectively. Further, if these were calculated to reflect only % S, the quantities would be 0.38 and 0.35%, respectively. Once again, this study clearly demonstrated that the values of % S intrinsically depend on the length of exposure to the acid during digestion. Finally, these studies highlight that elemental analysis is a reproducible method to determine the crude sulfur content of CNCs, but is not specific enough to determine the actual sulfur ester concentrations.
On a different note, it is worth to observe that the % S quantified based on elemental analysis is very high for the samples made up of predominantly dextran sulfate. It may be surprising this was not detected nor affected the infrared (IR) quantification that will be discussed in the coming section. The main reason for this is that the peaks that were targeted for IR quantification were specific to the ester and –O–S functionality and not the total sulfur content.
Infrared characterization
Infrared spectroscopy was used to determine the sulfate ester content of the different samples. A few infrared spectra for the different CNC + DS samples are presented in Fig. 7. The key peaks analyzed are highlighted. The estimation was based on the ratio of intensities for C–O–S (810–820 cm−1) relative to the total C–H signature (3000–2820 cm−1—alkyl chains of carboxylic compounds). The DS peaks were studied and reported by Cakic et al. (2005) when they investigated the synthesis and FTIR characterization of DSs. The rationale for selecting these two peaks was because of the increasing C–H with increasing DS content and plausible increase in % C–O–S linearly with this trend (assuming homogenous mixing and high DS sample purity). The different ratios were then plotted against the amount of CNC per sample, as shown in Fig. 8. Based on these calculations, the % sulfate ester bonds for CNC_SD, CNC_FD, and CNC_Std are 0.75 ± 0.08, 0.72 ± 0.07, and 0.70 ± 0.06, respectively.

Infrared spectra for the different samples, with different percentage of dextran sulfate (DS) and CNC

Plots of the varying % S estimated using infrared spectroscopy against the different percentages of CNC to DS for a CNC_SD, b CNC_FD, and c CNC_Std
Gu et al. (2013) studied two IR absorbance bands used for characterizing sulfated carbohydrate polymers. They focused on the asymmetrical S=O vibration at 1250 cm−1 and the symmetrical C–O–S (associated with C–O–SO3 group) vibration at about 820–830 cm−1. In their study, they were not able to quantify the area under the latter peak because it was too low in intensity. Additionally, the authors were not able to differentiate among the different samples when considering these peaks because of the interference resulting from the O–H stretching vibration (3000–3650 cm−1) and C–H stretch (2905 cm−1). Nevertheless, the authors stated that the broad O–H peak could be used to highlight the different cellulose crystal structures (I α and I β) within each sample type. In fact, they concluded that after acid hydrolysis process, the cellulose retained its I β crystalline structure.
General discussion based on method development
One of the main research questions concerning cellulose material is the chemical state of sulfur in native cellulose. What is known is that sulfur is essential for growth (Gu et al. 2013). In fact, it has been previously reported that the production of carbohydrates, proteins, and enzymes requires the presence of sulfur (Leustek et al. 2000). Additionally, sulfur is a key for the production of chlorophyll in plants, and depending on the plant genus, the sulfur content can vary from 0.1 to 1.1% (Leitao et al. 2001; Jackson et al. 1985). In fact, the sulfur content or the sulfate ester content after sulfuric acid hydrolysis of these plant materials is variable. In fact, reactions parameters of acid concentration, length of treatment time, amount of agitation, and cleanup methods have been shown to significantly affect the sulfate percentage (Beck-Candanedo et al. 2005). This might be the reason why the samples were characterized with higher sulfur content when estimated using elemental analysis versus the other methods.
From the current results, it can be seen that the XPS and IR results are very comparable. Given that elemental analysis combusts the entire sample, there is the chance of quantifying sulfur content not functioning as esters. On the other hand, XPS exploits the surface of these materials, which is where 90% of the sulfate ester groups are localized. Additionally, the IR method was performed on the bulk sample, but only a signature band characteristic of the sulfate esters was examined and extrapolated.
Conductometric titrations are another quick method to determine the sulfate ester content, but a few key aspects limit it. Firstly, it estimates the neutralization of all possible groups in solutions, i.e., carboxyl groups, which can contribute to the overall values. Hence, if the groups are not fully protonated, the values can be affected. Secondly, the method depends on rigorous calibration of the conductometer and the accurate preparation of reagents, factors that can vary among research laboratories. The % S as estimated by the different methods is summarized in Table 2.
Studying Table 2, it is difficult to determine whether there are any similarities among methods and samples. Hence, a radar plot was used to display the mean of the different methods for each sample in Fig. 9.

Radar plot of the % S for the different methods and samples (EA elemental analysis, CT conductometric titration)
Based on the data presented in Fig. 9, it is clear that CNC_FD and CNC_SD have higher sulfur content based on elemental analysis. On the other hand, IR gave a tight representation for the different samples. XPS gave a wider spread, and this may have been as a result of the sample preparation, inherent S present in samples, and the subjective curve fitting.
Effect of sample purification
The values obtained for each sample subjected using the different techniques after ion exchange purification are presented in Table 3. It should be noted these samples are just CNC and not CNC + DS. That is, instead of adding the different ratios of DS, more CNC (0.1 g per test) sample was used to improve the resolution for any given technique. These values obtained will facilitate comparison between the methods developed with the standardized methods used. In addition, the use of this strong acid resin enabled complete protonation of the samples prior to titration or any other analysis.
The results obtained were in agreement with those reported in Table 2, with one exception (italicized) being significantly lowered when compared to the value before sample cleanup. These correlations clearly demonstrate that the developed method can be used to determine the % S of CNC samples. In addition, the variations noticed among the different techniques were observed even after the samples were cleaned up or analyzed in the absence of DS. This clearly points to the ambiguity when considering what techniques to use when measuring the % S of CNC samples. The ultimate choice will come down to the time needed for analysis, the reproducibility required, and the availability of the necessary equipment.
In the end, it is clear the samples obtained from the different sources can be subjected to the developed method without further purification. This was confirmed with the similar conductometric titration values.
Measuring % S of different desulfated samples
For this set of experiments, only CNC was used in the absence of DS. The samples were subjected to the same purification method discussed in the previous section. After thorough purification, the samples were treated to reduce the % S over time. The change in % S after different times of desulfation is presented in Fig. 10.

Change in % S after different periods of desulfation: a CNC_SD, b CNC_FD, and c CNC_Std. Note, time zero are the values presented in Table 3 (note, the % S for the EA analyzed samples is fitted with data labels to illustrate the reduction over time)
The main aim of this section was to monitor the changes in % S for the different samples and investigate whether the different methods responded favorably to these changes in % S. Based on the results presented, it is clear that the IR method is comparable with the other methods presented. In fact, as the % S decreased, IR was in sync with the other methods, especially, the conductometric titration results.
In summary, the two previous sections clearly highlight that the developed method can be used to accurately predict the % sulfate ester groups of different CNC samples. Based on the extensive cleanup and no difference between results obtained, it can be finally said that IR may be the fastest method to determine the % S of different CNC samples.
Conclusion
A robust infrared method was successfully developed to determine the sulfate ester content of cellulose nanocrystals. The method was compared to three other established methods and compared favorably. In fact, the method was more precise than the others, when considering that all samples were produced from similar types of pulp and accepted under processing conditions. The method avoids the laborious sample preparation associated with elemental analysis, the calibrations for conductometric titration, and the sensitivity and subjectivity associated with curve fitting for XPS. The % S values based on IR estimation for CNC_SD, CNC_FD, and CNC_Std were 0.75 ± 0.08, 0.72 ± 0.07, and 0.70 ± 0.06, respectively. These values are higher than most reported in the literature because of the longer reaction time employed by Alberta Innovates Technology Futures pilot plant during production. For the CNC_Std samples produced by NRC, the values were within comparable ranges, despite variations in processing parameter.
References
Abitbol T, Kloser E, Gray DG (2013) Estimation of the surface sulfur content of cellulose nanocrystals prepared by sulfuric acid hydrolysis. Cellulose 20(2):785–794
Beck S, Bouchard J, Berry R (2012) Dispersibility in water of dried nanocrystalline cellulose. Biomacromolecules 13(5):1486–1494
Beck-Candanedo S, Roman M, Gray DG (2005) Effect of reaction conditions on the properties and behavior of wood cellulose nanocrystal suspensions. Biomacromolecules 6(2):1048–1054
Cakić M, Nikolić G, Ilić L, Stanković S (2005) Synthesis and FTIR characterization of some dextran sulphates. Chem Ind Chem Eng Q 5(2):74–78
de Castro DO, Bras J, Gandini A, Belgacem N (2016) Surface grafting of cellulose nanocrystals with natural antimicrobial rosin mixture using a green process. Carbohydr Polym 137:1–8
Flauzino Neto WP, Silvério HA, Dantas NO, Pasquini D (2013) Extraction and characterization of cellulose nanocrystals from agro-industrial residue—soy hulls. Ind Crops Prod 42:480–488
George M, Mussone PG, Bressler DC (2014) Surface and thermal characterization of natural fibres treated with enzymes. Ind Crops Prod 53:365–373
Gu J, Catchmark JM, Kaiser EQ, Archibald DD (2013) Quantification of cellulose nanowhiskers sulfate esterification levels. Carbohydr Polym 92(20):1809–1816
Habibi Y, Lucia LA, Rojas OJ (2010) Cellulose nanocrystals: chemistry, self-assembly, and applications. Chem Rev 110(6):3479–3500
Jackson LL, Engleman EE, Peard JL (1985) Determination of total sulfur in lichens and plants by combustion-infrared analysis. Environ Sci Technol 19(5):437–441
Jiang F, Esker AR, Roman M (2010) Acid-catalyzed and solvolytic desulfation of H2SO4-hydrolyzed cellulose nanocrystals. Langmuir 26(23):17919–17925
Kiziltas A, Nazari B, Kiziltas EE, Gardner DJS, Han Y, Rushing TS (2016) Cellulose NANOFIBER-polyethylene nanocomposites modified by polyvinyl alcohol. J Appl Polym Sci 133(6):42933
Kloser E, Gray DG (2010) Surface grafting of cellulose nanocrystals with poly(ethylene oxide) in aqueous media. Langmuir 26(16):13450–13456
Leitao JMM, Costa FS, Tack FMG (2001) Determination of total sulfur in soils and plants by an automated dry combustion method. Int J Environ Anal Chem 80(3):219–226
Leustek T, Martin MN, Bick J, Davies JP (2000) Pathways and regulation of sulfur metabolism revealed through molecular and genetic studies. Annu Rev Plant Physiol Plant Mol Biol 51:141–165
Qiao C, Chen G, Zhang J, Yao J (2016) Structure and rheological properties of cellulose nanocrystals suspension. Food Hydrocoll 55:19–25
Ranby BG (1949) Aqueous colloidal solutions of cellulose micelles. Acta Chem Scand 3:649–650
Acknowledgements
The authors acknowledge the Government of Alberta for the financial support. The authors are grateful to Alberta Innovates Technology Futures (AITF) for kindly supplying the CNC samples needed for this study. Also, the authors are thankful to National Research Council Canada for their in-kind supply of the CNC standard to AITF, from whom we were able to source a sample.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
George, M., Montemagno, C. Estimation of the sulfur ester content of cellulose nanocrystals prepared by sulfuric acid hydrolysis: a reproducible and fast infrared method. Wood Sci Technol 51, 535–556 (2017). https://doi.org/10.1007/s00226-016-0885-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00226-016-0885-2