
Abstract
The ultimate hope of researchers and patients is a pathway to development of treatments for osteoarthritis to modify the disease process in addition to the symptoms. However, development of disease modifying drugs requires objective endpoints such as measures of joint structure, joint tissue homeostasis and/or joint survival–measures such as provided by imaging biomarkers, molecular biomarkers and joint replacement frequency, respectively. Although biomarkers supporting investigational drug use and drug approval include surrogate endpoints that may not necessarily reflect or directly correlate with the clinical outcome of interest, a formal biomarker qualification process currently exists that is a rigorous three stage process that yields biomarker approvals (or denials) for specific contexts of use. From a cost perspective, biochemical biomarkers are the ‘ones to beat’; however, even well-validated biomarkers may not cross the translation gaps for eventual use in healthcare unless they offer an advantage in terms of cost per quality adjusted life year. This review summarizes the case FOR and AGAINST biomarkers in drug development and highlights the current data for a subset of biomarkers in the osteoarthritis research field informing on cartilage homeostasis, joint inflammation and altered subchondral bone remodeling.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
The Case FOR Biomarkers
Given the lack of treatments to prevent the incidence and progression of osteoarthritis (OA), disease modification is the holy grail—the thing most earnestly sought by patients [1] and arthritis care providers alike. Development of drugs to modify structural deterioration of the joint (disease modifying OA drugs or DMOADs) requires endpoints other than the traditional patient-reported outcomes (PROs)—an assessment of how an individual feels, functions, or survives. Rather, disease modification requires objective endpoints such as measures of joint structure, joint tissue homeostasis and/or joint survival–measures such as provided by imaging biomarkers, molecular biomarkers and joint replacement frequency, respectively. There is an especially strong rationale for the use of biomarkers in drug development; the chance of successfully transitioning from phase I of drug development to regulatory approval of a drug is three-fold increased (26% vs 8%) with vs without a biomarker [2]. The availability of a biomarker for patient selection also offers the prospect of personalized and thereby potentially safer treatment due to directing therapy to those most likely to benefit and sparing non-responders from potential side effects [3]. Although it is widely anticipated that disease modifiers for OA would also provide symptom modification, the highly heterogeneous nature of OA symptoms, and joint pain in particular, causes symptoms to be confounded outcomes. For development of disease modifying drugs, measurable, definable, non-confounded outcome measures are crucial.
A focus on pain and its treatment in the US increased as a consequence of increased numbers of disabled veterans in the 1940s and 1960s [4]. In the 1990s, opioid use was extended from cancer and acute pain to chronic noncancer pain such as arthritis [4]. Although pharmacological treatment of pain with opioids was intended to be part of multimodal care, it became unidimensional care in most settings with reliance almost completely on pharmacologic solutions to pain [4]. The emphasis on PROs as outcomes in trials for regulatory approval of OA drugs may have had the unintended consequence of contributing to a wealth of analgesics for OA and lack of DMOADs due to lack of FDA approved outcomes for disease modification. The focus on pain relief, without the risk of side effects and addiction linked to opioid analgesics, drove the development of the nerve growth factor (NGF) inhibitors. In contrast to opioids, which alter pain perception by targeting opioid receptors, NGF inhibitors block signaling of a pathway activated in response to injury, inflammation, or chronic pain. Although NGF inhibitors improve OA symptoms and function, a subset of treated individuals experience rapidly progressive osteoarthritis, particularly when taken together with non-steroidal anti-inflammatory drugs [5]. A debate currently exists concerning whether the joint structural damage associated with the NGF inhibitors is due to increased voluntary weight-bearing in the context of joint analgesia [6] and/or due to direct toxic effects on the joint since both chondrocytes and synoviocytes express NGF and Tropomyosin receptor kinase A (TrkA), the high affinity nerve growth factor receptor [5]. This suggests that highly effective analgesia, devoid of disease modifying effects that directly target the underlying biological processes, has the risk of worsening the disease in the long-term. Obviously, an ideal treatment would provide both symptom and disease modifying benefits; however, which could occur first and the timing of onset of these two benefits are currently not at all clear.
It is an interesting and important characteristic of soluble biomarkers that they are more reflective of disease activity than current disease status [7]. While a radiographic or magnetic resonance image (MRI) may provide a sensitive assessment of joint status, biochemical biomarkers are often generated as part of a pathophysiological process. In particular, the class of biomarkers referred to as neo-epitopes, which are generated when proteases degrade the proteins of the tissue, can provide a particularly relevant reflection of disease activity [8, 9]. Consequently, imaging may not always be directly correlated to a soluble biomarker, as disease activity and status may be disconnected, in particular in a slowly progressing disease such as OA that is associated with long periods of inertia followed by progression [10]. As a corollary, early and end stage disease may both be associated with periods of high and low disease activity, reflected by high and low levels of biomarkers (Fig. 1).

Osteoarthritis phasic disease activity is reflected by soluble biomarkers. Progression of OA is not linear. In times of high disease activity, biomarkers increase, and the disease subsequently progresses based on anatomic imaging. This may be followed by periods of low biomarker levels, even at later more severe stages of disease
In response, molecular biomarkers of disease processes may be used together with imaging, to provide additive value. For example, if we wish to predict how much water will be present in a bathtub tomorrow, we need three measures: how much water is currently present in the bathtub (analogous to an anatomic image of a joint); how much water is running into the bathtub (analogous to a tissue formation biomarker); and how much water is running out of the bathtub (analogous to a degradation biomarker).
The Case AGAINST Biomarkers
The 21st Century Cures Act (December 13, 2016) formally established in the US an updated, multistage process for qualification (a formal clinical validation linking a biomarker with biological and clinical end points) of biomarkers as drug development tools. A drug development tool may be used by any person for the qualified context of use. This process was designed to shorten the time and reduce the failure rate in drug development and bring new innovations and advances faster and more efficiently to patients who need them. Some concern has been raised regarding the potential, under the twenty-first Century Cures Act, for the FDA to approve drugs and devices on the basis of less rigorous, that is, biomarker-related rather than patient-reported data, and thereby weaken the traditional standards required by the FDA, with an unpredictable long-term effect on drug safety and efficacy.
In response to this concern about the relative rigor of biomarker compared with patient-reported data, it is important to recognize that formal biomarker qualification by the FDA is a rigorous three stage process that yields approvals (or denials) for specific contexts of use usually based on data from a minimum of two studies [11]. Moreover, a qualification determination may be rescinded or modified if new information calls into question the basis for such qualification. Drugs approved on the basis of a biomarker surrogate are generally required to verify clinical efficacy in a post-marketing study; the approval is expected to be withdrawn if post-approval trials fail to verify clinical benefit or do not demonstrate sufficient clinical benefit to justify the risks associated with the drug [12]. Moreover, given that the discovery, validation and qualification of objective measures of disease are of critical importance if we hope to achieve substantive changes in the field of OA pharmacological treatment, it does not seem fair to generally consider biomarker-related data as less rigorous than patient-report data; in contrast, in some circumstances, PROs may not even be an option. For instance, both early osteoporosis [13] and liver fibrosis [14] are silent disease processes so are unapproachable from the perspective of PROs. However, the asymptomatic early stages are likely the most opportune for therapeutic intervention for reverting fibrosis or preventing further progression to irreversible clinical organ failure, namely fracture or liver cirrhosis, respectively. Like these diseases, OA is also silent in its early stages [15]. Therefore, PROs would not be expected to be informative for these critical early stages; rather, biomarkers reflecting tissue turnover and preclinical disease activity would be critically important for development of DMOADs to target reversible stages of disease.
Another concern regards the potential inability of biomarkers supporting investigational drug use and drug approval to reflect or directly correlate with the clinical outcome of interest. However, PROs in OA, particularly joint pain symptoms, may originate from disease related and disease unrelated or remotely related phenomena. Pain PROs in OA have significantly higher placebo response rates than patient-reported function and objectively measured function [16]. Given that molecular and objectively measured biomarkers are generally less susceptible to placebo effects than PROs, as demonstrated by objective pulmonary function tests versus PROs in asthma [16], and use of gait speed as outcomes in pulmonary arterial hypertension trials [12], biomarkers might enable—with fewer participants exposed in a trial and therefore with a lower risk—the discernment of treatment efficacy earlier in the drug development process than PROs.
Finally, another concern relates to the potential difficulty of systemic biomarkers to reliably report on local cell or tissue level phenomena. For instance, development of systemic biomarkers in the OA field may be particularly difficult as a consequence of the small affected area within the joint capsule and the need for diffusion of analytes through the joint capsule into the systemic body fluid. However, as technologies are emerging, allowing increased sensitivity and increased understanding of endotypes that are systemic, we truly need to investigate, understand and focus on the correct context of use for each biomarker—recognizing that one biomarker may certainly not fit all purposes. For example, CTX-I is not a diagnostic biomarker of osteoporosis as it does not relate to the current amount of bone, but rather it is highly prognostic and an excellent efficacy of intervention biomarker for osteoporosis therapy as it reflects the level of bone resorption activity [17].
Molecular Biomarkers for OA
The joint is comprised of three major tissues: the articular cartilage, the synovium and the bone; all three are affected by the disease, which manifests as articular cartilage breakdown, synovial inflammation, proliferation and thickening, osteophyte formation and subchondral sclerosis [18]. Abnormalities of cartilage homeostasis (balance of anabolism and catabolism), synovial inflammation and subchondral bone remodeling are identified as potential targets for OA therapy [19]. Therefore, we focus here on molecular biomarkers that may inform each of these domains of disease and therapy.
Biomarkers Providing Insights into Altered Joint Tissue Homeostasis in OA
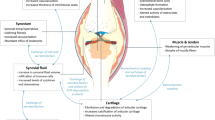
An imbalance of tissue homeostasis in OA has been a longstanding hypothesis explaining incident and progressive disease [20] (Fig. 2). As summarized below, high systemic catabolic markers and low systemic anabolic markers have been associated with OA progression (Fig. 3). As a biomarker of tissue homeostasis, type II collagen has been of special interest as it is the main component of articular cartilage and among joint tissues, is likely the most articular cartilage specific. An abundance of biomarker epitopes inform on the turnover (catabolism and anabolism) of type II collagen [21] (Fig. 4). For purposes of illustrating altered joint homeostasis in OA, we focus here on systemic biomarkers of type II collagen that can be quantified by commercially available tools and that inform on type II collagen catabolism and anabolism, namely, urinary (u) C-telopeptide fragments of type II collagen (uCTX-II), serum (s) N-propeptide of collagen IIA (PIIANP) and serum N-propeptide of collagen IIB (PIIBNP also termed PRO-C2), another measure of type II collagen formation.

An imbalance of cartilage tissue homeostasis is a characteristic of OA. OA presence and progression are characterized by high cartilage degradation, reflected by high urinary CTX-II, and low cartilage synthesis, reflected by low serum PIIANP or PIIBNP. Histological analysis of OA cartilage in the human knee (right panel) reveals the presence of CTX-II in the damaged and eroded surface of articular cartilage, the bone-cartilage interface and the tidemark. Adapted from “Alpha C‐telopeptide of type I collagen is associated with subchondral bone turnover and predicts progression of joint space narrowing and osteophytes in osteoarthritis”, Huebner et al. (2014); used by permission of Wiley, Order Number: 4722730414354

The role of cartilage catabolism and anabolism in OA progression. This theoretical construct, supported by literature, depicts OA progressors compared to non-progressors as a those with higher and/or rising catabolic biomarker concentrations, and b lower and/or more slowly rising anabolic biomarker concentrations. The greater the difference in concentration between progressors and non-progressors, the greater the ease with which the two groups can be distinguished and the greater possible effect size attainable in a clinical trial (the greater the therapeutic window based on a biomarker)

Key biomarkers of type II collagen degradation and synthesis. PIIANP and PIIBNP (PRO-C2) are N-terminal propeptide sequences indicative of type II collagen synthesis (anabolism). CTX-II is a C-terminal telopeptide sequence indicative of type II collagen degradation (catabolism). N- and C-terminal telopeptides of type I collagen, indicative of bone resorption, are available for osteoporosis monitoring, termed NTX-I and CTX-I, respectively. Adapted from “Biochemical markers and the FDA Critical Path: How biomarkers may contribute to the understanding of pathophysiology and provide unique and necessary tools for drug development”, Karsdal et al. (2009), Biomarkers 14(3):181–202; used by permission of Taylor & Francis, Order Number: 4740400891077
CTX-II is a product of type II collagen degradation and one of the most studied markers in OA [22]. CTX-II is an MMP-generated (by both MMP-9 and MMP-13) neoepitope. Although the tissue origin of CTX-II has been disputed [23], CTX-II is strongly present in areas corresponding to proteoglycan depletion in TNF-α- and oncostatin M treated cartilage explants [24] and is localized to the bone-cartilage interface, the tidemark, and damaged articular cartilage of the OA knee in the human [23] (Fig. 2). High baseline uCTX-II predicts increased odds (OR 1.29, p = 0.005) of clinically relevant (combination of symptom and radiographic worsening) knee OA progression over 4 years [25]. In a post hoc analysis of 640 individuals with knee OA from two clinical trials investigating oral salmon calcitonin, CSMC021C2301 (NCT00486434) and CSMC021C2302 (NCT00704847), high baseline uCTX-II predicts total joint replacements (TJR) in OA trial participants over 2 years [26]. High baseline uCTX-II is statistically significantly associated with a 3.08 times higher risk of undergoing a total joint replacement (TJR) of the knee or hip during the study period, and 8.94 times higher risk specifically of knee arthroplasty.
N-terminal propeptides of type II collagen reflect cartilage formation. Two main splice variants exist, termed type IIA (PIIANP, peptide sequence QDVQEAGSCV) and type IIB (PIIBNP or PRO-C2, peptide sequence QDVRQPGPKG) collagen propeptides [27]. The PIIANP assay specifically detects exon 2 sequence (a specific splice form of type II collagen) in the N-terminal propeptide of type II collagen, whereas the high sensitivity PRO-C2 assay specifically recognizes the exon 1/3 junction of type II collagen [27]. PIIANP is considered a product of chondroprogenitor cells [28]. In striking contrast to CTX-II, a high serum PIIANP predicts decreased odds (OR 0.83, p = 0.049 for 12-month time-integrated concentration) of clinically relevant (combination of symptom and radiographic worsening) knee OA progression over 4 years [25] (Fig. 3). Moreover, higher cartilage collagen synthesis, as reflected in systemic PIIANP concentrations, is associated with a lesser burden of OA features in lower extremity joints (knees and hips), even accounting for the OA burden in the hands and spine, age, sex and body mass index. Serum concentrations of both PIIANP and PIIBNP are lower in individuals with established knee OA compared to controls [27].
Interestingly, two large scale genome-wide association studies for OA [29, 30] brought to 90 the number of genetic risk loci for OA; three of the OA associated genes (TGFB1, GDF5, FGF18) encode growth factors. Articular cartilage explants stimulated in vitro by FGF18, IGF-1 and TGFB1 increase secretion of PRO-C2; whereas, only IGF-1 increases PIIANP and only minimally under the same culture conditions [27]. These data are all consistent with a low cartilage repair endotype with a genetic predisposition as a cause for OA.
Congruent with these findings are results showing that high PIIBNP/PRO-C2 predicted cartilage loss by MRI and radiographic joint space narrowing in the phase III clinical studies with calcitonin [31]. Additionally, recent proteomic analysis of human lower limb joint cartilages revealed the existence of an innate anabolic process, upregulated in OA and regulated by miRNA known to control limb regeneration in animals [32]. A combination of high catabolism and low anabolism characterized progressors in the FNIH cohort [25]. In the phase I FNIH study, serum PIIANP contributed modestly but significantly to predictions of clinically relevant knee OA progression by magnetic resonance imaging (MRI) biomarkers [33]. Taken together, these results show the importance of an imbalance of tissue homeostasis, the catabolic and anabolic responses, in OA progression and disease burden.
Biomarkers Providing Insights into Joint Inflammation in OA
Although the role of inflammation in OA has been heavily debated, cumulative evidence from ultrasound and MRI demonstrates inflammation in the majority of individuals with radiographic knee OA illustrated by a 70–81% frequency of effusions [34, 35], a 34–50% frequency of synovial thickening [34], a 35–40% frequency of popliteal (Baker’s) cysts [34], a 76% frequency of activated immune cells (macrophages and neutrophils), synovial tissue and synovial fluid cell analyses [36, 37]) and etarfolatide imaging [38]. Inflammation corresponds to joint pain [38]. Both MRI effusion synovitis and Hoffa’s synovitis predict incident radiographic OA one year later (OR 3.23 and OR 2.47) [39]. A subset of six synovial fluid (SF) biomarkers (MMP-3, sVCAM-1, sICAM-1, VEGF, TIMP-1, and MCP-1) was recently shown to be associated with synovial inflammation in OA, as well as radiographic and symptom severity [40]. These six OA-related SF biomarkers were specifically linked to indicators of activated macrophages and neutrophils. Products of activated macrophages and neutrophils, namely TGF-β1 and elastase, respectively, in synovium and SF, are significantly associated with knee synovitis based on in vivo etarfolatide imaging; at baseline they predict knee OA progression with areas under the curve from Receiver Operating Characteristic analyses of 0.95 (for TGF-β1) and 0.90 (for elastase), with greater stability of prediction when both are utilized [37]. Compared to SF biomarkers, it has been harder to identify systemic biomarkers that reflect localized joint inflammation in OA. In the FNIH phase I study, 24 M TIC serum hyaluronan (sHA) yielded odds ratio (OR) 1.22 for prediction of clinically relevant progression. At baseline, sHA and serum metalloproteinase-3 (sMMP-3) were associated with moderate to large (score ≥ 2, n = 117) effusion synovitis by MRI, with OR 1.35 and 1.30 per 1 standard deviation difference in biochemical markers providing evidence that it is possible for select systemic biomarkers to reflect localized synovitis [41].
There has been increasing recognition of a connection between the gut microbiome and OA leading to a new domain of biomarkers such as lipopolysaccharide (LPS), other gut microbe related molecules, and LPS binding protein (LBP). In a small study, serum LPS and LBP were associated with the abundance of activated macrophages in the knee joint capsule and synovium [42]; likewise, SF LPS and LBP were associated with the abundance of activated macrophages in the synovium. SF LPS was positively associated with severity of knee joint space narrowing and total WOMAC pain score [42]. In two large independent Dutch cohorts, a significant association has been recently reported between Streptococcus species abundance in stool microbiome samples, knee WOMAC pain and knee inflammation (effusion by MRI) [43].
With regard to inflammation, some lessons learned may be taken from the rheumatoid arthritis (RA) field in which joint inflammation drives joint erosion, and anti-inflammatory treatments block progression [44]. Type I collagen degradation by MMPs (C1M) [45] and type IV collagen degradation (C4M) [46] have been shown to be highly predictive for joint erosion and responsive to anti-inflammatory treatments such as anti-IL6R and anti-TNF-α. C1M was shown to be released from human synovium ex vivo and to respond to efficacious RA treatments [47]. Moreover, translational biomarkers are a potential tool for early assessment and decision-making in drug development as shown for RA treatment [48]. Taken together, joint inflammation has been strongly associated with OA severity and progression; moreover, an important link of OA to the gut microbiome is being elucidated consistent with local priming of macrophages by products of the gut microbiome resulting in inflammation of the synovial lining [49].
Biomarkers Providing Insights into Altered Subchondral Bone Remodeling in OA
Because of the differential adaptive capacity of the bone, abnormalities of bone in OA occur more rapidly and are more readily discernible than cartilage abnormalities [50]; this has been referred to as the “canary in the mine” phenomenon. For this reason, bone biomarkers are attractive for their potential to detect OA, including in its early stages [51]. Genetic studies in OA have observed enrichment for genes underlying monogenic forms of bone development diseases [30] further underscoring an association of bone and cartilage in the pathogenesis of OA. Major changes in subchondral bone gene expression are revealed in a comparison of degenerated medial vs unaffected lateral tibial plateau compartments of the OA knee [52]; a total of 972 differentially expressed genes were identified (based on fold change ≥ ± 2, P ≤ 0.05) and novel pathways such as Periostin (POSTN) and Leptin (LEP), which are implicated in bone remodeling by osteoblasts. Using a novel method for isolating site-matched overlying articular cartilage and underlying subchondral bone, we observed a strong coordinate (both up and down) regulation of gene expression of multiple genes (ADAMTS1, ASPN, BMP6, BMPER, CCL2, CCL8, COL5A1, COL6A3, COL7A1, COL16A1, FRZB, GDF10, MMP3, OGN, OMD, POSTN, PTGES, TNFSF11 and WNT1) in cartilage and bone in association with the severity of cartilage degeneration [53].
Based upon analysis of radiographic subchondral bone trabecular texture in OA, risk of knee OA progression is characterized by thickening of horizontal trabeculae (an early change) followed by thinning of vertical trabeculae (a later change) in a process known as stress shielding [54]. It therefore should come as no surprise that bone biomarkers traditionally employed for monitoring osteoporosis, show promise in OA [18]. In the phase I FNIH study, increased odds of clinically relevant knee OA progression were predicted by higher baseline uCTX-Iα (OR 1.20), and higher 24 M TIC of sCTX-I (OR 1.28), sNTX-I (OR 1.25), uNTX-I (OR 1.29), uCTX-Iα (OR 1.32), and uCTX-Iβ (OR 1.27). CTX-Iα localizes primarily to high bone turnover areas in subchondral bone in human knee OA [23] and is taken to indicate turnover of new bone and therefore new bone formation; uCTX-Iβ is taken to indicate turnover of older bone [23]. In the post hoc analysis of 640 individuals with knee OA from two clinical trials investigating oral salmon calcitonin (described above), high baseline sCTX-I was statistically significantly associated with 3.4 times higher risk of undergoing an arthroplasty of the knee or hip, but did not reach statistical significance for risk of knee arthroplasty alone [26]. In a subset of 216 women from the Chingford study, high uCTX-I and uNTX-I were associated with progressive knee OA; concentrations of these biomarkers were higher in OA progressors than controls and knee OA non-progressors, and comparable to levels observed in individuals with osteoporosis [55]. Taken together, these results indicate that risk of incident OA is, in part, associated with bone morphometric abnormalities and risk of OA progression is characterized by high subchondral bone turnover.
OA Biomarkers—The Way Forward
According to a recent informative review on imaging biomarkers in the cancer field by a consensus group of the Cancer Research UK (CRUK) and the European Organisation for Research and Treatment of Cancer (EORTC), biomarkers must cross two ‘translational gaps’ before they can be used to guide clinical decisions [56] (Fig. 5). In OA, effective technical validation and clinical qualification overcome the first translational gap enabling biomarkers to become useful ‘medical research tools’. The biomarkers that cross the second translational gap are relevant and useful in patient clinical care because they improve clinical outcomes sufficiently to justify the additional costs of testing and treatment and therefore warrant their consideration as ‘clinical decision-making tools’. Some biomarkers that have only crossed the first translational gap are nevertheless highly useful in the development of therapies [56]. The consensus group recommended parallel (rather than sequential) tracks of technical (assay) validation, biological/clinical validation and assessment of cost-effectiveness [56].

‘Translational gaps’ in biomarker development. Inspired by O’Connor et al. (2017) [56]
To overcome these translational gaps, and in conformity with the recommendation for ongoing parallel analyses, the FNIH OA Biomarkers Consortium, in collaborative synergy with the Osteoarthritis Research Society International and Arthritis Foundation (providing input on patients’ perspectives) have combined efforts toward biomarker qualification and encouraging an evidence-based revision of the regulatory guidance for OA clinical trials (Fig. 6). The FNIH biomarker phase I study toward qualification of MRI, radiographic and biochemical biomarkers is completed [25, 33, 54, 57,58,59]. The phase II FNIH study is expected to be initiated in 2020 with analysis of the most promising imaging and biochemical biomarkers from Phase I in extant samples from the placebo arms of multiple completed OA clinical trials. The aim of these studies is to qualify prognostic biomarkers for OA progression under the following COUs (examples from letters of intent submitted to FDA for soluble biomarkers):

Status of soluble biomarker qualification in OA—Synergy of current endeavors. The work and papers cited include the following: the OARSI FDA Initiative in which OARSI coordinated seven working groups (including an OA biomarker working group with publication of recommendations related to soluble biomarkers in 2011 [61]) to provide a critical appraisal of the science related to the design of clinical development programs for human drugs, biological products, and medical devices for the treatment and prevention of OA to assist the FDA as they work to finalize the draft guidance originally issued in July 1999, culminating in the submission of the white paper to the FDA in August 2010; OARSI published recommendations related to design and conduct of trials for OA at a variety of joint locations, for pharmacological and non-pharmacologic trials, for imaging and soluble biomarker assessments [62], and for performance and patient-reported outcomes; OA as a Serious Disease white paper (submitted to the FDA December 2016) [63]; the Arthritis Foundation sponsored patient-focused drug development meeting March 2017 on the OA patient perspective on current treatments [1]; the Arthritis Foundation formed the OA Centers of Excellence to develop a clinical trial paradigm for acute joint injury (2018); an OARSI initiative white paper suggesting post-marketing approval trial designs for OA drugs approved on the basis of a surrogate endpoint (2019) [12]; and the ongoing FDA Biomarkers Consortium qualification endeavor. The culmination of all these efforts is to engender an evidence-based new OA Clinical Trial Guidance from the FDA that could facilitate development of DMOADs
Primary COU
Prognostic enrichment molecular biomarkers for use in phase 2 and 3 clinical trials to identify individuals with a diagnosis of knee osteoarthritis who are likely to experience disease progression within the subsequent 48 months based on the WOMAC pain subscale and/or radiographic joint space width loss and/or joint replacement.
Secondary or Allied COU
Prognostic biomarkers based on time-integrated concentrations (TICs) from baseline to 12 months, to provide a method for early identification of osteoarthritis patients to define who are likely to experience disease progression within the subsequent 48 months based on the WOMAC pain subscale and/or radiographic joint space width loss and/or joint replacement.
The phase III FNIH study is anticipated to involve analysis of the most informative markers from phase II with the aim of qualifying biomarkers as pharmacodynamic response markers (per the Biomarkers, Endpoints, and other Tools (BEST) categories [60]) utilizing samples/images from treatment and placebo arms of OA trials. In addition, it is anticipated that second generation biomarkers will be emerging that will leverage new multi-omics technologies, that will augment and in some cases, replace existing markers on the basis of greater sensitivity, specificity and/or predictive capability. Ultimately, it will be important to link the magnitude of change in a biomarker to a clinically meaningful change in a clinical outcome.
From a cost perspective, biochemical biomarkers are the ‘ones to beat’. Imaging biomarkers must provide good ‘value for money’ and compare favorably with biospecimen-derived biomarkers; in the research setting, the value added by imaging biomarkers should be greater than the cost of performing the study [56]. The CRUK/EORTC consensus group underscored the difficulty of crossing translational gap 2 to achieve use in healthcare in that even well-validated biomarkers may not cross this gap unless they offer an advantage in terms of cost per quality adjusted life year (QALY) gained [56].
Conclusions
OA biomarkers are more likely to be disease related than subjective PROs and therefore appropriate and necessary for development of DMOADs. Biomarkers create a potential path for treating early OA—before illness—when disease is more likely modifiable. Systemic biomarkers potentially report on the overall burden of disease and therefore provide holistic endpoints for generalized disease analyses. Molecular biomarkers provide the potential for identifying direct biomarkers, in the pathway of disease, that could facilitate drug development of agents that could modify both symptoms and structure without unintended adverse joint consequences associated with symptom modification alone. Molecular biomarkers improve chances of drug program success and create potential means for developing personalized medicine strategies for OA. However, given that is unlikely that any single biomarker can be sufficiently sensitive and specific to fulfill all needs such as early disease detection, prediction of disease progression and monitoring response to therapy as an efficacy of intervention marker, it is likely that a variety of biochemical markers will ultimately be used serially and in combination to optimize OA drug development and patient therapy in OA.
References
The Voice of the Patient: Osteoarthritis Report PC, Copenhaver C, Dernier D, Geller K, Hanson B, Harvey K, Johnson F, Kraus V, Lester G, McGrath E, Shuey D, Tuan R, Tucci A, Wyatt J (2017) Osteoarthritis patient-focused drug development: voice of the patient report. In: The voice of the patient. Arthritis Foundation, Washington, DC
Thomas D, Burns J, Audette J, Carroll A, Dow-Hygelund C, Hay M (2016) Clinical development success rates 2006–2015. In: Biotechnology Innovation Organization (BIO), biomedtracker and amplion, pp 1–26
Karsdal MA, Christiansen C, Ladel C, Henriksen K, Kraus VB, Bay-Jensen AC (2014) Osteoarthritis—a case for personalized health care? Osteoarthr Cartil 22:7–16
Bernard SA, Chelminski PR, Ives TJ, Ranapurwala SI (2018) Management of pain in the United States—a brief history and implications for the opioid epidemic. Health Serv Insights 11:1178632918819440
Miller RE, Block JA, Malfait AM (2017) Nerve growth factor blockade for the management of osteoarthritis pain: what can we learn from clinical trials and preclinical models? Curr Opin Rheumatol 29:110–118
LaBranche TP, Bendele AM, Omura BC, Gropp KE, Hurst SI, Bagi CM, Cummings TR, Grantham LE 2nd, Shelton DL, Zorbas MA (2017) Nerve growth factor inhibition with tanezumab influences weight-bearing and subsequent cartilage damage in the rat medial meniscal tear model. Ann Rheum Dis 76:295–302
Sand JM, Knox AJ, Lange P, Sun S, Kristensen JH, Leeming DJ, Karsdal MA, Bolton CE, Johnson SR (2015) Accelerated extracellular matrix turnover during exacerbations of COPD. Respir Res 16:69
Karsdal MA, Henriksen K, Leeming DJ, Woodworth T, Vassiliadis E, Bay-Jensen AC (2010) Novel combinations of post-translational modification (PTM) neo-epitopes provide tissue-specific biochemical markers—are they the cause or the consequence of the disease? Clin Biochem 43:793–804
Siebuhr AS, He Y, Gudmann NS, Gram A, Kjelgaard-Petersen CF, Qvist P, Karsdal MA, Bay-Jensen AC (2014) Biomarkers of cartilage and surrounding joint tissue. Biomark Med 8:713–731
Felson D, Niu J, Sack B, Aliabadi P, McCullough C, Nevitt MC (2013) Progression of osteoarthritis as a state of inertia. Ann Rheumatol Dis 72:924–929
FDA (2019) Drug development tool qualification process: transparency provisions
Kraus VB, Simon LS, Katz JN, Neogi T, Hunter D, Guermazi A, Karsdal MA (2019) Proposed study designs for approval based on a surrogate endpoint and a post-marketing confirmatory study under FDA's accelerated approval regulations for disease modifying osteoarthritis drugs. Osteoarthr Cartil 27:571–579. https://doi.org/10.1016/j.joca.2018.1011.1002
Kraus V, Hsueh M-F (2020) Biomarkers and Osteoarthritis. In: Ginsburg G, Willard H, Tsalik E, Woods C (eds) Genomic and Precision Medicine. Elsevier, p 429–444
Karsdal MA, Krarup H, Sand JM, Christensen PB, Gerstoft J, Leeming DJ, Weis N, Schaffalitzky de Muckadell OB, Krag A (2014) Review article: the efficacy of biomarkers in chronic fibroproliferative diseases—early diagnosis and prognosis, with liver fibrosis as an exemplar. Aliment Pharmacol Ther 40:233–249
Kraus VB, Blanco FJ, Englund M, Karsdal MA, Lohmander LS (2015) Call for standardized definitions of osteoarthritis and risk stratification for clinical trials and clinical use. Osteoarthr Cartil 23:1233–1241
Huang Z, Chen J, Hu QS, Huang Q, Ma J, Pei FX, Shen B, Kraus VB (2019) Meta-analysis of pain and function placebo responses in pharmacological osteoarthritis trials. Arthritis Res Ther 21:173
Henriksen K, Bohren KM, Bay-Jensen AC, Karsdal MA (2010) Should biochemical markers of bone turnover be considered standard practice for safety pharmacology? Biomarkers 15:195–204
Bay-Jensen AC, Sondergaard BC, Christiansen C, Karsdal MA, Madsen SH, Qvist P (2010) Biochemical markers of joint tissue turnover. Assay Drug Dev Technol 8:118–124
Zhang W, Robertson WB, Zhao J, Chen W, Xu J (2019) Emerging trend in the pharmacotherapy of osteoarthritis. Front Endocrinol 10:431
Kraus V, Vincent T (2020) Osteoarthritis. In: Goldman L, Schafer A (eds) Goldman-Cecil Medicine. Elsevier, Philadelphia, PA, pp 1698–1703
Karsdal MA, Woodworth T, Henriksen K, Maksymowych WP, Genant H, Vergnaud P, Christiansen C, Schubert T, Qvist P, Schett G, Platt A, Bay-Jensen AC (2011) Biochemical markers of ongoing joint damage in rheumatoid arthritis–current and future applications, limitations and opportunities. Arthritis Res Therapy 13:215
Bay-Jensen AC, Reker D, Kjelgaard-Petersen CF, Mobasheri A, Karsdal MA, Ladel C, Henrotin Y, Thudium CS (2016) Osteoarthritis year in review 2015: soluble biomarkers and the BIPED criteria. Osteoarthr Cartil 24:9–20
Huebner JL, Bay-Jensen AC, Huffman KM, He Y, Leeming DJ, McDaniel GE, Karsdal MA, Kraus VB (2014) Alpha C-telopeptide of type I collagen is associated with subchondral bone turnover and predicts progression of joint space narrowing and osteophytes in osteoarthritis. Arthritis Rheumatol (Hoboken, NJ) 66:2440–2449
Sondergaard BC, Henriksen K, Wulf H, Oestergaard S, Schurigt U, Brauer R, Danielsen I, Christiansen C, Qvist P, Karsdal MA (2006) Relative contribution of matrix metalloprotease and cysteine protease activities to cytokine-stimulated articular cartilage degradation. Osteoarthr Cartil 14:738–748
Kraus VB, Collins JE, Hargrove D, Losina E, Nevitt M, Katz JN, Wang SX, Sandell LJ, Hoffmann SC, Hunter DJ (2017) Predictive validity of biochemical biomarkers in knee osteoarthritis: data from the FNIH OA Biomarkers Consortium. Ann Rheum Dis 76:186–195
Bjerre-Bastos J, Bay-Jensen A-C, Karsdal M, Byrjalsen I, Andersen J, Riis B, Christiansen C, Bihlet A (2019) Biomarkers of bone and cartilage turnover CTX-I and CTXII predict total joint replacements in osteoarthritis. Osteoarthr Cartil 27(S31):12
Luo Y, He Y, Reker D, Gudmann NS, Henriksen K, Simonsen O, Ladel C, Michaelis M, Mobasheri A, Karsdal M, Bay-Jensen AC (2018) A novel high sensitivity type II collagen blood-based biomarker, PRO-C2, for assessment of cartilage formation. Int J Mol Sci 19:3485
Oganesian A, Zhu Y, Sandell LJ (1997) Type IIA procollagen amino propeptide is localized in human embryonic tissues. J Histochem Cytochem 45:1469–1480
Styrkarsdottir U, Lund SH, Thorleifsson G, Zink F, Stefansson OA, Sigurdsson JK, Juliusson K, Bjarnadottir K, Sigurbjornsdottir S, Jonsson S, Norland K, Stefansdottir L, Sigurdsson A, Sveinbjornsson G, Oddsson A, Bjornsdottir G, Gudmundsson RL, Halldorsson GH, Rafnar T, Jonsdottir I, Steingrimsson E, Norddahl GL, Masson G, Sulem P, Jonsson H, Ingvarsson T, Gudbjartsson DF, Thorsteinsdottir U, Stefansson K (2018) Meta-analysis of Icelandic and UK data sets identifies missense variants in SMO, IL11, COL11A1 and 13 more new loci associated with osteoarthritis. Nat Genet 50:1681–1687
Tachmazidou I, Hatzikotoulas K, Southam L, Esparza-Gordillo J, Haberland V, Zheng J, Johnson T, Koprulu M, Zengini E, Steinberg J, Wilkinson JM, Bhatnagar S, Hoffman JD, Buchan N, Suveges D, Yerges-Armstrong L, Smith GD, Gaunt TR, Scott RA, McCarthy LC, Zeggini E (2019) Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK Biobank data. Nat Genet 51:230–236
Luo Y, Higgins N, He Y, Byrjalsen I, Andersen J, Bihlet A, Karsdal M, Bay-Jensen A (2019) Identification of superior responders to a bone and cartilage centric treatment in osteoarthritis: low levels of cartilage formation may provide an opportunity to stimulate formation. Osteoarthr Cartil 27:S61
Hsueh MF, Onnerfjord P, Bolognesi MP, Easley ME, Kraus VB (2019) Analysis of "old" proteins unmasks dynamic gradient of cartilage turnover in human limbs. Sci Adv 5:e3203
Hunter D, Deveza L, Collins J, Losina E, Nevitt M, Roemer F, Guermazi A, Bowes M, Dam E, Eckstein F, Lynch J, Katz J, Kwoh C, Hoffman S, Kraus V (2019) Multivariable modelling of biomarker data from the phase 1 Foundation for NIH Osteoarthritis Biomarkers Consortium. Ann Rheum Dis 76:186–195
Tarhan S, Unlu Z (2003) Magnetic resonance imaging and ultrasonographic evaluation of the patients with knee osteoarthritis: a comparative study. Clin Rheumatol 22:181–188
Song IH, Althoff CE, Hermann KG, Scheel AK, Knetsch T, Schoenharting M, Werner C, Burmester GR, Backhaus M (2008) Knee osteoarthritis. Efficacy of a new method of contrast-enhanced musculoskeletal ultrasonography in detection of synovitis in patients with knee osteoarthritis in comparison with magnetic resonance imaging. Ann Rheum Dis 67:19–25
Hsueh M-F, Lu Y-J, Wellman S, Bolognesi M, Kraus VB (2018) Functional folate receptor cell-associated inflammatory cytokines predict the progression of knee osteoarthritis. Osteoarthr Cartil 26(S121):228
Hsueh M-F, Zhang X, Wellman S, Bolognesi M, Kraus V (2019) Synergistic roles of macrophages and neutrophils in osteoarthritis progression. Arhthritis Rheum (in review)
Kraus VB, McDaniel G, Huebner JL, Stabler TV, Pieper CF, Shipes SW, Petry NA, Low PS, Shen J, McNearney TA, Mitchell P (2016) Direct in vivo evidence of activated macrophages in human osteoarthritis. Osteoarthr Cartil 24:1613–1621
Atukorala I, Kwoh CK, Guermazi A, Roemer FW, Boudreau RM, Hannon MJ, Hunter DJ (2016) Synovitis in knee osteoarthritis: a precursor of disease? Ann Rheumatol Dis 75:390–395. https://doi.org/10.1136/annrheumdis-2014-205894
Haraden CA, Huebner JL, Hsueh MF, Li YJ, Kraus VB (2019) Synovial fluid biomarkers associated with osteoarthritis severity reflect macrophage and neutrophil related inflammation. Arthritis Res Ther 21:146
Deveza LA, Kraus VB, Collins JE, Guermazi A, Roemer FW, Nevitt MC, Hunter DJ (2018) Is synovitis detected on non-contrast-enhanced magnetic resonance imaging associated with serum biomarkers and clinical signs of effusion? Data from the osteoarthritis initiative. Scand J Rheumatol 47:235–242
Huang ZY, Stabler T, Pei FX, Kraus VB (2016) Both systemic and local lipopolysaccharide (LPS) burden are associated with knee OA severity and inflammation. Osteoarthr Cartil 24:1769–1775
Boer CG, Radjabzadeh D, Medina-Gomez C, Garmaeva S, Schiphof D, Arp P, Koet T, Kurilshikov A, Fu J, Ikram MA, Bierma-Zeinstra S, Uitterlinden AG, Kraaij R, Zhernakova A, van Meurs JBJ (2019) Intestinal microbiome composition and its relation to joint pain and inflammation. Nat Commun 10:4881
Panagopoulos PK, Lambrou GI (2018) Bone erosions in rheumatoid arthritis: recent developments in pathogenesis and therapeutic implications. J Musculoskel Neuronal Interact 18:304–319
Siebuhr AS, Bay-Jensen AC, Leeming DJ, Plat A, Byrjalsen I, Christiansen C, van de Heijde D, Karsdal MA (2013) Serological identification of fast progressors of structural damage with rheumatoid arthritis. Arthritis Res Therapy 15:R86
Gudmann NS, Junker P, Juhl P, Thudium CS, Siebuhr AS, Byrjalsen I, Karsdal MA, Bay-Jensen AC (2018) Type IV collagen metabolism is associated with disease activity, radiographic progression and response to tocilizumab in rheumatoid arthritis. Clin Exp Rheumatol 36:829–835
Kjelgaard-Petersen C, Siebuhr AS, Christiansen T, Ladel C, Karsdal M, Bay-Jensen AC (2015) Synovitis biomarkers: ex vivo characterization of three biomarkers for identification of inflammatory osteoarthritis. Biomarkers 20:547–556
Kjelgaard-Petersen CF, Platt A, Braddock M, Jenkins MA, Musa K, Graham E, Gantzel T, Slynn G, Weinblatt ME, Karsdal MA, Thudium CS, Bay-Jensen AC (2018) Translational biomarkers and ex vivo models of joint tissues as a tool for drug development in rheumatoid arthritis. Arthritis Rheumatol 70:1419–1428
Huang Z, Kraus VB (2016) Does lipopolysaccharide-mediated inflammation have a role in OA? Nat Rev Rheumatol 12:123–129
Goldring MB, Goldring SR (2010) Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann N Y Acad Sci 1192:230–237
Engbersen M, Huang Z, Kraus V (2016) Bone biomarkers related to osteoarthritis. In: Preedy V (ed) Biomarkers in disease: methods, discoveries and applications. Springer, Dordrecht, pp 1–29
Chou CH, Wu CC, Song IW, Chuang HP, Lu LS, Chang JH, Kuo SY, Lee CH, Wu JY, Chen YT, Kraus VB, Lee MT (2013) Genome-wide expression profiles of subchondral bone in osteoarthritis. Arthritis Res Ther 15:R190
Chou CH, Lee CH, Lu LS, Song IW, Chuang HP, Kuo SY, Wu JY, Chen YT, Kraus VB, Wu CC, Lee MT (2013) Direct assessment of articular cartilage and underlying subchondral bone reveals a progressive gene expression change in human osteoarthritic knees. Osteoarthr Cartil 21:450–461
Kraus VB, Collins JE, Charles HC, Pieper CF, Whitley L, Losina E, Nevitt M, Hoffmann S, Roemer F, Guermazi A, Hunter DJ (2018) Predictive validity of radiographic trabecular bone texture in knee osteoarthritis: the osteoarthritis research society international/foundation for the National Institutes of Health Osteoarthritis Biomarkers Consortium. Arthritis Rheumatol 70:80–87
Bettica P, Cline G, Hart DJ, Meyer J, Spector TD (2002) Evidence for increased bone resorption in patients with progressive knee osteoarthritis: longitudinal results from the Chingford study. Arthritis Rheum 46:3178–3184
O'Connor JP, Aboagye EO, Adams JE, Aerts HJ, Barrington SF, Beer AJ, Boellaard R, Bohndiek SE, Brady M, Brown G, Buckley DL, Chenevert TL, Clarke LP, Collette S, Cook GJ, deSouza NM, Dickson JC, Dive C, Evelhoch JL, Faivre-Finn C, Gallagher FA, Gilbert FJ, Gillies RJ, Goh V, Griffiths JR, Groves AM, Halligan S, Harris AL, Hawkes DJ, Hoekstra OS, Huang EP, Hutton BF, Jackson EF, Jayson GC, Jones A, Koh DM, Lacombe D, Lambin P, Lassau N, Leach MO, Lee TY, Leen EL, Lewis JS, Liu Y, Lythgoe MF, Manoharan P, Maxwell RJ, Miles KA, Morgan B, Morris S, Ng T, Padhani AR, Parker GJ, Partridge M, Pathak AP, Peet AC, Punwani S, Reynolds AR, Robinson SP, Shankar LK, Sharma RA, Soloviev D, Stroobants S, Sullivan DC, Taylor SA, Tofts PS, Tozer GM, van Herk M, Walker-Samuel S, Wason J, Williams KJ, Workman P, Yankeelov TE, Brindle KM, McShane LM, Jackson A, Waterton JC (2017) Imaging biomarker roadmap for cancer studies. Nat Rev Clin Oncol 14:169–186
Collins JE, Losina E, Nevitt MC, Roemer FW, Guermazi A, Lynch JA, Katz JN, Kent Kwoh C, Kraus VB, Hunter DJ (2016) Semiquantitative Imaging Biomarkers of Knee Osteoarthritis Progression: Data From the Foundation for the National Institutes of Health Osteoarthritis Biomarkers Consortium. Arthritis Rheumatol (Hoboken, NJ) 68:2422–2431
Hunter D, Nevitt M, Losina E, Kraus V (2014) Biomarkers for osteoarthritis: current position and steps towards further validation. Best Pract Res 28:61–71
Kraus V, Hargrove D, Hunter D, Renner J, Jordan J (2014) Establishment of reference intervals for osteoarthritis related biomarkers—the FNIH/OARSI OA Biomarkers Consortium. Osteo Cartilage 22:podium presentation
Food and Drug Administration-NIH Biomarker Working Group (2016) BEST (Biomarkers, EndpointS, and other Tools) Resource. In: FDA, Silver Spring, MD
Kraus VB, Burnett B, Coindreau J, Cottrell S, Eyre D, Gendreau M, Gardiner J, Garnero P, Hardin J, Henrotin Y, Heinegard D, Ko A, Lohmander LS, Matthews G, Menetski J, Moskowitz R, Persiani S, Poole AR, Rousseau JC, Todman M (2011) Application of biomarkers in the development of drugs intended for the treatment of osteoarthritis. Osteoarthr Cartil 19:515–542
Kraus VB, Blanco FJ, Englund M, Henrotin Y, Lohmander LS, Losina E, Onnerfjord P, Persiani S (2015) OARSI Clinical Trials Recommendations: Soluble biomarker assessments in clinical trials in osteoarthritis. Osteoarthritis Cartilage 23:686–697
March L, Cross M, Lo C, Arden N, Gates L, Leyland K, Hawker G, King L (2016) Osteoarthritis: a serious disease. Access Date Access 2016
Author information
Authors and Affiliations
Contributions
Both VBK and MAK drafted and critically revised the important intellectual content of this manuscript; both approved the final version for submission and both take responsibility for the accuracy of the article.
Corresponding author
Ethics declarations
Disclosures
Dr. Morten Karsdal is the Chief Executive Officer of Nordic Bioscience. Nordic Bioscience has pending patents related to several biomarkers described in this review (PIIBNP/PRO-C2, C1M and C4M). Dr. V Kraus has no conflicts to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kraus, V.B., Karsdal, M.A. Osteoarthritis: Current Molecular Biomarkers and the Way Forward. Calcif Tissue Int 109, 329–338 (2021). https://doi.org/10.1007/s00223-020-00701-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-020-00701-7