
Abstract
Phosphorus, a 5A element with atomic weight of 31, comprises just over 0.6% of the composition by weight of plants and animals. Three isotopes are available for studying phosphorus metabolism and kinetics. 31P is stable, whereas the radioactive isotope 33P has a half-life of 25 days and 32P has a half-life of 14 days. Phosphate ester and phosphoanhydride are common chemical linkages and phosphorus is a key element in organic molecules involved in a wide variety of essential cellular functions. These include biochemical energy transfer via adenosine triphosphate (ATP), maintenance of genetic information with nucleotides DNA and RNA, intracellular signaling via cyclic adenosine monophosphate (cAMP), and membrane structural integrity via glycerophospholipids. However, this review focuses on the metabolism of inorganic phosphorus (Pi) acting as a weak acid. Phosphoric acid has all three hydrogens attached to oxygen and is a weak diprotic acid. It has 3 pKa values: pH 2.2, pH 7.2, and pH 12.7. At physiological pH of 7.4, Pi exists as both H2PO4(−) and HPO4(2−) and acts as an extracellular fluid (ECF) buffer. Pi is the form transported across tissue compartments and cells. Measurement of Pi in biological fluids is based on its reaction with ammonium molybdate which does not measure organic phosphorus. In humans, 80% of the body phosphorus is present in the form of calcium phosphate crystals (apatite) that confer hardness to bone and teeth, and function as the major phosphorus reservoir (Fig. 1). The remainder is present in soft tissues and ECF. Dietary phosphorus, comprising both inorganic and organic forms, is digested in the upper gastrointestinal tract. Absorbed Pi is transported to and from bone, skeletal muscle and soft tissues, and kidney at rates determined by ECF Pi concentration, rate of blood flow, and activity of cell Pi transporters (Fig. 2). During growth, there is net accretion of phosphorus, and with aging, net loss of phosphorus occurs. The bone phosphorus reservoir is depleted and repleted by overall phosphorus requirement. Skeletal muscle is rich in phosphorus used in essential biochemical energy transfer. Kidney is the main regulator of ECF Pi concentration by virtue of having a tubular maximum reabsorptive capacity for Pi (TmPi) that is under close endocrine control. It is also the main excretory pathway for Pi surplus which is passed in urine. Transcellular and paracellular Pi transports are performed by a number of transport mechanisms widely distributed in tissues, and particularly important in gut, bone, and kidney. Pi transporters are regulated by a hormonal axis comprising fibroblast growth factor 23 (FGF23), parathyroid hormone (PTH), and 1,25 dihydroxy vitamin D (1,25D). Pi and calcium (Ca) metabolism are intimately interrelated, and clinically neither can be considered in isolation. Diseases of Pi metabolism affect bone as osteomalacia/rickets, soft tissues as ectopic mineralization, skeletal muscle as myopathy, and kidney as nephrocalcinosis and urinary stone formation.

Content of phosphorus in human adult: skeleton, soft tissue, and extracellular fluid (grams, log scale). Corresponding data for calcium are shown for comparison

Phosphate (Pi) transport to and from tissue compartments in mg/24 h. At a dietary phosphorus of 1400 mg, 1120 mg is absorbed in upper intestine to the ECF, 210 mg returned to intestine by endogenous secretion, resulting in 910 mg net Pi absorption and 490 mg fecal excretion. At bone, 180 mg is deposited by bone formation and 180 mg return to the ECF by bone resorption. At kidney, 5040 mg is filtered at the glomerulus and 4130 mg return to the ECF by tubular reabsorption with 910 mg excreted in the urine. In soft tissue, Pi is exchanged between ECF and cells
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Phosphorus, a 5A element with atomic weight of 31, comprises just over 0.6% of the composition by weight of plants and animals. Three isotopes are available for studying phosphorus metabolism and kinetics. 31P is stable, whereas the radioactive isotope 33P has a half-life of 25 days and 32P has a half-life of 14 days. Phosphate ester and phosphoanhydride are common chemical linkages and phosphorus is a key element in organic molecules involved in a wide variety of essential cellular functions. These include biochemical energy transfer via adenosine triphosphate (ATP), maintenance of genetic information with nucleotides DNA and RNA, intracellular signaling via cyclic adenosine monophosphate (cAMP), and membrane structural integrity via glycerophospholipids. However, this review focuses on the metabolism of inorganic phosphorus (Pi) acting as a weak acid. Phosphoric acid has all three hydrogens attached to oxygen and is a weak diprotic acid. It has 3 pKa values: pH 2.2, pH 7.2, and pH 12.7. At physiological pH of 7.4, Pi exists as both H2PO4(−) and HPO4(2−) and acts as an extracellular fluid (ECF) buffer. Pi is the form transported across tissue compartments and cells. Measurement of Pi in biological fluids is based on its reaction with ammonium molybdate which does not measure organic phosphorus. In humans, 80% of the body phosphorus is present in the form of calcium phosphate crystals (apatite) that confer hardness to bone and teeth, and function as the major phosphorus reservoir (Fig. 1). The remainder is present in soft tissues and ECF. Dietary phosphorus, comprising both inorganic and organic forms, is digested in the upper gastrointestinal tract. Absorbed Pi is transported to and from bone, skeletal muscle and soft tissues, and kidney at rates determined by ECF Pi concentration, rate of blood flow, and activity of cell Pi transporters (Fig. 2). During growth, there is net accretion of phosphorus, and with aging, net loss of phosphorus occurs. The bone phosphorus reservoir is depleted and repleted by overall phosphorus requirement. Skeletal muscle is rich in phosphorus used in essential biochemical energy transfer. Kidney is the main regulator of ECF Pi concentration by virtue of having a tubular maximum reabsorptive capacity for Pi (TmPi) that is under close endocrine control. It is also the main excretory pathway for Pi surplus which is passed in urine. Transcellular and paracellular Pi transports are performed by a number of transport mechanisms widely distributed in tissues, and particularly important in gut, bone, and kidney. Pi transporters are regulated by a hormonal axis comprising fibroblast growth factor 23 (FGF23), parathyroid hormone (PTH), and 1,25 dihydroxy vitamin D (1,25D). Pi and calcium (Ca) metabolism are intimately interrelated, and clinically neither can be considered in isolation. Diseases of Pi metabolism affect bone as osteomalacia/rickets, soft tissues as ectopic mineralization, skeletal muscle as myopathy, and kidney as nephrocalcinosis and urinary stone formation.
Phosphorus in Circulation and ECF
The circulation and ECF are responsible for the transport of Pi to and from the organs involved in Pi metabolism (Fig. 2). In plasma, the phosphorus content is about 12 mg/dL (3.87 mmol/L) with about a third present as inorganic phosphorus (Pi) (Fig. 3). Over 80% of plasma Pi is non-protein bound and comprises three protonated species along with various complexes with Ca, magnesium, and sodium [1]. Although plasma Pi is about 20% protein bound, over 95% is ultrafilterable because of Donnan membrane effects. Thus, the transport of Pi, as into glomerular filtrate, is commonly assessed from a measure of total Pi concentration in plasma. Only in conditions of marked changes in acid–base this assumption does not hold. Pi in plasma and other biological fluids is measured by reacting with ammonium molybdate to form phosphomolybdate whose absorbance at 340 nm is directly proportional to the concentration of Pi. Plasma Pi ranges from 2.5 to 4.5 mg per dl [0.81–1.45 mmol/L] in adults. In children it is higher due to a higher TmPi and, with age decreases progressively from about 6 mg/100 mLGF in the neonate to the adult reference range [2] (Fig. 4). Plasma Pi concentration has a heritable component [3, 4], increases with PI intake [5], and has a diurnal variation [5, 6] (Fig. 5). The adult reference range is the same in men and women and is not affected by age. Pi concentration in the circulation is an essential measure for assessing Pi metabolism. Hypophosphatemia and hyperphosphatemia always reflect underlying disease. Both acute hypophosphatemia and hyperphosphatemia are common findings in intensive care units and contribute to the high morbidity of severely sick patients [7, 8] (Table 1). Chronic hypophosphatemia causes musculoskeletal disorders (Table 1). It leads to delayed bone mineralization producing rickets in children and osteomalacia in adults and to proximal muscle weakness reflecting the key role Pi plays in the chemical energy transfer in skeletal muscle. In contrast, chronic hyperphosphatemia manifests as soft tissue mineralization in subcutaneous, vascular, and nervous tissues (Table 1). Renal failure is the commonest cause of chronic hyperphosphatemia and has a complex pathophysiology [9]. An underlying abnormality is over saturation of the ECF with calcium phosphate and the plasma total Pi x Ca concentration product has been used to estimate this risk. Plasma is metastable with respect to precipitation of calcium phosphate as assessed by the octacalcium ion product. At normal total plasma Ca concentrations of about 10 mg/dL, spontaneous precipitation of calcium phosphate is not expected at plasma concentrations of Pi below 14 mg/dL, i.e., at a product of about 140. In fact, mineralization is found in patients with a product of 70 mg2/dL2 which is half the expected value, clearly indicating that various tissue promoters of crystal formation are also involved. However, the product is no better a predictor than plasma Pi concentration alone [10].

Content of phosphorus in human plasma. Total phosphorus is about 12 mg/dL with 72% as organic and 28% as inorganic; inorganic phosphorus [phosphate] comprises 20% protein bound and 80% free, comprising various ion species. Data redrawn from (1)

Maximum tubular reabsorption phosphate [TmPi] mg/100 ml glomerular filtrate [GF] after an overnight fast, versus age, 6 -18 years, in 291 girls and 273 boys. Data redrawn from (2)

Diurnal variation, % change from baseline, in serum phosphate (Pi), ionized calcium [Ca + +], parathyroid hormone [PTH], and fibroblast growth factor 23 [FGF23] over 48 h in eight healthy men. Range in serum Pi is 40% compared with 8% in Ca + +; range in FGF23 is 10% compared with 50% in PTH. Data redrawn from (6)
Dietary Phosphorus
Diet is the primary source of body phosphorus. Estimation of phosphorus nutrition is assessed from diet history and nutrient databases. For the purposes of nutritional requirements and metabolic balance studies, dietary intake of phosphorus, both organic and inorganic, is usually based on food consumed over a 24-h day/night period. Phosphorus is present in most foods and closely parallels protein content and about 15 mg of phosphorus is ingested with every gram of protein in the USA [11]. About 30% of dietary phosphorus is consumed as animal flesh and as vegetables. A further 30% is consumed as dairy products, about 70% of which is inorganic phosphorus. The remaining 40% is ingested as Pi in foods, food additives for food processing, and as dietary supplements [11] (Fig. 6). Phosphorus content is often absent or poorly documented in food labels, oral supplements, and medications. This uncertainty is further compounded by major underestimates of dietary phosphorus content using nutrient databases as compared with chemical analysis [12]. The recommended daily allowance (RDA) of 700 mg per day and the estimated average requirement (EAR) of 580 mg per day [13, 14] are usually exceeded by about a factor of two in the American diet (Table 2) [15]. The EAR is somewhat arbitrary since chronic dietary phosphorus insufficiency causing osteomalacia/rickets is not readily assessed. The tolerable upper dietary limit (UL) of 4000 mg per day is also questionable since there is evidence that such high intakes may, in some situations, promote bone and cardiovascular disease [16]. Phosphorus is absorbed as Pi. However, bioavailability of phosphorus for absorption is not easily assessed since it depends on completeness of digestion, presence in the diet of various Pi-binding agents, and also on the food source. For example, intake of dietary phytate (inositol hexaphosphate) which has a high content of phosphorus is about 1 g per day and may be much higher in vegetarian diets [17]. Phosphorus in phytate is available to plants and ruminants through digestion by phytase. In humans, however, bioavailability is variable because cooking destroys plant phytase, human digestive enzymes lack phytase, and phytate is largely in the solid state at the pH of the human small intestine where the bulk of Pi is absorbed. Further, some oral medications are strong Pi-binding agents. Aluminum hydroxide, for example, ingested chronically for gastric acidity binds dietary Pi, causes Pi depletion and leads to osteomalacia [18]. Indeed, in chronic renal failure, oral Pi-binding agents are ingested therapeutically to reduce Pi bioavailability and prevent hyperphosphatemia [10, 19].

Dietary phosphorus food sources (11)
Gut: Pi Absorption
The small intestine has a large capacity for Pi transport and is responsible for the bulk of dietary phosphorus that is absorbed. Efficiency of absorption is around 80% with about 20% of the absorbed Pi returning to gut lumen from ECF as endogenous secretions (Fig. 2). Absorption occurs by both paracellular diffusion and active, saturable, transcellular mechanisms that act at low phosphorus intakes [20, 21]. Because dietary phosphorus is usually in surplus of needs, diffusion predominates in humans. However, all the mechanisms involved and their relative magnitude are not yet fully elucidated. It is known that Pi transport from gut lumen into the enterocyte occurs through two families of sodium-dependent solute carriers (Fig. 7). Type II transporter, NaPi2b [SLC34A2], actively transports Pi transcellularly [22]. It is present in the apical membrane of the enterocyte, functions at low luminal concentrations of Pi, and its activity is regulated by 1,25D/VDR. However, the importance of this pathway quantitatively is still uncertain since inactivating mutations of SLC34A2 in humans do not result in major disturbances of phosphate metabolism, although they do result in pulmonary alveolar calcium phosphate microlithiasis [23]. PiT1 [SLC20A1] and PiT 2 [SLC20A2] are also involved in the intracellular transport of Pi by the enterocyte [24]. On the other hand, paracellular Pi transport is regulated by the proton concentration in the enterocyte which is regulated by the sodium hydrogen exchanger NHE3, encoded by SCLC9A3. Studies with Tenapanor, a small molecule designed to act locally in the gut, indicate that the paracellular route is a major gut Pi transporting mechanism that can be manipulated pharmacologically [25]. By inhibiting the activity of NHE3, Tenapanor reduces sodium transport into, and increases proton concentration in the enterocyte. The increase in cell pH selectively increases the intercellular tight junction resistance to Pi transport and reduces Pi gut absorption (Fig. 7) [26]. In patients with renal failure and Pi retention, Tenapanor treatment produces a substantial reduction in serum Pi [27]. Pi absorption can be measured in humans by a radioactive labeled absorption test [28]. It is decreased in a number of diseases including, chronic renal failure, small bowel malabsorption, vitamin D-deficient osteomalacia, hypophosphatemic osteomalacia, and increased in primary hyperparathyroidism, and idiopathic renal stone formation (Fig. 8) [20]. In these diseases, Pi absorption mirrors low and high 1,25D serum concentrations, respectively, emphasizing the central role 125D/VDR in Pi absorption. VDR knockout in the gut of mice reduces Pi absorption efficiency by 50% [29, 30]. However, the effect of low dietary Pi intake to increase absorption [31, 32] appears to be independent of 1,25D/VDR axis [29]. The effect of FGF23 on Pi absorption in humans is largely indirect through a major effect on 1,25D secretion [33]. Endogenous secretion of Pi can be assessed from balance and kinetic studies and is largely determined by ECF Pi concentration (Fig. 2). Unabsorbed phosphorus appearing in feces is measured chemically but provides little useful clinical information except in the context of a metabolic phosphorus balance [12].

Phosphate [Pi] in digestive juice from a meal is absorbed by transcellular and paracellular transport mechanisms in the small intestine. NaPi2b and PiT1 transporters regulate transcellular transport in the enterocyte. 1,25D/VDR promotes Pi transport. NHE3 transporter regulates paracellular Pi transport by setting intracellular proton concentration which controls activity of paracellular channels. Role of FGF23/Klotho-FGFR in Pi absorption, if any, is unknown

Radio phosphorus absorption [32P in 50 mg Pi, fractional rate/hour] in normal subjects, [n = number subjects], and in patients with chronic renal failure, small bowel malabsorption, vitamin D-deficient osteomalacia, hypophosphatemic osteomalacia, primary hyperparathyroidism (PHPT), and idiopathic renal calcium stone disease. Redrawn from (20)
Bone: Pi Transport
The skeleton is the largest reservoir of Pi and importantly is also the source of the major hormone regulating Pi transport, FGF 23. Bone mineral Pi is exchangeable with ECF Pi but this flux does not contribute to ECF Pi homeostasis [34, 35]. There are two fundamental transport pathways for Pi in bone. The first involves the transport of ECF Pi into and out of bone. The second is the transport of Pi within bone from the osteoblast to the apatite crystal. Daily net transport of Pi from ECF to new bone formation and back to ECF by bone resorption is less than that of kidney and gut (Fig. 2). Bone turnover transfers Pi between bone and ECF and controls the formation of new bone and resorption of unwanted or damaged bone. It enables bone growth, fracture repair, and adaptation to mechanical needs. Bone formation, regulated by the sclerostin pathway, and bone resorption, regulated by the RANKL pathway, are normally closely coupled [36, 37]. About 180 mg Pi is deposited in formation and 180 mg is removed by resorption every 24 h. The removal of bone, which initiates the remodeling cycle, is performed by the osteoclast. It resorbs osteoid and by dissolving apatite releases Pi back to the ECF pool. In states in which bone formation and resorption are partly uncoupled, such as with growth or skeletal immobilization, there is net gain or loss, respectively, of Pi between bone and ECF. Mechanisms other than bone turnover have been proposed that allow the bone reservoir to transfer Pi to and from the ECF. In states that require large amounts of Pi and Ca to simultaneously exit the bone reservoir over relatively prolonged periods, such as pregnancy and lactation, mineral surrounding osteocytes is thought to dissolve by a process named osteolytic osteolysis and pass via the canaliculi to the ECF [38, 39]. Once pregnancy and lactation are complete, the process reverses and the depleted mineral is totally replaced. However, not all studies corroborate that such changes occur [40], and mechanisms underlying osteolytic osteolysis still remain to be elucidated. Both the ECF Pi concentration and the internal transport of Pi in bone are essential for the formation of apatite. The Pi concentration in ECF determines, in part, the rate of Pi transported to and from the osteocyte, the cell responsible for maintenance of bone health, the osteoblast, the cell responsible for bone formation, and the lining cell responsible for enveloping bone tissue. In severe chronic hypophosphatemia such as occurs in tumor-induced osteomalacia [41], XLH [42], and vitamin D-deficient osteomalacia [43], the Pi from ECF to the mineralization front is reduced to such an extent that the mineralization rate is severely slowed resulting in rickets in children and osteomalacia in adults (Table1). Within bone, Pi also needs to be generated and transported by the osteoblast for apatite formation. Mature crystals of apatite [Ca10 (PO4)6 (OH)2], about 3 nm thick and about 50 by 25 nm in length and width, are layered into a collagen scaffold [44]. They are in close anatomical contact also with the osteoid proteins and the osteocytes and their canaliculi (Fig. 9). Active Pi transport to and from bone cells is regulated by PiT1&2 [45,46,47,48]. In the osteoblast, Pi is transported to matrix vesicles that contain the machinery required for forming amorphous Ca–Pi [49, 50] (Fig. 10). Matrix vesicles contain orphan phosphatase 1(PHOSPH1), an enzyme essential for Pi production [51]. Microvesicles, about 200 nm in size, are exocytosed to the extracellular matrix where nascent amorphous Ca–Pi crystals accrete more mineral to form mature apatite crystals. Osteoid matrix fluid is oversaturated with respect to Ca–Pi and unwanted mineralization is kept in check by a number of inhibitors. Alkaline phosphatase and ectonucleotide pyrophosphatase phosphodiesterase [ENPP1] regulate the concentration of pyrophosphate, a key inhibitor, in the osteoid matrix and simultaneously regulate the availability of Pi for transport to the matrix vesicle [52,53,54]. In the absence of PHOSPH1 or alkaline phosphatase activity, bone fails to mineralize at a normal rate leading to rickets and osteomalacia, whereas in the absence of ENPP1, generalized arterial calcification of infancy occurs. The sibling proteins, regulated by metalloendopeptidase PEX [52], are also key crystal inhibitors acting at several sites to prevent crystal formation at unwanted locations, such as the osteocytic canaliculi [55, 56]. Thus, both inadequate supply of Pi from the ECF or a failure of Pi transport by the osteoblast lead to a mineralization defect resulting in osteomalacia and rickets.

Phosphate [Pi] to and within bone. Pi is transported from ECF to the osteoblast by Pit1 and PiT2. In the osteoblast Pi along with calcium [Ca] is packaged in microvesicles [MV] and exocytosed to bone matrix. Nascent apatite crystals align themselves with newly formed collagen fibers. Further mineral accretion into mature apatite crystals is regulated by phosphate/pyrophosphate [Pi/PPi] ratio and inhibitory proteins, including dentine matrix protein 1[DMP] and osteopontin [OPN]. The newly formed matrix is intimately associated with osteocytes and their canalicular network

Matrix vesicle phosphate [Pi] arises from three sources. Within the vesicle, orphan phosphatase 1[PHOSPH1] releases Pi from phosphocholine [PC]. Outside the vesicle, Pi released by the activity of tissue nonspecific alkaline phosphatase [TNAP] and by ectonucleotide pyrophosphatase phosphodiesterase [NPP1] on adenosine triphosphate [ATP] is transported into the vesicle via PiT1. The latter increases pyrophosphate [PPi] concentration in the bone matrix
Muscle: Pi Transport
Voluntary muscle is not involved in the overall transport and metabolism of Pi (Fig. 2). Nevertheless, it is included here because proximal myopathy is a prominent feature of chronic hypophosphatemia and rhabdomyolysis results in acute hyperphosphatemia. Lean mass measured by dual x-ray absorptiometry (DXA) in men is about 20 kg/m2 and in women about 15 kg/m2 [57]. Because of its large bulk, voluntary muscle contains a major fraction of the soft tissue phosphorus. It is mainly in the form of organic phosphorus, particularly, ATP and phosphoryl creatinine. Intracellular-free Pi in muscle is about 1–2 mg/dL [3–5 mmol] [58] and is linearly correlated with ECF Pi [59]. Pi transport into muscle cell is regulated by Pit1 and PIT2 transporters [60]. Normal ECF and muscle cell Pi concentrations are essential for maintaining the stores of creatinine phosphate and the functioning of ATP as the energy source for the mechanical activity of muscle [61, 62]. The myopathy due to vitamin D-deficient osteomalacia and tumor-induced osteomalacia rapidly responds to treatment [43], suggesting that an impaired Pi supply to muscle is a primary etiological factor.
Kidney: Pi Reabsorption and Urine Excretion
Kidney handles the major fraction of daily Pi transport (Fig. 2), and is also the source of 1,25D (Fig. 11), a key hormone regulating Pi transport. Renal glomerular filtration transports over 5,000 mg Pi every 24 h to the proximal tubule which reabsorbs over 80% of this back to ECF (Fig. 2). Kidney is the primary organ controlling Pi concentration in the circulation [63]. It performs this function via both the glomerular filtration rate (GFR), and the rate of reabsorption of Pi in the proximal tubule. Children have higher Pi concentration than adults because of a higher TmPi. As GFR decreases, as in chronic renal failure, there is a proportionate decrease in the rate of delivery of Pi to the proximal tubule. Unless there is a compensatory decrease in Pi gut absorption, and/or decrease in Pi tubular reabsorption, and/or increase in net tissue Pi accretion, there is an inevitable rise of Pi concentration in the circulation. Because dietary phosphorus is usually in excess, the gut is unable to compensate for a chronic decrease in GFR. Accretion of Pi in soft tissue and bone is limited except during growth. Thus, compensation for a decrease in GFR largely falls on the tubule’s ability to decrease reabsorption. The rate of Pi reabsorption from the renal filtrate is determined largely by the activity of sodium-phosphate cotransporters type 2 (NaPi2a/c) and type 3 (PiT2) [60, 64,65,66,67] (Fig. 11). Studies using a specific inhibitor of NaPi 2a indicate that NaPi2a plays the major role [68]. Pi is secreted back from the tubule cell to ECF by transporters including XPR1 [69]. The proximal tubule is the main site of Pi reabsorption. It has a tubular maximum capacity for Pi reabsorption (TmPi) of about 2 mg/dL, above which any increase in filtered Pi is excreted in the urine [70]. There is a splay with a range of about 1 mg/100 mL GF over which tubular reabsorption gradually increases before achieving full TmPi. Maximum tubular reabsorption of Pi can best be calculated clinically by measuring plasma Pi and plasma creatinine (Cr), and the corresponding urine Pi and urine Cr in a collection over a suitable time interval of not more than 1 to 2 h with blood collected at midpoint of the urine collection. A 24-h time interval should not be used because blood Pi concentration varies substantially over 24 h leading to incorrect low TmPi. Blood and urine are collected after an overnight fast to reduce variable effects of diet and GFR and optimize reproducibility in the individual. Tubular reabsorption can be expressed as fractional excretion of Pi in relation to creatinine clearance (plasma Pi × urine Cr/plasma Cr × urine Pi). Because GFR varies within and between individuals expressing TmPi, per unit of glomerular filtrate (TmPi/GFR) provides an estimate of TmPi that can be more accurately compared within and between individuals. Based on data from phosphate infusions in healthy subjects TmPi can be derived from nomograms [71] or from equations such as TmPi = P − PE/1–0.1 loge(P/PE) where Pi = plasma Pi, PE = urine Pi x plasma Cr/urine Cr [63]. TmPi is high in the neonate and decreases progressively with age until the adult value is achieved as a teenager (Fig. 4). Urine Pi can be expressed in a number of ways. As a Pi/Cr ratio, it is part of the calculation of TmPi. For nutritional studies, metabolic balance studies, and estimation of urinary risk of stone formation, Pi in a urine collection over a 24-h period is used. In an adult in phosphorus balance, a 24-h urine Pi provides an estimate of dietary phosphorus intake [72, 73]. This is not the case in growing children in positive Pi balance [12], or in subjects ingesting Pi binders [19] where the 24-h urine Pi can almost be reduced to zero. A 24-h urine Pi is only weakly positively related to dietary calcium intake. However, when large amounts of Ca supplements are ingested, as for example, in patients with osteoporosis or hypoparathyroidism, Ca acts as a Pi binder and 24-h urine Pi is reduced [74]. In children there is no sex difference in 24-h urine Pi until skeletal maturity is achieved. Thereafter, because of a higher dietary Pi and GFR, men have higher absolute 24-h urine Pi than women, although they have the same urine Pi/Cr ratio. A 24-h urine Pi is measured as a concentration in assessment of urinary risk factors for renal stone formation [75]. In healthy subjects, Ca–Pi crystalluria is common, is passed without incident, and reflects the major effect of urine pH on oversaturation with Ca–Pi. Thus, although Pi is a common component of urinary stones, its presence is pH dependent and does not reflect an abnormality in Pi metabolism. Calcium phosphate stones predominate in patients with primary hyperparathyroidism, mixed calcium oxalate/phosphate stones in patients with idiopathic stone disease, and magnesium ammonium phosphate stones in patients with infected urine [76]. Importantly, Ca–Pi either as a crystal aggregate in a collecting tubule or as a subepithelial deposit (Randall plaque) in a renal papilla plays a key role as the nidus for renal stone formation [77].

About 80% of glomerular filtrate phosphate [Pi] is reabsorbed by transcellular mechanisms in the proximal tubule. NaPi2a and NaPi2c regulate Pi transport into the tubule cell which is translocated through the cell and transported to ECF by XPR1. The proximal tubule has receptors for both PTHR1 and FGFR/Klotho. The distal tubule has receptors for FGFR/Klotho and the enzymes, CYP27B1 and CYP24A1, responsible for metabolism of 25 hydroxy vitamin D to 1, 25 dihydroxy vitamin D and 24, 25 dihydroxy vitamin D
Endocrine Regulation of Pi: FGF 23, PTH, 1,25D.
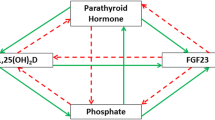
The endocrine regulation of Pi metabolism is carried out by the highly integrated actions of three hormones, FGF 23, PTH, and 1,25D [78, 79]. The central role is played by FGF 23 [80] secreted by osteocytes [81, 82] (Fig. 12). Increases in ECF Pi concentration over days [72, 73] but not acute increases over hours [83] upregulates FGF 23 secretion which in turn via the FGFR/Klotho receptor in the proximal tubule [84, 85] decreases activity of NaP2a and NaP2c. Transcellular Pi transport from luminal fluid back to ECF decreases, resulting in reduced TmPi and decreased ECF Pi [86]. Simultaneously, renal 1,25D production falls resulting in decreased Pi gut absorption [80, 87]. The overall effect is to prevent hyperphosphatemia and maintain normophosphatemia. Conversely, decrease in Pi ECF downregulates FGF 23 secretion resulting in increased TmPi and increased Pi absorption. The overall effect is to prevent hypophosphatemia and maintain normophosphatemia (Fig. 12). The increase in FGF23 due to high Pi intake is more marked than that due to decreased intake suggesting that the primary function of FGF23 is to prevent hyperphosphatemia and ectopic mineralization [72]. The mechanism(s) by which the osteocyte senses ECF Pi concentration remains unestablished [88, 89]. It does not involve a rapidly acting positive-feedback loop [82] but to chronic (over days) changes in ECF Pi [72, 73]. Diurnal variation in serum Pi is not associated with increases in serum FGF23, contrasting with diurnal variation in serum Ca which inversely correlates with changes in serum PTH [6] (Fig. 5). This relatively slow positive-feedback loop response probably lies in the Pi sensing mechanisms. There is evidence from knockout studies in bone cells that FGFR1, which is phosphorylated by high Pi concentration, may be part of the Pi sensing mechanism [90]. In addition, PiT 2 [91] and AMP-activated kinase may be involved in the sensing mechanism [92]. Non-hormonal stimulated FGF23 secretion occurs with low iron status (Fig. 12) [93,94,95] by acting on the posttranslational cleavage of pre-secreted FGF 23 into active and inactive fragments [96]. This O-glycosylation-regulated cleavage may also be involved in the relatively slow response to changes in ECF Pi. Iron status clinically has important implications for FGF23 secretion in common diseases such as chronic renal failure [97]. Whereas FGF23 regulates Pi metabolism, and PTH and 1,25D regulate both Pi and Ca metabolism, Pi ECF concentration does not directly affect PTH secretion [98], although high dietary phosphorus intake [98], oral Pi supplements [72] do so indirectly by reducing ECF Ca concentration. FGF 23 and PTH both decrease TmPi via NaPi2a and NaPi2c, but in contrast to FGF 23 which decreases 1,25D secretion, PTH increases 1,25D secretion (Fig. 12). Thus, these three hormones function as a highly integrated hormonal axis aimed at maintaining both Pi and Ca metabolism and the bone mineral reservoirs. In addition, these three hormones are capable of modulating one another’s secretion in vitro and in vivo animal models. FGF23 decreases PTH secretion [99, 100], 1,25D decreases PTH secretion [101, 102] and increases FGF 23 secretion [103], and PTH increases FGF 23 secretion [104] (Fig. 13). The relative importance of these latter pathways in normal physiology and disease pathophysiology still requires some elucidation. It should be noted that hypophosphatemia caused by severe phosphate depletion [18] is associated with low FGF23 resulting in high TmPi and 1,25D, and low PTH. On the other hand, hypophosphatemia due to a primary increase in FGF 23 secretion from disease, such as TIO and XLH [33], has low TmPi, 1,25D, and high normal PTH.

Increase in ECF phosphate concentration [Pi] stimulates secretion of FGF23 by the osteocyte. Increased FGF 23 acts on renal tubule cell to reduce phosphate reabsorption [TmPi] and decreases ECF Pi, and to reduce 1,25 dihydroxy vitamin D [1,25D] and Pi gut absorption. Decrease in circulating Pi concentration [Pi] reduces secretion of FGF23 which increases TmPi and 125D secretion. Low iron status via non-endocrine mechanism increases FGF 23 secretion

FGF 23, PTH, and 1,25D function as a highly integrated hormonal axis that maintains Pi and Ca metabolism and regulates Pi and Ca mineral reservoirs in the skeleton
Interactions Between Pi And Ca Metabolism
Metabolism of Pi and Ca do not work in isolation from each other. They closely interact at transport mechanisms in ECF, gut, bone, and kidney. Further, changes in ECF concentration of Pi and Ca independently regulate secretion of the hormones, FGF 23, PTH, and 1,25D that maintain Pi and Ca homeostasis. In ECF, oversaturation with octacalcium phosphate leads to soft tissue mineralization, whereas under saturation leads to bone demineralization. Healthy bone formation requires the requisite supply of both Pi and Ca together from the ECF, and when bone resorbs, Pi and Ca are released together to the ECF in amounts equal to their 3/5 molar ratio in apatite. In the diet, Pi and Ca are intimately related. Dairy products are a major source of both dietary Pi and Ca. Breast milk supplies about 150 mg Pi and 180 mg Ca/100 g which combined with casein as milk micelles is a highly absorbable supply for the growing infant. On the other hand, dietary Pi bioavailability is markedly reduced by high intake of Ca supplements, an interaction used therapeutically in chronic renal failure and hypoparathyroidism to reduce increased ECF Pi concentration. In contrast, a high intake of Pi supplements ingested for treatment of hypophosphatemic states impairs calcium absorption, reduces circulating ECF Ca, and leads to secondary hyperparathyroidism. At the kidney, an increase in Ca ECF concentration reduces TmPi, even in the absence of PTH, and in urine oversaturation with Ca–Pi increases the risk of stone formation. These multiple interactions between Pi and Ca emphasize that Pi metabolism cannot be fully understood in isolation from Ca metabolism [105] in either health or disease.
References
Marshall RW (1976) Plasma Fractions. In: Nordin BEC (ed) Calcium, Phosphate, and Magnesium Metabolism. Churchill Livingstone, Edinburgh, pp 162–185
Kruse K, Kracht U, Gopfert G (1982) Renal threshold phosphate concentration (TmPO4/GFR). Arch Dis Child 57:217–223
Whitfield JB, Martin NG (1984) The effects of inheritance on constituents of plasma: a twin study on some biochemical variables. Ann Clin Biochem 21(Pt 3):176–183
Hunter DJ, Lange M, Snieder H, MacGregor AJ, Swaminathan R, Thakker RV, Spector TD (2002) Genetic contribution to renal function and electrolyte balance: a twin study. Clin Sci (Lond) 103:259–265
Portale AA, Halloran BP, Morris RC Jr (1987) Dietary intake of phosphorus modulates the circadian rhythm in serum concentration of phosphorus. Implications for the renal production of 1,25-dihydroxyvitamin D. J Clin Invest 80:1147–1154
Martin KJ, Bell G, Pickthorn K, Huang S, Vick A, Hodsman P, Peacock M (2014) Velcalcetide (AMG 416), a novel peptide agonist of the calcium-sensing receptor, reduces serum parathyroid hormone and FGF23 levels in healthy male subjects. Nephrol Dial Transplant 29:385–392
Shiber JR, Mattu A (2002) Serum phosphate abnormalities in the emergency department. J Emerg Med 23:395–400
Subramanian R, Khardori R (2000) Severe hypophosphatemia: Pathophysiologic implications, clinical presentations, and treatment. Medicine 79:1–8
Block GA, Ix JH, Ketteler M, Martin KJ, Thadhani RI, Tonelli M, Wolf M, Juppner H, Hruska K, Wheeler DC (2013) Phosphate homeostasis in CKD: report of a scientific symposium sponsored by the national kidney foundation. Am J Kidney Dis 62:457–473
Block GA, Hulbert-Shearon TE, Levin NW, Port FK (1998) Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 31:607–617
Calvo MS, Uribarri J (2013) Contributions to total phosphorus intake: all sources considered. Semin Dial 26:54–61
Vorland CJ, Martin BR, Weaver CM, Peacock M, Gallant KMH (2018) Phosphorus balance in adolescent girls and the effect of supplemental dietary calcium. JBMR Plus 2:103–108
Medicine IO (ed) (1997) DRI dietary reference intakes for calcium, phosphorus, magnesium, vitamin D, and fluoride. The National Academies Press, Washington, DC, pp 1–413
Yates AA, Schlicker SA, Suitor CW (1998) Dietary Reference Intakes: the new basis for recommendations for calcium and related nutrients, B vitamins, and choline. J Am Diet Assoc 98:699–706
Fulgoni VL 3rd, Keast DR, Bailey RL, Dwyer J (2011) Foods, fortificants, and supplements: where do americans get their nutrients? J Nutr 141:1847–1854
Calvo MS, Uribarri J (2013) Public health impact of dietary phosphorus excess on bone and cardiovascular health in the general population. Am J Clin Nutr 98:6–15
Schlemmer U, Frolich W, Prieto RM, Grases F (2009) Phytate in foods and significance for humans: food sources, intake, processing, bioavailability, protective role and analysis. Mol Nutr Food Res 53(Suppl 2):S330–375
Lotz M, Zisman E, Bartter FC (1968) Evidence for a phosphorus-depletion syndrome in man. N Engl J Med 278:409–415
Kazama JJ (2009) Oral phosphate binders: history and prospects. Bone 45(Suppl 1):S8–12
Wilkinson R (1976) Absorption of calcium, phosphorus and magnesium. In: Nordin BEC (ed) Calcium phosphate and magnesium metabolism. Churchill Livingstone, Edinburgh, pp 36–112
Fleet JC, Peacock M (2014) Physiology of vitamin D, calcium, and phosphate absorption. In: Morris AH, Anderson P, Nordin BEC (eds) The physiological basis of metabolic bone disease. CRC Press, Boca Raton, pp 13–40
Sabbagh Y, Giral H, Caldas Y, Levi M, Schiavi SC (2011) Intestinal phosphate transport. Adv Chronic Kidney Dis 18:85–90
Stokman L, Nossent EJ, Grunberg K, Meijboom L, Yakicier MC, Voorhoeve E, Houweling AC (2016) A case of pulmonary alveolar microlithiasis associated with a homozygous 195 kb deletion encompassing the entire SLC34A2 gene. Clin Case Rep 4:412–415
Candeal E, Caldas YA, Guillen N, Levi M, Sorribas V (2017) Intestinal phosphate absorption is mediated by multiple transport systems in rats. Am J Physiol Gastrointest Liver Physiol 312:G355–G366
Labonte ED, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Dy E, Black D, Zhong Z, Langsetmo I, Spencer AG, Bell N, Deshpande D, Navre M, Lewis JG, Jacobs JW, Charmot D (2015) Gastrointestinal inhibition of sodium-hydrogen exchanger 3 reduces phosphorus absorption and protects against vascular calcification in CKD. J Am Soc Nephrol 26:1138–1149
King AJ, Siegel M, He Y, Nie B, Wang J, Koo-McCoy S, Minassian NA, Jafri Q, Pan D, Kohler J, Kumaraswamy P, Kozuka K, Lewis JG, Dragoli D, Rosenbaum DP, O'Neill D, Plain A, Greasley PJ, Jonsson-Rylander AC, Karlsson D, Behrendt M, Stromstedt M, Ryden-Bergsten T, Knopfel T, Pastor Arroyo EM, Hernando N, Marks J, Donowitz M, Wagner CA, Alexander RT, Caldwell JS (2018) Inhibition of sodium/hydrogen exchanger 3 in the gastrointestinal tract by tenapanor reduces paracellular phosphate permeability. Sci Transl Med 10:eaam6474
Block GA, Rosenbaum DP, Leonsson-Zachrisson M, Astrand M, Johansson S, Knutsson M, Langkilde AM, Chertow GM (2017) Effect of tenapanor on serum phosphate in patients receiving hemodialysis. J Am Soc Nephrol 28:1933–1942
Walker GS, Peacock M, Marshall DH, Giles GR, Davison AM (1980) Factors influencing the intestinal absorption of calcium and phosphorus following renal transplantation. Nephron 26:225–229
Segawa H, Kaneko I, Yamanaka S, Ito M, Kuwahata M, Inoue Y, Kato S, Miyamoto K (2004) Intestinal Na-P(i) cotransporter adaptation to dietary P(i) content in vitamin D receptor null mice. Am J Physiol Renal Physiol 287:F39–47
Capuano P, Radanovic T, Wagner CA, Bacic D, Kato S, Uchiyama Y, St-Arnoud R, Murer H, Biber J (2005) Intestinal and renal adaptation to a low-Pi diet of type II NaPi cotransporters in vitamin D receptor- and 1alphaOHase-deficient mice. Am J Physiol Cell Physiol 288:C429–434
Fox J, Care AD (1978) Effect of low calcium and low phosphorus diets on the intestinal absorption of phosphate in intact and parathyroidectomized pigs. J Endocrinol 77:225–231
Danisi G, Caverzasio J, Trechsel U, Bonjour JP, Straub RW (1990) Phosphate transport adaptation in rat jejunum and plasma level of 1,25-dihydroxyvitamin D3. Scand J Gastroenterol 25:210–215
Zhang X, Imel EA, Ruppe MD, Weber TJ, Klausner MA, Ito T, Vergeire M, Humphrey J, Glorieux FH, Portale AA, Insogna K, Carpenter TO, Peacock M (2016) Pharmacokinetics and pharmacodynamics of a human monoclonal anti-FGF23 antibody (KRN23) in the first multiple ascending-dose trial treating adults with X-linked hypophosphatemia. J Clin Pharmacol 56:176–185
Armstrong WD (1955) Radioisotope studies of the physiology of calcified tissues. Minn Med 38:618–622
Dias RS, Kebreab E, Vitti DM, Roque AP, Bueno IC, France J (2006) A revised model for studying phosphorus and calcium kinetics in growing sheep. J Anim Sci 84:2787–2794
Koide M, Kobayashi Y, Yamashita T, Uehara S, Nakamura M, Hiraoka BY, Ozaki Y, Iimura T, Yasuda H, Takahashi N, Udagawa N (2017) Bone formation is coupled to resorption via suppression of sclerostin expression by osteoclasts. J Bone Miner Res 32:2074–2086
Langdahl B, Ferrari S, Dempster DW (2016) Bone modeling and remodeling: potential as therapeutic targets for the treatment of osteoporosis. Ther Adv Musculoskelet Dis 8:225–235
Belanger LF (1969) Osteocytic osteolysis. Calcif Tissue Res 4:1–12
Wysolmerski JJ (2012) Osteocytic osteolysis: time for a second look. BoneKEy Rep 229:1–7
Wittig NK, Birkbak ME, Bach-Gansmo FL, Pacureanu A, Wendelboe MH, Bruel A, Thomsen JS, Birkedal H (2019) No Signature of osteocytic osteolysis in cortical bone from lactating NMRI Mice. Calcif Tissue Int 105:308–315
Minisola S, Peacock M, Fukumoto S, Cipriani C, Pepe J, Tella SH, Collins MT (2017) Tumour-induced osteomalacia. Nat Rev Dis Primers 3:17044
Imel EA, Peacock M (2010) X-linked hypophosphatemia: understanding and management. Drugs Fut 35:755–763
Peacock M (1993) Osteomalacia and rickets. In: Nordin BEC, Need AG, Morris HA (eds) Metabolic bone and stone disease. Churchill Livingstone, London, pp 83–118
Wang L, Nancollas GH, Henneman ZJ, Klein E, Weiner S (2006) Nanosized particles in bone and dissolution insensitivity of bone mineral. Biointerphases 1:106–111
Cowan CM, Zhang X, James AW, Kim TM, Sun N, Wu B, Ting K, Soo C (2012) NELL-1 increases pre-osteoblast mineralization using both phosphate transporter Pit1 and Pit2. Biochem Biophys Res Commun 422:351–357
Zoidis E, Ghirlanda-Keller C, Gosteli-Peter M, Zapf J, Schmid C (2004) Regulation of phosphate (Pi) transport and NaPi-III transporter (Pit-1) mRNA in rat osteoblasts. J Endocrinol 181:531–540
Albano G, Moor M, Dolder S, Siegrist M, Wagner CA, Biber J, Hernando N, Hofstetter W, Bonny O, Fuster DG (2015) Sodium-dependent phosphate transporters in osteoclast differentiation and function. PLoS ONE 10:e0125104
Suzuki A, Ghayor C, Guicheux J, Magne D, Quillard S, Kakita A, Ono Y, Miura Y, Oiso Y, Itoh M, Caverzasio J (2006) Enhanced expression of the inorganic phosphate transporter Pit-1 is involved in BMP-2-induced matrix mineralization in osteoblast-like cells. J Bone Miner Res 21:674–683
Bottini M, Mebarek S, Anderson KL, Strzelecka-Kiliszek A, Bozycki L, Simao AMS, Bolean M, Ciancaglini P, Pikula JB, Pikula S, Magne D, Volkmann N, Hanein D, Millan JL, Buchet R (2018) Matrix vesicles from chondrocytes and osteoblasts: Their biogenesis, properties, functions and biomimetic models. Biochim Biophys Acta Gen Subj 1862:532–546
Bolean M, Simao AMS, Barioni MB, Favarin BZ, Sebinelli HG, Veschi EA, Janku TAB, Bottini M, Hoylaerts MF, Itri R, Millan JL, Ciancaglini P (2017) Biophysical aspects of biomineralization. Biophys Rev 9:747–760
Dillon S, Staines KA, Millan JL, Farquharson C (2019) How To Build a Bone: PHOSPHO1, Biomineralization, and Beyond. JBMR Plus 3:e10202
McKee MD, Hoac B, Addison WN, Barros NM, Millan JL, Chaussain C (2000) Extracellular matrix mineralization in periodontal tissues: Noncollagenous matrix proteins, enzymes, and relationship to hypophosphatasia and X-linked hypophosphatemia. Periodontol 2013(63):102–122
Millan JL (2013) The Role of Phosphatases in the Inititiation of Skeletal Mineralization. Calcif Tiss Int 93:299–306
Hatch NE, Franceschi RT (2009) Osteoblast differentiation stage-specific expression of the pyrophosphate-generating enzyme PC-1. Cells Tissues Org 189:65–69
Foster BL, Ao M, Salmon CR, Chavez MB, Kolli TN, Tran AB, Chu EY, Kantovitz KR, Yadav M, Narisawa S, Millan JL, Nociti FH Jr, Somerman MJ (2018) Osteopontin regulates dentin and alveolar bone development and mineralization. Bone 107:196–207
Qin C, Baba O, Butler WT (2004) Post-translational modifications of sibling proteins and their roles in osteogenesis and dentinogenesis. Crit Rev Oral Biol Med 15:126–136
Kelly TL, Wilson KE, Heymsfield SB (2009) Dual energy X-Ray absorptiometry body composition reference values from NHANES. PLoS ONE 4:e7038
Kemp GJ, Meyerspeer M, Moser E (2007) Absolute quantification of phosphorus metabolite concentrations in human muscle in vivo by 31P MRS: a quantitative review. NMR Biomed 20:555–565
Bevington A, Mundy KI, Yates AJ, Kanis JA, Russell RG, Taylor DJ, Rajagopalan B, Radda GK (1986) A study of intracellular orthophosphate concentration in human muscle and erythrocytes by 31P nuclear magnetic resonance spectroscopy and selective chemical assay. Clin Sci (Lond) 71:729–735
Kavanaugh MP, Kabat D (1996) Identification and characterization of a widely expressed phosphate transporter/retrovirus receptor family. Kidney Int 49:959–963
Pesta DH, Tsirigotis DN, Befroy DE, Caballero D, Jurczak MJ, Rahimi Y, Cline GW, Dufour S, Birkenfeld AL, Rothman DL, Carpenter TO, Insogna K, Petersen KF, Bergwitz C, Shulman GI (2016) Hypophosphatemia promotes lower rates of muscle ATP synthesis. FASEB J 30:3378–3387
Wallimann T, Tokarska-Schlattner M, Schlattner U (2011) The creatine kinase system and pleiotropic effects of creatine. Amino Acids 40:1271–1296
Peacock M (2018) Hypoparathyroidism and the Kidney. Endocrinol Metab Clin North Am 47:839–853
Virkki LV, Biber J, Murer H, Forster IC (2007) Phosphate transporters: a tale of two solute carrier families. Am J Physiol Renal Physiol 293:F643–654
Biber J, Hernando N, Forster I (2013) Phosphate transporters and their function. Annu Rev Physiol 75:535–550
Forster IC (2019) The molecular mechanism of SLC34 proteins: insights from two decades of transport assays and structure-function studies. Pflugers Arch 471:15–42
Forster IC, Hernando N, Biber J, Murer H (2013) Phosphate transporters of the SLC20 and SLC34 families. Mol Aspects Med 34:386–395
Thomas L, Xue J, Murali SK, Fenton RA, Dominguez Rieg JA, Rieg T (2019) Pharmacological Npt2a inhibition causes phosphaturia and reduces plasma phosphate in mice with normal and reduced kidney function. J Am Soc Nephrol 30:2128–2139
Ansermet C, Moor MB, Centeno G, Auberson M, Hu DZ, Baron R, Nikolaeva S, Haenzi B, Katanaeva N, Gautschi I, Katanaev V, Rotman S, Koesters R, Schild L, Pradervand S, Bonny O, Firsov D (2016) Renal fanconi syndrome and hypophosphatemic rickets in the absence of xenotropic and polytropic retroviral receptor in the nephron. J Am Soc Nephrol 28:1073–1078
Bijvoet OL, Morgan DB, Fourman P (1969) The assessment of phosphate reabsorption. Clin Chim Acta 26:15–24
Walton RJ, Bijvoet OL (1975) Nomogram for derivation of renal threshold phosphate concentration. Lancet 2:309–310
Burnett SM, Gunawardene SC, Bringhurst FR, Juppner H, Lee H, Finkelstein JS (2006) Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res 21:1187–1196
Ferrari SL, Bonjour JP, Rizzoli R (2005) Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab 90:1519–1524
Lau AH, Kuk JM, Franson KL (1998) Phosphate-binding capacities of calcium and aluminum formulations. Int J Artif Organs 21:19–22
Robertson WG, Peacock M, Heyburn PJ, Marshall DH, Clark PB (1978) Risk factors in calcium stone disease of the urinary tract. Br J Urol 50:449–454
Hodgkinson A, Peacock M, Nicholson M (1969) Quantitative analysis of calcium-containing urinary calculi. Invest Urol 6:549–561
Coe FL, Evan AP, Worcester EM, Lingeman JE (2010) Three pathways for human kidney stone formation. Urol Res 38:147–160
Lederer E (2014) Regulation of serum phosphate. J Physiol 592:3985–3995
Blau JE, Collins MT (2015) The PTH-Vitamin D-FGF23 axis. Rev Endocr Metab Disord 16:165–174
Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T (2004) FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 19:429–435
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE (2006) Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 38:1310–1315
Feng JQ, Ye L, Schiavi S (2009) Do osteocytes contribute to phosphate homeostasis? Curr Opin Nephrol Hypertens 18:285–291
Ito N, Fukumoto S, Takeuchi Y, Takeda S, Suzuki H, Yamashita T, Fujita T (2007) Effect of acute changes of serum phosphate on fibroblast growth factor (FGF)23 levels in humans. J Bone Miner Metab 25:419–422
Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444:770–774
Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro M (2006) Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281:6120–6123
Baum M, Schiavi S, Dwarakanath V, Quigley R (2005) Effect of fibroblast growth factor-23 on phosphate transport in proximal tubules. Kidney Int 68:1148–1153
Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T (2004) Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 113:561–568
Bergwitz C, Juppner H (2011) Phosphate sensing. Adv Chronic Kidney Dis 18:132–144
Sabbagh Y (2013) Phosphate as a sensor and signaling molecule. Clin Nephrol 79:57–65
Takashi Y, Kosako H, Sawatsubashi S, Kinoshita Y, Ito N, Tsoumpra MK, Nangaku M, Abe M, Matsuhisa M, Kato S, Matsumoto T, Fukumoto S (2019) Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc Natl Acad Sci USA 116:11418–11427
Bon N, Frangi G, Sourice S, Guicheux J, Beck-Cormier S, Beck L (2018) Phosphate-dependent FGF23 secretion is modulated by PiT2/Slc20a2. Mol Metab 11:197–204
Glosse P, Feger M, Mutig K, Chen H, Hirche F, Hasan AA, Gaballa MMS, Hocher B, Lang F, Foller M (2018) AMP-activated kinase is a regulator of fibroblast growth factor 23 production. Kidney Int 94:491–501
Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ (2011) Iron Modifies Plasma FGF23 Differently in Autosomal Dominant Hypophosphatemic Rickets and Healthy Humans. J Clin Endocrinol Metab 96:3541–3549
Wolf M, Koch TA, Bregman DB (2013) Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J Bone Miner Res 28:1793–1803
Clinkenbeard EL, Farrow EG, Summers LJ, Cass TA, Roberts JL, Bayt CA, Lahm T, Albrecht M, Allen MR, Peacock M, White KE (2014) Neonatal iron deficiency causes abnormal phosphate metabolism by elevating FGF23 in normal and ADHR mice. J Bone Miner Res 29:361–369
White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ (2001) Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int 60:2079–2086
Wolf M, White KE (2014) Coupling fibroblast growth factor 23 production and cleavage: iron deficiency, rickets, and kidney disease. Curr Opin Nephrol Hypertens 23:411–419
Estepa JC, Aguilera-Tejero E, Lopez I, Almaden Y, Rodriguez M, Felsenfeld AJ (1999) Effect of phosphate on parathyroid hormone secretion in vivo. J Bone Miner Res 14:1848–1854
Krajisnik T, Bjorklund P, Marsell R, Ljunggren O, Akerstrom G, Jonsson KB, Westin G, Larsson TE (2007) Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol 195:125–131
Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro M, Mohammadi M, Sirkis R, Naveh-Many T, Silver J (2007) The parathyroid is a target organ for FGF23 in rats. J Clin Invest 117:4003–4008
Chertow BS, Baylink DJ, Wergedal JE, Su MH, Norman AW (1975) Decrease in serum immunoreactive parathyroid hormone in rats and in parathyroid hormone secretion in vitro by 1,25-dihydroxycholecalciferol. J Clin Invest 56:668–678
Jaaskelainen T, Huhtakangas J, Maenpaa PH (2005) Negative regulation of human parathyroid hormone gene promoter by vitamin D3 through nuclear factor Y. Biochem Biophys Res Commun 328:831–837
Kolek OI, Hines ER, Jones MD, LeSueur LK, Lipko MA, Kiela PR, Collins JF, Haussler MR, Ghishan FK (2005) 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol 289:G1036–G1042
Rhee Y, Bivi N, Farrow E, Lezcano V, Plotkin LI, White KE, Bellido T (2011) Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone 49:636–643
Peacock M (2010) Calcium metabolism in health and disease. Clin J Am Soc Nephrol 5(Suppl 1):S23–30
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Munro Peacock declare that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Peacock, M. Phosphate Metabolism in Health and Disease. Calcif Tissue Int 108, 3–15 (2021). https://doi.org/10.1007/s00223-020-00686-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-020-00686-3