
Abstract
Data have accumulated to show that various types of collagen crosslinking are implicated in the health of individuals, as well as in a number of disease states, such as osteoporosis, diabetes mellitus, chronic kidney disease, inflammatory bowel disease, or in conditions of mild hyperhomocysteinemia, or when glucocorticoid use is indicated. Collagen crosslinking is a posttranslational modification of collagen molecules and plays important roles in tissue differentiation and in the mechanical properties of collagenous tissue. The crosslinking of collagen in the body can form via two mechanisms: one is enzymatic crosslinking and the other is nonenzymatic crosslinking. Lysyl hydroxylases and lysyl oxidases regulate tissue-specific crosslinking patterns and quantities. Enzymatic crosslinks initially form via immature divalent crosslinking, and a portion of them convert into mature trivalent forms such as pyridinoline and pyrrole crosslinks. Nonenzymatic crosslinks form as a result of reactions which create advanced glycation end products (AGEs), such as pentosidine and glucosepane. These types of crosslinks differ in terms of their mechanisms of formation and function. Impaired enzymatic crosslinking and/or an increase of AGEs have been proposed as a major cause of bone fragility associated with aging and numerous disease states. This review focuses on the effects of collagen crosslinking on bone material properties in health and disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bone is a two-phase composite material in which the mineral phase provides stiffness and the collagen fibers provide tensile strength and the postyield property of ductility [1]. The function of bone collagen is to provide protection against external mechanical loads and to act as a scaffold for bone cells. The determinants of bone quality include its microarchitecture, degree of mineralization, accumulated microdamage, and collagen crosslink formation [2]. The degrees of mineralization and microarchitecture are mainly regulated by bone turnover, whereas collagen crosslink formation is regulated not only by bone turnover, but also by the extent of oxidative stress and glycation (Fig. 1). Oxidative stress deteriorates bone formation via cellular dysfunction [3]. Recently, data have accumulated to indicate that enzymatic and nonenzymatic crosslinking of collagen affect the mineralization process and bone strength. Impaired enzymatic crosslinking, involving immature divalent and mature trivalent crosslinks, and/or an increase in nonenzymatic crosslinking including advanced glycation end products (AGEs) have been proposed as a major cause of bone fragility associated with aging and disease states such as diabetes mellitus, chronic kidney disease (CKD), and mild hyperhomocysteinemia (Fig. 1). In this review, we summarize the mechanisms responsible for the formation of bone collagen crosslinking and their effects on bone material properties in health and disease.

Plausible mechanisms for reduced bone strength in aging and disease states. BMD depending on degree of mineralization and bone volume is mainly regulated by bone turnover, whereas collagen crosslink formation is regulated by not only bone turnover rate, but also the activities of lysyl oxidases and lysine hydroxylases secreted from osteoblasts, the levels of oxidative stress, and the amount of glycation. Impairment of immature divalent and mature trivalent enzymatic crosslinking and/or an increase in AGEs resulting from nonenzymatic crosslinking have been proposed as major causes of bone fragility in states of estrogen deficiency, progressive aging, hyperhomocysteinemia, and chronic disease states such as diabetes and renal failure
Posttranslational Modifications—Collagen Crosslinking
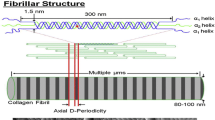
Type I collagen is the major organic constituent in bone. Type I collagen is composed of two nonhelical domains called telopeptides at the amino-terminal (N) and carboxyl-terminal (C), and a triple helical region. Crosslinking is a major posttranslational modification of collagen. Newly secreted collagen molecules are stabilized by the formation of crosslinks between neighboring collagen molecules. Covalent crosslinks form enzymatically between the nonhelical domains and the helical domain of adjacent collagen molecules, and they are the covalent bonds.
Collagen crosslinking can be divided into two distinct types based on the key mechanism by which it originates: (1) lysyl hydroxylase- and lysyl oxidase-mediated enzymatic crosslinks; and (2) glycation- or oxidation-induced nonenzymatic AGE crosslinks. Enzymatic and nonenzymatic AGE crosslinks exhibit unique functions in terms of the structural stabilization they provide.
Enzymatic Crosslink Formation—Immature and Mature Crosslinks
Enzymatic crosslink formation in collagen fibers is regulated by the action of two types of enzymes: (1) the lysyl hydroxylases (LHs or procollagen-lysine, 2-oxyglutarate, 5-dioxigenases [PLODs]) and (2) lysyl oxidases (LOX, or protein-lysine 6-oxidases) [4, 5]. The extent of lysine (Lys) hydroxylation of precursor crosslinks is regulated intracellularly by the expression of PLODs, and the resulting tissue-specific pattern of collagen crosslinks. After hydroxylation of Lys, collagen molecules are secreted into the extracellular space. LOX only acts on extracellular collagen fibers and specifically binds to Lys or hydroxylysine (Hyl) residues in the telopeptides to regulate total enzymatic crosslinking (Fig. 2).

The pathways of enzymatic (immature and mature) and nonenzymatic crosslink formations in collagen. The intracellular enzyme lysine hydroxylase and extracellular enzyme lysyl oxidase control the pattern and the quantity of tissue-specific enzymatic crosslinking. AGEs are formed via nonenzymatic glycation, oxidation, or glycoxidation between the helical Lys or Hyl residues. DeH-DHLNL dehydro-dihydroxylysinonorleucine, de-H-HLNL dehydro-hydroxylysinonorleucine, de-H-LNL dehydro-lysinonorleucine, CML carboxyl methyl lysine
Lysine Hydroxylation Regulates Tissue-Specific Enzymatic Crosslinking Patterns
The specific crosslinking sites in collagen molecules are at telopeptidyl Lys or Hyl and helical Lys or Hyl residues. Tissue-specific patterns of crosslinking are predominantly dependent on the differential activity of the lysyl hydroxylases which determines the conversion of Lys into Hyl (Fig. 2). Although type I collagen in all tissues is genetically identical, the tissue-specific ratio of Hyl/Lys-derived enzymatic crosslinking during the mineralization process [4–8, 9], and in woven bone [10], mature bone [10–14], tendon [4, 15–18], ligament [7, 15, 17–19], and skin [4, 9, 20, 21], relies on the extent of Lys hydroxylation. Such posttranslational modifications play important roles in the expressions of physiological distinctive functions of collagen during the differentiation process in tissues [21–23]. Thus, the Hyl aldehyde (Hylald)-derived crosslinks, dehydro-dihydroxylysinonorleucine (deH-DHLNL) and dehydro-hydroxylysinonorleucine (deH-HLNL), as well as their mature forms which include pyridinium and pyrrole crosslinks, represent the major types of crosslinking present in bone.
Various isoforms of LH have been identified (e.g., LH1, 2, and 3) [22]. LH2 and its spliced variant of LH2b are thought to be crucial enzymes for catalyzing hydroxylation of the nontriple helical regions or the telopeptide domain, of collagen molecules [6, 8, 24]. The distinctive expression of LH2b in osteoblastic cells is crucial to the induction of the type of enzymatic crosslinking pattern that influences mineralization of the matrix in bone (Fig. 2) [5, 6, 8, 25]. Helical Lys hydroxylation regulated by LH1 determines the ratio of pyridinoline (PYD) to deoxypyridinoline (DPD) and the ratio of pyrrololine (PYL) to deoxypyrrololine (DPL). Lys hydroxylation of telopeptides and its associated influence over the formation of Hylald-derived crosslinks are due to an upregulation of LH2 expression and occur before the onset of the mineralization [5, 6, 8, 25]. Interestingly, an overmodification of Hyl, and an excessive induction of aldehyde-derived crosslink formation has been shown to result in an inhibition of matrix mineralization and collagen fibrillogenesis in vitro [7, 8].
Pyridinium collagen crosslinks, such as PYD, predominate in tendon, ligament, and cartilage [9, 11, 15, 17, 18, 26]. In contrast, DPD is relatively specific for calcified tissue such as human bone [9, 11], dentin [27–29], and mineralized matrix in osteoblast cell cultures [5, 6, 25, 30, 31]. It has been demonstrated using X-ray diffraction that the types of collagen crosslinking profiles are associated with the type of molecular packing that occurs within fibrils [19]. For instance, nucleation of calcium apatite crystals starts in gap regions, or in areas adjacent to crosslinking sites [19].
Lysyl Oxidase and Total Enzymatic Crosslinking
Tissue-specific enzymatic crosslinking patterns are regulated by LHs, whereas the total amount of enzymatic crosslinking is controlled by the action of LOX. After collagen molecules are secreted extracellularly, the initiation of crosslinking is via conversion of the epsilon (ε)-amino groups of specific Lys and Hyl residues located in the telopeptide domains, into their active aldehyde derivatives, allysine and hydroxyallysine, respectively (Fig. 2). LOX aggregates collagen molecules into fibers [32]. Under physiological conditions, excessive formation of enzymatic crosslinking cannot occur in vitro [6, 8, 25, 30] or in vivo [9–11, 13, 14, 16, 29] because formation of enzymatic crosslinking is regulated by the expression of LOX. LOX is a copper metalloenzyme that requires pyridoxal phosphate (vitamin B6) and tyrosyl-lysine quinone as essential cofactors [33, 34]. We have previously demonstrated that a vitamin B6 deficiency in normal rats induced a 25% reduction in the formation of LH- and LOX-mediated enzymatic immature crosslinks in bone compared with rats fed a vitamin-B6-replete diet [35]. It is well known that a vitamin B6 deficiency commonly occurs in people with diabetes and has adverse effects on crosslink formation in this population [36]. However, there is little information available regarding the regulation of expression and activation of LOX. Transforming growth factor beta (TGF-β) [37], connective tissue growth factor (CTGF) [38], and insulin-like growth factor 1 (IGF-1) [39] act as positive regulators of LOX. Estrogen is also a positive regulator of LOX [40]. LOX activity was shown to decrease by 75 % 3 days after ovariectomy (OVX) in a mouse model, but LOX activity was completely rescued by estradiol injections. Active vitamin D3 such as 1,25-dihydroxyvitamin D3 (1,25[OH]2D3) elevated the expression of LOX in an osteoblastic cell culture system [41]. The other active vitamin D, alfacalcidol, induced enzymatic crosslinking in the bones of OVXed rats [10]. It is generally thought that the conversion of enzymatic crosslinks from immature divalent to mature trivalent crosslinks occurs via a spontaneous reaction in a time-dependent manner. Mechanical stress may accelerate this conversion rate. Despite the recent findings of an in vitro study using osteoblastic MC3T3-E1 cells, low-intensity pulsed ultrasound (LIPUS) [6] and gravitational load [30] increased not only the lysyl oxidase mRNA expression and total amount of enzymatic crosslinks, but also the conversion rate of immature divalent to mature trivalent pyridinium crosslinks. These results suggest that various types of mechanical stress accelerate the stabilization of collagen through the conversion of immature crosslinks to mature pyridinium compounds. Therefore, it is possible that some as yet unknown factor, which is regulated by alterations in the mechanical environment, including the therapeutic levels of LIPUS and proper gravitational loading, may thus affect the crosslink maturation pathway. However, this hypothesis remains to be proven and thus requires further experimentation.
Reports also exist for compounds that can inhibit LOX. Homocysteine (HCY) acts as a negative regulator of LOX [42, 43]. Furthermore, HCY binds to the aldehydic groups of allysine and hydroxyallysine, which are the precursors of enzymatic crosslinking, and may inhibit enzymatic crosslinking [44]. Thus, HCY inhibits LOX activities at both the gene and protein level. Interestingly, a mildly hyperhomocysteinemic population was recognized as having reduced bone quality in terms of collagen crosslink formation [45, 46]. Other negative regulators of LOX are fibroblast growth factor (FGF) [47], high doses of prostaglandin E2 [48], and tumor necrosis factor alpha (TNF-α) [49]. Impaired enzymatic crosslinking induced by β-aminopropionitrile (BAPN) treatment has inhibited the differentiation of the osteoblastic cell line MC3T3-E1 and collagen crosslink formation [50, 51], indicating that enzymatic crosslinking itself can adversely affect osteoblastic differentiation.
Initially, enzymatic crosslinks form as divalent immature covalent bonds that include deH-DHLNL, deH-HLNL, and deH-LNL [52]. Specific crosslinking sites in collagen molecules are at the telopeptidyl Lys aldehydes or allysine and Hyl aldehydes or hydroxyallysin) located at position 9 in the N-terminal telopeptide and position 16 in the C-terminal telopeptide, respectively; they react and condensate with Lys or Hyl residues located at positions 930 and 87, respectively, in the triple helical region of an adjacent collagen molecule. A portion of immature divalent crosslinks undergo a spontaneous reaction with another telopeptidyl Lys or Hyl aldehyde to form mature trivalent pyridinium or pyrrole crosslinks (Fig. 2). Divalent crosslinking has been shown to markedly decrease in association with the aging in human bone [53], tendon, and skin [54], as well as in bovine specimens [55].
Pyridinium crosslinking types such as PYD [56] and DPD [57] are formed via the hydroxyallysine pathway. The proposed mechanism of pyridinium crosslinking formation is derived from two divalent immature crosslinks, or via a rearrangement between a divalent crosslink and a free hydroxyallysine in another collagen telopeptide [57–59]. In contrast, the proposed mechanism of pyrrole crosslinking formation is via condensation of de-HLNL/de-DHLNL and a reaction with another allysine [60, 61], or by interaction of two divalent immature crosslinks including deH-HLNL [62]. The concentration of pyrrole crosslinking varied from about half to an equivalent concentration to that of PYD in bone [63]. Enzymatic mature pyrrole crosslinks are equally important as pyridinium crosslinks. Pyrrole crosslinks are unstable during acid hydrolysis, and therefore we cannot measure them using the conventional HPLC method. Because of the difficulty of the pyrrole measurement, its function is not clear. Regarding the effect of pyrrole crosslinks on bone strength, we describe in the section of “Collagen crosslinking and bone strength”. Since the major determinant of the total amount of immature, lysinonorleucine-type and their mature forms such as pyridinium and pyrrole is lysyl oxidase activity, overall formation of pyrrole crosslinks might be similar to other lysyl oxidase-controlled crosslinking.
To elucidate the function of enzymatic crosslinking, several investigations have focused only on mature trivalent crosslinks without accounting for the presence of divalent immature crosslinks [64, 65]. Immature crosslinking, as well as the mature form of crosslinking, affects the physiological function of bone [5–8, 10–14, 30, 35, 36, 41, 45, 53–55, 66, 67]. Immature crosslinks are found in bone most frequently, because the conversion rate of immature into mature forms may be lower than that for other connective tissues, such as tendons and ligaments [4, 9, 15–19, 21]. The result is that immature crosslinks are present at 2–4 times the content of mature pyridinium crosslinks. It was reported that the immature crosslinks in bone are an independent determinant of bone strength in an OVXed monkey model [14]. Thus, the immature divalent enzymatic crosslinks are the major type of collagen crosslink found in bone tissue. In addition, the relative amounts of immature crosslinking can reflect changes in bone metabolic turnover and the effect of an administered drug(s) with a higher sensitivity than is the case with mature crosslinking [14, 35, 36, 67]. These observations have led to the proposal that a simultaneous estimation of both immature and mature crosslinks is important for elucidating the actual dynamic state of enzymatic crosslink formation in bone collagen. Furthermore, the precise estimation of Lys hydroxylation and its associated distinctive ratio of Hyl- to Lys-derived enzymatic crosslinking is also important to elucidate the function of enzymatic crosslinking in bone.
Enyzmatic immature divalent and mature trivalent crosslinks and AGEs crosslinking contribute independently to bone mechanical property. Each crosslinking concentration is normalized by collagen concentration (mol/ mol of collagen). Quantitative analysis of immature crosslinks such as DHLNL, HLNL, and LNL is performed by high-performance liquid chromatography (HPLC) or liquid chromatography-electrospray ionization mass spectrometry after reduction by radiolabeled [68, 69] or nonradioactive [9, 70, 71] borohydride for stabilization of immature reducible crosslinks, followed by acid hydrolysis. In HPLC analysis without radioisotopes, immature divalent crosslinks are detected by post-column labeling with ninhydrin [70] or O-phthalaldehyde [9]. Pyridinium crosslinks and AGEs crosslinking PEN are simultaneously determined by the same HPLC system using natural fluorescence after acid hydrolysis of tissue [9, 72–74]. Pyrrole crosslinks are unstable during acid hydrolysis and therefore cannot be measured using the conventional HPLC methods. In general, detection of pyrroles was achieved by treating with Ehrlich’s reagent (p-dimethylaminobenzaldehyde) [75]. Determination of the precise concentration of pyrrole in bone is difficult because of its instability. Because the major determinant of the total amount of immature divalent and mature crosslinks is LOX activity, overall formation of pyrrole crosslinks might be similar to other lysyl oxidase-controlled crosslinking. Indeed, human bone seems to contain pyrrole to the same extent as pyridinium crosslinks [75]. Furthermore, the actual concentration of each crosslink is used to estimate collagen maturity. The ratio of total pyridinium crosslinks (Pyr + Dpyr) to immature crosslinks (DHLNL + HLNL + LNL) was reported as collagen maturation index [13, 36, 45]. In terms of collagen maturity, the relative crosslinking ratio of PYD/DHLNL estimated by the wavelength ratio 1660/1690 cm−1 using raman spectrometry and Fourier transform-infrared (FTIR) microspectroscopy has been reported to estimate collagen maturation without the actual content of each enzymatic and AGEs crosslink being indicated. Recently, Farlay et al. [76] evaluated whether the ratio of PYD/DHLNL determined by 1660/1690 cm−1 using spectrometry such as Raman analysis and FTIR reflected the actual collagen crosslinking ratio. PYD/DHLNL crosslinks were measured biochemically using conventional HPLC and compared with the 1660/1690 cm−1 band ratio. They concluded that the 1660/1690 cm−1 ratio was not related to the PYD/DHLNL crosslinking ratio, but increased with bone mineral maturity. The change in this ratio could be due to a modification of the collagen’s secondary structure related to the mineralization process. To date, there have been no reports to contradict this theory. Therefore, it has become necessary to investigate human bone biopsies biochemically using HPLC in order to measure the content of each enzymatic crosslink. A further consideration may be needed regarding the estimation of collagen maturity using FTIR [77].
Nonenzymatic Crosslink Formation—AGEs
Crosslinking in collagen that accumulates via nonenzymatic AGEs in bone is indicative of the adverse effects on both cellular functions, via activation of the receptors for AGEs (RAGEs), and on bone material properties [78] (Figs. 1, 2). AGEs are classified as either noncrosslinking or crosslinking types (Fig. 2).
Lysine or arginine residue in collagen molecule is a precursor of noncrosslinking and crosslinking AGEs [79]. The initial step of AGE formation involves the aldehyde of an open chain form of glucose, ketose, or some other metabolic intermediates (e.g., glyoxal, methylglyoxal, and 3-deoxyglucosone) reacting with a free ε-amino group of a collagen-bound Lys or Hyl to form a chemically unstable Schiff base, glycosyl-Lys [80]. The hexosyl Lys or Hyl is subsequently made more stable by a spontaneous Amadori rearrangement. A carbonyl group is then generated by the oxidation of Amadori adduct. If this carbonyl group reacts with a hydroxyl group, such as lysine and arginine, then noncrosslinking type AGEs, such as carboxyl methyl lysine (CML), may be generated in the collagen molecule. In contrast, when a carbonyl group binds to amino acids in adjacent collagen molecules, then crosslinking AGEs, such as pentosidine (PEN), will be formed [81, 82]. To date, the determinant generating noncrosslinking and crosslinking remains to be elucidated.
PEN is a well-established fluorescent intermolecular AGE that forms crosslinks in various connective tissues as well as in bone collagen [83, 84]. There are no data available to definitely confirm whether other AGEs are formed in human bone collagen. These include vesperlysine [85] identified from bovine serum albumin in which AGEs were induced by the Maillard reaction with a sugar; nonfluorescent component-1 (NFC-1) identified from aorta [86]; and nonfluorescent glucosepane [87].
Although PEN has been used as a surrogate marker of total AGEs, estimation of accumulated AGEs based on PEN content has a limitation. PEN forms one crosslink per several hundred collagen molecules; therefore, it may have little effect on the mechanical properties of the collagen fibers. Although PEN is just one of many AGEs in bone, the measurement of PEN in bone is a common quantification method, because PEN can be easily and precisely quantified by HPLC in small specimens [9, 72–74]. Karim et al. [88] reported a significant relationship between the PEN content and the bulk fluorescence used to detect all AGEs by immunohistochemistry; they also described a fluorescence detection method which allowed for determination of the total AGE content, including both the crosslinking and noncrosslinking types, in bone. We also showed the similar relationship between the concentrations of total AGEs and PEN in primate vertebral bone [89]
This result suggested that the pathway for their formation may be similar, and PEN may be a useful surrogate marker of all fluorescent types of AGEs in bone. PEN is a Lys-arginine (Arg) covalent bond between adjacent collagen fibers, and its content is often used as a surrogate marker of the estimated total amount of AGEs in mechanical testing studies [13, 14, 36, 45, 64, 65, 90–93]. In contrast, the collagen crosslink glucosepane, a prominent nonfluorescent AGE, is an acid-labile Lys-Arg. Because structural similarities exist between PEN and glucosepane, the crosslinks of these two AGEs are formed by a parallel mechanism [94]. However, to date, there is no information on whether glucosepane forms in human bone. The AGE PEN is formed between helical Lys and Arg without involvement of the telopeptides. An increase in interhelical crosslinking via AGE-derived bonds such as PEN results in tissue brittleness and resistance to solubilization by pepsin [95, 96]. PEN has also been implicated in the induction of osteoblastic dysfunction [97]. Thus, crosslinking AGEs may have unfavorable effects on bone strength via the induction of cellular dysfunction and also due to collagen structural aspects. Crosslinking AGEs such as PEN, crossline, and pyrropyridine, have natural fluorescence, whereas noncrosslinking AGEs do not fluoresce except GA pyridine. Because there is no report regarding the existence of GA pyridine in bone, the determination of concentration of total fluorescent AGEs in bone is suitable for estimating accumulation of crosslinking AGEs in bone.
In contrast to crosslinking AGEs, there is no direct experimental evidence linking noncrosslinking AGEs with changes in the collagen fibril mechanical properties. In general, noncrosslinking AGEs cause a reduced bone strength via cellular dysfunction rather than due to structural aspects. Noncrosslinking AGEs such as carboxymethyl lysine (CML) have been shown to adversely affect the osteoblastic function via their interaction with the cell surface receptor for AGEs (RAGE) [98–100]. CML directly downregulates the activity of LOX.
Using primary osteoblast cultures, Khosravi et al. [101] determined that the lysyl oxidase protein was regulated by type I collagen and collagen was modified by CML-collagen. Data indicate that nonglycated collagen upregulates lysyl oxidase concentrations in primary osteoblast cultures, while CML-collagen fails to regulate lysyl oxidase in these cells. An important finding showed that collagen glycation disrupted collagen binding to, and activation of, the Discoidin Domain Receptor-2 (DDR2), representing a mechanism for the diminished levels of lysyl oxidase and consequently impaired enzymatic crosslinking.
The roles of AGEs on bone resorption by osteoclastic activity are controversial. A nonhistone nuclear protein, high-mobility group box 1 (HMGB1), is released from bone marrow macrophages in response to the receptor activator of nuclear factor kappa-B ligand (RANKL) stimulation. Predominantly via RAGE, the extracellular HMGB1 regulates osteoclastic actin cytoskeleton remodeling, differentiation, and function [102]. Thus, AGEs may increase osteoclast activity [103]. These results are consistent with other evidence showing that an excessive accumulation of AGEs occurs in the bone of patients with postmenopausal osteoporosis [13, 45] and chronic renal failure [104] who have a high turnover of bone. However, an opposite conclusion was reported in an in vitro study using mature osteoclasts from rabbits and humans [105]. This result seems to explain the increased formation of AGEs in the bones from animal models of types 1 and 2 diabetes which show a low rate of bone turnover [36, 92], and in patients with type 2 diabetes [106], who also exhibit a low rate of bone turnover. The accumulation of AGEs in bone occurs in low turnover bone, such as in models of diabetes. Because in vivo and in vivo studies have demonstrated that an increase in AGEs in bone occurs both in low turnover bone and also in high turnover bone, AGEs may not play a crucial role in the regulation of bone remodeling.
The accumulation of AGEs in the matrix of tissues depends on the lifespan of the tissue which is regulated by turnover rates under physiological conditions [91, 107, 108]. Furthermore, a chronic increase in the circulating concentrations of blood sugars as a consequence of poor glycemic control, such as occurs in individuals with diabetes mellitus, can increase AGE formation in bone [36, 92]. Not only poor glycemic control and prolonged tissue life span, but also oxidative stress can result in excessive accumulation of AGE crosslinks in bone [109]. We have previously demonstrated that bone from Sod1 −/− mice deficient in cytoplasmic copper/zinc superoxide dismutase (CuZn-Sod, encoded by Sod1) exhibit impaired enzymatic crosslinking and a marked accumulation of AGEs [110] (Fig. 3). Because hyperhomocysteinemia contributes to increased oxidative stress [111], hyperhomocysteinemia may also induce the excessive formation of AGEs in bone. Hyperhomocysteinemia in rabbits, induced by feeding them a methionine-enriched diet, exhibited a significant increase in the formation of AGEs in their bones [112].

Impaired enzymatic crosslinking and excessive formation of the AGE crosslink pentosidine in Sod1—deficient mice. a The concentration of enzymatic mature pyridinium crosslinking in the femoral bone of Sod1 −/− and Sod1 +/+ male mice. b The concentration of pentosidine in the femoral bone of Sod1 −/− and Sod1 +/+ male mice. c The total collagen content (percentage of tissue dry weight) in the femoral bone of Sod1 −/− and Sod1 +/+ male mice. d Bone stiffness measurement using a bone strength tester in the femoral bone of a male rat. (Nojiri et al. [110] with permission)
Because AGE formation is regulated by the concentration of glycation products and/or oxidative stress, excessive crosslinking by AGEs occurs even when the bone turnover rate is not low and euglycemia is achieved [13, 14, 45, 104]. We demonstrated that cortical [13] and cancellous [45] bone samples from patients with primary osteoporosis, but without diabetes or renal failure, exhibiting a high rate of bone turnover as estimated by urinary DPD concentrations, contained significantly higher concentrations of AGEs; PEN crosslinks had increased in newly formed young osteons as well as in old osteons. PEN was also markedly elevated in both the plasma protein and skin collagen fibers of uremic patients regardless of their glycemic status or tissue turnover rate, likely due to the increased oxidative stress induced by renal dysfunction [113].
Glycated hemoglobin cannot reflect the AGEs induced by oxidative stress. Kalninova et al. [114] reported the levels of AGEs to remain significantly elevated in comparison with healthy controls, despite the presence of good glycemic compensation as estimated by the glycated hemoglobin level in patients with type 2 diabetes mellitus. Furthermore, Okazaki et al., [115] demonstrated that AGEs, but not high glucose, may inhibit the osteoblastic differentiation of stromal cells by decreasing the osterix expression and partly by increasing the RAGE expression, as well as inhibiting cell growth and increasing cell apoptosis, thereby indicating that AGEs directly impair the cellular function independent of high glucose levels. When evaluating AGE accumulation in bone, glycated hemoglobin, which is an average measure of blood glucose over the past 2–3 months, both the amount of oxidative stress and the signaling cascades activated after AGE–RAGE interactions should be taken into consideration.
Collagen Crosslinking and Bone Strength
Enzymatic and nonenzymatic crosslinks are determinants of the tensile strength, toughness, and postyield properties in bone. A reduction of LOX activity instigated by lathyrogens such as BAPN, a vitamin B6 deficiency, or a copper deficiency, impairs enzymatic crosslinking, resulting in a decrease of bone strength. Recently, McNerny et al. [116] demonstrated that lathyrism in C57B16 mice altered enzymatic collagen crosslinking, which is important to bone quality as evidenced by losses of bone strength following LOX inhibition via treatment with 150 or 350 mg kg−1 BAPN for 3 weeks. These young growing mice exhibited both significantly reduced pyridinium crosslinking and cortical bone fracture toughness. Ratios reflecting relative crosslink maturity were positive regressors of fracture toughness (PYD/[DHLNL + HLNL]: r 2 = 0.208, p < 0.05; [PYD + DPD/[DHNL + HLNL]: r 2 = 0.196, p < 0.1], whereas the content of mature pyridinium crosslinking correlated significantly with tissue strength (PYD: r 2 = 0.159, p = 0.014; DPD: r 2 = 0.112, p < 0.05). Because there was no information of the AGEs in this study, the differential contribution of various AGEs to bone strength is not clear.
Oxlund et al. [117] had a similar result using growing 10-weeks-old rats. Treatment with BAPN for 1 month induced a 45 % reduction in pyridinium crosslinking, resulting in a 26 and 30 % reduction in the bending strength and elastic modulus, respectively, of rat femoral diaphyses compared with nontreated control rats; however, no information regarding the immature crosslinks and AGEs was made available.
Another similar result was reported in copper-deficient chicks; a reduction in the ultimate torsional strength of their bones appeared to be related to a decrease in LOX activity and a decrease in the concentrations of immature divalent crosslinks such as deH-DHLNL, deH-HLNL, and deH-LNL, although there was no information disclosed about mature trivalent crosslinking [118]. The role of immature divalent crosslinking on bone strength as well as mature crosslinks was reported in a preclinical study using a type 2 diabetic rat model. We noted a marked reduction in enzymatic immature divalent crosslinking without a corresponding reduction in the mature trivalent pyridinium crosslinks, and these findings were accompanied by a 25 % decrease in stiffness and elastic modulus that were independent of bone mineral density (BMD) in femoral bone [36]. Therefore, immature divalent crosslinking, which is the predominant type of crosslink in bone collagen, may play an important role in the mechanical properties of bone. Knott et al. [119] demonstrated that pyrrole crosslinking, as well as the immature divalent crosslinks, correlated positively to bone strength in avian cortical bones. However, pyrrole crosslinking does not necessarily affect the strength of bone in humans, while the concentration of pyridinium crosslinks and the ratio of PYD/DPD correlated positively with the stiffness and ultimate strain in bone [63]. The ratio of PYD/DPD may be explained by the different extent of Lys hydroxylation as described previously. However, pyrrole crosslinking may affect the material properties [63] because its concentration can vary, ranging from about half to the equivalent of that of PYD in bone.
We have reported that enzymatic crosslink formation is regulated not only by LOX activity, but also by noncollagenous proteins that bind collagen and regulate collagen fibrillogenesis [120–122]. Periostin, the noncollagenous matrix protein, seems to promote the formation of regular enzymatic crosslinks. Recently, we demonstrated that the binding of periostin to type I collagen regulated the formation of enzymatic crosslinks and the subsequent fibrillogenesis of collagen molecules [120]. However, periostin knockout mice exhibit significantly thinner collagen fibril diameters and impaired enzymatic crosslinking, resulting in decreased tissue mechanical integrity. Moreover, other types of type I collagen binding proteins, such as the small leucine-rich proteoglycans (SLRPs) that regulate collagen assembly, also act as regulators of enzymatic crosslinking [123, 124].
It has been clearly demonstrated that the excessive formation and accumulation of AGE crosslinks in bone induced via incubation with a sugar solution imparts brittleness to collagen fibers; this can lead to the accumulation of microdamage which results in deterioration of the postyield properties and toughness of bone [79, 125, 126]. Because these in vitro incubation studies exclude the effect of AGEs on tissue cellular activities and maintain a constant bone architecture and BMD, it seems to suggest that AGE crosslinking itself may deteriorate bone mechanical strength at a material level. In vivo studies from human and animal specimens also demonstrate that AGE crosslinking is an independent determinant of bone mechanical property, particularly of the postyield properties and toughness [64, 91, 93, 107, 127, 128]. Studies that have investigated the correlation between bone strength and collagen crosslinking were limited to acquisition of data related to the actual concentration of mature enzymatic and/or AGE crosslinks and did not account for the presence of immature divalent crosslinks. Some studies only perform an evaluation of AGEs.
Because immature divalent and mature trivalent pyridinium and pyrrole crosslinks, as well as AGE crosslinks such as PEN and glucosepane, may also affect the mechanical functions of bone, measurement of these crosslinks may therefore help to elucidate the actual role of collagen crosslinking as a determinant of bone strength. To clarify the independent contribution made by crosslinking, mineralization, and bone architecture to bone strength were analyzed simultaneously. Parameters that were determinants of the strength and mechanical properties of bone were measured after once-weekly treatment with hPTH 1–34 for 18 months in an OVXed primate model [14]. The most powerful determinant of both ultimate load and breaking energy was bone volume, followed in second place by the concentration of immature enzymatic divalent crosslinks. The most powerful determinant of bone stiffness was the concentration of the immature enzymatic divalent crosslinks.
We analyzed the relationships between collagen crosslink parameters and bone mechanical properties in the diabetic WBN/Kob rats [36]. The actual concentration of total enzymatic crosslinks, (the sum of immature and mature crosslinks) was modestly associated with stiffness and the elastic modulus in bone, whereas the concentration of PEN and the ratio of PEN to total enzymatic crosslinks were significantly associated not only with bone stiffness and elastic modulus, but also its energy absorption and maximum load. Interestingly, based on the regression analysis, the ratio of PEN to total enzymatic crosslinks as an indicator of imbalance between AGEs and enzymatic crosslink formation was more consistent with bone mechanical properties than were the total enzymatic crosslinks or PEN alone. Therefore, analysis of bone strength and its various determinants in the same bone specimen is advisable to enable elucidation of the contribution of the bone architecture and material properties to bone strength.
Age-Related Changes in Enzymatic and AGE Crosslinks
Age-related alterations that affect crosslinking, characterized by increased stability during thermal denaturation and solubilization with pepsin, may be the cause of the increase in bone fragility that occurs with aging [64, 66, 96, 129, 130]. Collagen content (% of tissue weight) in human bone reaches a maximum in adolescence and gradually decreases thereafter [96, 131]. Fujii et al. [53] have reported an age-related decrease in immature divalent crosslinking (the sum of deH-DHLNL, deH-HLNL, and deH-LNL) in human femoral bone ranging from 3 to 89 years in age. A portion of immature crosslinks convert gradually into mature trivalent crosslinks. Eyre at al. [11] have also reported similar changes attributable to the aging of immature divalent crosslinks and mature pyridinium crosslinking in femoral cortical and cancellous bone from human subjects ranging in age from 1 month to 80 years. Although immature divalent crosslink content was relatively lower at or below 25 years of age than younger age, significant amounts remain throughout adult life. Moreover, the amount of immature divalent crosslinking was significantly higher than that of mature pyridinium crosslinking. The amount of mature pyridinium crosslinking reached a maximum by the age of 10–15 years and then remained in the same range throughout adult life. Bailey et al. [131] have shown no significant age-related changes in the collagen content of pyrrole crosslinks in human cancellous bone from the iliums of females aged from 18 to 96 years and males aged from 23 to 92 years with no significant difference detected between genders. Interestingly, the same group and Zioupos et al. [129] demonstrated that age-related reductions in bone strength, such as maximum stress, Young’s modulus, energy absorption, and toughness did not correlate with the amount of mature trivalent enzymatic crosslinking. However, these studies were limited to estimation of enzymatic crosslinks in bone. To characterize and elucidate the role of age-related changes in bone strength, senescent crosslinks of collagen, or AGEs, should also be estimated in the same bone specimen.
Because enzymatic crosslinking alone cannot explain the age-related reduction in bone strength during the aging process, an analysis of crosslinking by AGEs such as PEN, is needed. In our first report of an age-related increase in PEN, an assessment was made in human femoral bone from healthy male subjects ranging in age from 1 month to 83 years old, and we determined the PEN content ranged from 4 to 10 times higher at 50 years of age than it was at 10 years of age [9]. Odetti et al. [132] also showed a significant age-related increase in PEN content in cortical and cancellous bone from female and male subjects. However, in their study, the age-related accumulation of PEN in cancellous bone did not reach statistical significance. Because they obtained bone specimens from joints showing signs of osteoarthritis and from subjects that had fractures at the time, such samples may have displayed differences in terms of the local bone turnover rate and crosslink formation [133]. In contrast, other reports in the literature of human bone biopsies used bone specimens without subchondral bone, specimens from subjects with renal failure, diabetes, and/or osteoarthritis.
Age-related increases in the PEN content of human bones may be a determinant of bone strength, particularly in relation to postyield properties and toughness. Wang et al. [64] reported a correlation between collagen crosslinks composed of enzymatically formed mature pyridinium crosslinks as well as the nonenzymatically formed PEN in the femoral bones from 30 human cadavers that ranged from 19 to 89 years of age, and measured their bone strength. PEN formation correlated with a decrease in strength, work to fracture, and the fracture toughness of bone. Nyman et al. [128] demonstrated that the content of PEN and collagen in bone provided an explanatory mechanism for the age-related decrease in postyield energy dissipation. Among AGEs, PEN concentration made the greatest contribution to plastic strain energy and provided the best explanation for damage accumulation, indicating that crosslinking resulting from AGE formation may be an independent determinant of the age-related increase in bone fragility.
Collagen crosslinking may differ according to location and may be related to the distinct function of bone at each site. There is a significant difference in collagen crosslink formation profiles among long bones, mandibular bones, and flat bones such as calvaria. This difference seems to be attributable to the different bone turnover rates at these sites. A comparison study of the calvarial flat bones and long bones in mice revealed that calvarial collagen is more soluble during chemical digestion by pepsin, matrix metalloproteinase (MMP), and cathepsin K, probably as a result of differences in the crosslinking profiles [134]. Mature pyridinium crosslinks were formed in higher amounts in collagen from long bones than in calvarial collagen, whereas the reverse was evident for PEN. Although the calvariae had a twofold higher concentration of PEN than that found in the long bone, calvarial collagen was more soluble than long bone, and this was an unexpected finding. A plausible explanation for this phenomenon may be that the contribution of PEN to chemical digestion may be less than that of pyridinium crosslinks in rodents because the PEN concentration in rodent bone is lower than that observed in human bone [9, 36, 64, 92, 110].
Mandibular bones have been shown to contain more immature collagen crosslinks, and fewer mature crosslinks, than long bones, which is due to a higher rate of bone turnover in the mandible than at long bone sites [135].
Under physiological conditions, mechanical loading is well known to be affected by bone size and bone strength. Gravitational loading has been shown to increase the expression of LOX and increase its enzymatic activity, which induced the formation of mature pyridinium crosslinks in osteoblastic MC3T3-E1 cells. In contrast, MC3T3-E1 cells cultured under simulated microgravity have exhibited a reduction in LOX activity and enzymatic crosslink formation. Under both conditions, PEN was not detected in the cell layer matrix [30]. We have demonstrated that site-specific age-related changes occur in collagen content and crosslink formation in non-weightbearing and weightbearing bones from human subjects [78, 96]. Cortical bone was obtained from 40 healthy male subjects ranging in age from 0 month to 84 years. Non-weightbearing bones such as the proximal humerus, distal radius, and ilium, as well as weightbearing bone sites such as the femur neck, mid-tibia, and the fourth lumbar vertebral body were analyzed. Collagen content reached a maximum in the second decade of life in weightbearing bones, whereas in non-weightbearing bones, the maximum was reached in the third decade of life and decreased thereafter. A significant reduction in immature crosslinking was evident from birth up to the first decade of life in weightbearing bones and up to the second decade in non-weightbearing bones. Mature pyridinium crosslinking content was significantly higher in weightbearing bones than in non-weightbearing bones from the first to the fourth decades of life. In contrast, there was no difference in PEN contents between weightbearing and non-weightbearing bones. Physiological mechanical loading may accelerate the stabilization of collagen through the conversion of immature crosslinks to mature crosslinks, indicating the collagen metabolism inherent at different sites is influenced by mechanical stress associated with everyday functions. Isakasson et al. [136] used growing male mice to demonstrate that increased bone loading caused by running reduced bone collagen PEN content, although statistical significance was marginal. Therefore, physiological loading and exercise such as running may have a favorable effect on the formation of collagen crosslinks in bone.
Crosslinking Abnormalities Associated with Diseases
Data have accumulated to indicate that enzymatic and nonenzymatic collagen crosslinking abnormalities exist under pathological conditions such as osteoporosis [13, 66, 112, 137], mild hyperhomocysteinemia [45, 46, 112], diabetes mellitus [36, 92], chronic kidney disease [104, 138], inflammatory bowel disease (IBD) [139], and when glucocorticoids are used [67].
Osteoporosis
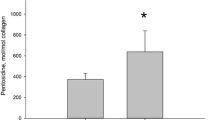
Postmenopausal women with primary osteoporosis are characterized by an unacceptably low BMD, and in cases of hip fracture, exhibit significantly reduced concentrations of immature and mature enzymatic crosslinks, including pyridinium, as well as excessive concentrations of PEN and an increase in the hydroxylation of Lys residues in bone compared with age-matched subjects without osteoporosis [13]. Similar crosslink abnormalities are induced by OVX in a monkey model [14]. Overhydroxylation of Lys residues and qualitative crosslinking abnormalities such as impaired enzymatic crosslinking and excessive accumulation of AGEs may be due to an estrogen deficiency that causes a higher rate of bone turnover and increases Lys hydroxylation [21] as estrogen acts as a positive regulator of LOX [40]. Furthermore, estrogen deficiency has been shown to reduce the activity of superoxide dismutase (SOD), resulting in an increase in oxidative stress [140]. We have demonstrated that AGE crosslinks such as PEN are formed excessively, and immature and mature enzymatic crosslinks such as pyridinium are impaired, causing a reduction in the bone strength of SOD knockout mice [110] (Fig. 3). Therefore, an increase in oxidative stress induced by aging, estrogen deficiency, and some diseases may play a central role in the deterioration of collagen crosslinking in body tissues. It is noteworthy that the PEN content in bones from patients with osteoporotic femoral neck fractures was significantly higher than in age-matched subjects without osteoporosis; this was the case even in younger osteons, suggesting that accelerated AGE crosslink formation occurred, even in the newly formed collagen [45]. These patients who sustained hip fractures did not have diabetes; however, because they had a higher rate of bone turnover, as estimated by urinary excretion of DPD, an increase in oxidative stress may have been the cause of the excessive formation of PEN in young osteons as well as older osteons. Not only estrogen deficiency, but also other factors that can increase oxidative stress such as hyperhomocysteinemia, may be a cause of the increase in PEN formation [141]. A moderately increased plasma concentration of homocysteine (HCY) in the general population may be prognostic of a fracture risk that is independent of BMD [4, 142–144]. Lubec proposed that hyperhomocysteinemia might interfere with normal enzymatic collagen crosslink formation based on the analysis of bone turnover markers [145]. We were the first to report a case-controlled study in which there was a significant reduction in the actual amount of enzymatic crosslinking, both immature and mature pyridinium crosslinks, and a significant increase in the AGE crosslink PEN, in the bones from patients who had postmenopausal osteoporotic hip fractures, moderately elevated plasma HCY, and a vitamin B6 insufficiency [45] (Fig. 4). However, this was a cross-sectional study, and therefore it could not be determined whether HCY caused collagen crosslinking abnormalities. To elucidate this issue, we investigated whether hyperhomocysteinemia affects the actual content of enzymatic and nonezymatic (PEN) crosslinks in bone in rabbits fed a methionine-enriched diet to induce hyperhomocysteinemia [112]. This study demonstrated that hyperhomocysteinemia reduced bone toughness, independent of BMD, via a reduction in enzymatic crosslinking and an increase in the PEN in bone, with or without an estrogen deficiency [112]. HCY interferes with enzymatic crosslinking by inhibiting the enzymatic activity of LOX [42, 44]. Because the HCY that accumulated specifically bound to the collagen in bone compared with heart tissue, the adverse effects of hyperhomocysteinemia on collagen crosslinking may also predominate in bone collagen using a hyperhomocysteinemia-induced rat [146]. These results support our previously reported clinical studies including a 5-years prospective study of 502 postmenopausal women with moderate hyperhomocysteinemia [142]. Moderate hyperhomocysteinemia induced by a TT genotype of the methylenetetrahydrofolate reductase (MTHFR) polymorphism (C677T) in postmenopausal women was independent of fracture risk [142].

Mild hyperhomocysteinemia and a vitamin B6 insufficiency can deteriorate collagen crosslinking in both young and old osteons. Twenty-five female intracapsular hip fracture cases (78 ± 6 years) and 25 age-matched postmortem controls (77 ± 6 years) were analyzed. The less mineralized, low-density bone fraction represents newly formed young bone, whereas the more highly mineralized, high-density bone fraction represents older bone. In cases with fractures, not only reduced enzymatic crosslinks in high-density bone and increased pentosidine in both low- and high-density bones, but also higher plasma homocysteine and lower pyridoxal levels were evident compared with the controls. Enzymatic crosslinking content: the sum of DHLNL + HLNL + LNL + PYD + DPD (Saito et al. [45] with permission)
Diabetes Mellitus
Fracture risks in patients with type 1 or 2 diabetes have been shown to be significantly increased by an increment higher than what can be explained by a reduction in BMD. Thus, both types of diabetes may be associated with a reduction in vertebral and femoral neck bone strength, which does not necessarily reflect what might be expected based on their BMD [147]. Therefore, diabetes may deteriorate bone collagen crosslinking as well as bone mass and BMD (Fig. 5). Recently, Zucker Diabetic Sprague-Dawley rats showed a different distribution of collagen D-spacing than nondiabetic rats in which distributions were more variable and shifted to higher values; however, no information was available with respect to collagen crosslinking [148]. Little is known about the abnormalities of enzymatic collagen crosslinks, including immature and mature crosslinks and AGEs in relation to the pathology of diabetes. While quantitative and qualitative changes are known to occur in collagen as a result of diabetes, these are influenced by many factors including age of onset, disease duration, insulin status, and glycemic control, suggesting a suitable animal model for type 1 and type 2 diabetes is required to analyze bone collagen crosslinking under these circumstances [36, 92, 149]. Tomasek et al. [149] were the first to report that streptozotocin-induced type 1 diabetes exhibited an increase in collagen-linked fluorescence (an indicator of AGEs) that was coincident with reduced bone strength, Silva et al. [92] demonstrated in streptozotocin-induced type 1 diabetic rats that increased PEN, trabecular bone loss, and impaired diaphyseal growth occurred without any alteration in the content of mature enzymatic pyridinium crosslinks or collagen content. An increase in the AGE crosslink PEN has been shown to reduce the material property of bone in type 1 diabetes in rats. With respect to type 2 diabetes, we have demonstrated that impaired enzymatic crosslinking, in immature divalent enzymatic crosslinks, without a change in pyridinium crosslinks but with excessive formation of PEN in bone, decreased femoral bone strength without any alteration of collagen content and BMD in spontaneously diabetic WBN/Kob rats [36]. Immature enzymatic divalent crosslinking was impaired, even in the prediabetic stage, and this resulted in a reduction in bone strength without a change in BMD and AGE crosslinks. It is generally thought that the conversion of crosslinks from immature divalent to mature trivalent crosslinks occurs via a spontaneous reaction in a time-dependent manner. While serum osteocalcin and urinary excretion of DPD were significantly lower in the WBN/Kob rats in a prediabetic state, bone turnover may have been lower in the WBN/Kob rats than in the nondiabetic Wistar rats used as a comparison. Thus, the prolonged lifespan of the bone collagen in the WBN/Kob rats may contribute to the time-dependent enzymatic maturation as immature forms react and form mature pyridinium crosslinks. The impairment of immature divalent crosslinking may be due to vitamin B6 deficiency. Vitamin B6 is essential to the activation of LOX [33]. Serum concentrations of vitamin B6, such as pyridoxal, pyridoxamine, and pyridoxine in WBN/Kob rats, have been shown to be decreased after they reach 6 months of age, and they were also significantly lower than those in the 6-months-old or older nondiabetic rats. The concentration of immature enzymatic crosslinks was significantly associated with vitamin B6 concentrations, whereas the concentration of PEN was only modestly associated with blood glucose and serum vitamin B6 concentrations in the prediabetic stage in these rats. A vitamin B6 deficiency has been shown to occur in response to an upregulation of gluconeogenesis in both prediabetic and diabetic stages, and a latent deficiency of vitamin B6 may cause impaired LOX-dependent immature crosslinking (Fig. 5). Gluconeogenesis is a pathway to generate glucose from noncarbohydrate carbon substrates. After the onset of frank diabetes, immature divalent crosslinking was continuously reduced, and PEN markedly increased; these changes were consistent with a significant reduction in bone strength parameters, including stiffness, energy absorption, and maximum load, compared with nondiabetic Wistar rats, despite no change in both collagen content and BMD. Type 2 diabetes also increases both plasma concentrations of HCY and oxidative stress which can result in arteriosclerosis [150, 151]. Thus, hyperhomocysteinemia and oxidative stress in a state of diabetes may induce the additive adverse effect of nonenzymatic crosslink formation. Aberrant crosslinking patterns, as have been shown to occur in rodent models of types 1 and 2 diabetes, may result in accelerated bone fragility without any accompanying change in BMD (Fig. 5; Table 1).

A plausible mechanism for poor bone material properties in cases of diabetes. In diabetes, a vitamin B6 insufficiency, and increases in oxidative stress, glycation, and homocysteine may decrease lysyl oxidase (LOX) activity and increase the formation of AGEs. Such collagen crosslinking abnormalities can induce cellular dysfunction
Chronic Kidney Disease
It is well known that the fracture risk of a patient with renal failure (chronic kidney disease) is increased [152, 153]. Both their BMD and bone material properties are attenuated in CKD. We have performed transiliac bone biopsies on 22 dialysis patients (mean age 56 ± 9 years) with CKD and severe secondary hyperparathyroidism and proceeded to measure immature and mature enzymatic crosslinks as well as the AGE PEN. At the same time, dynamic bone histomorphometry was evaluated [104]. Collagen crosslinking data were compared with 17 age-matched non-CKD subjects (mean age 58 ± 8 years). The concentration of immature divalent crosslinking in CKD patients was similar to non-CKD subjects, whereas the concentration of mature trivalent pyridinium forms in dialysis patients was significantly lower than in that in non-CKD subjects. Interestingly, the sum of immature and mature crosslinks was positively correlated with intact PTH in plasma. PTH increased the expression of LOX via platelet-derived growth factor-A (PDGFA) in the bones of subjects with hyperparathyroidism [154]. In end-stage renal disease, although increased oxidative stress caused by renal dysfunction accelerates the formation of AGE crosslinking. PEN has been shown to be remarkably increased in dialysis patients, and was inversely correlated with bone-formation rate/bone volume and mineral apposition rate, suggesting that collagen AGE crosslinking is strongly associated with disorders of bone metabolism in dialysis patients with hyperparathyroidism. Aoki et al. [138] have demonstrated that the suppression of osteoblast differentiation and the decreased activation of LOX proteins associated with accumulation of AGEs in osteoblasts caused both structural abnormalities in bone collagen fibrils and a severe mineralization disorder; however, no information was provided concerning the actual formation of LOX-controlled crosslinking in rats with hyperthyroidism induced via adenine. In contrast, there is no report about the actual content of enzymatic (immature divalent and mature trivalent) crosslinking, or of nonenzymatic (AGE) crosslinks in adynamic bones from patients with end-stage renal disease without hyperparathyroidism. Furthermore, the quantitative analyses of collagen and crosslink formation are needed at stages 2–4 of CKD as well as for end-stage of renal disease because oxidative stress, HCY, and fracture risk are increased as a result of this pathology [155–157]. Hyperhomocysteinemia and oxidative stress in CKD may show an additive adverse effect on crosslink formation.
Inflammatory Bowel Disease
Inflammatory bowel diseases (IBD), such as ulcerative colitis and Crohn’s disease, are often associated with severe osteoporosis in both children and adults. Although the basic etiology of IBD is unknown, there are several factors that may contribute to the pathogenesis of this disease, such as dysregulation of the immune system, oxidative stress, elevated HCY concentrations, and inflammatory mediators [139, 158, 159]. Krajcovicova et al. [159] demonstrated that treatment with an immunomodulator and anti-TNF-α increased the BMD in patients with IBD. In contrast, supplementation with vitamin D and calcium had no effect on BMD, indicating that TNF-α may be a key factor implicated in a low BMD, rather than the nutritional status in IBD patients. Furthermore, mild hyperhomocysteinemia [112], TNF-α, and oxidative stress [3, 110] in people with IBD may deteriorate collagen crosslinking. Gamsjaeger et al. [139] recently reported that the HLA-B27 transgenic rat is susceptible to both long-bone osteopenia and poor-bone material properties such as a lower ratio of mineral/matrix and higher relative proteoglycan and AGE concentrations (ϵ-N-Carboxymethyl-l-lysine); however, no information on the content of enzymatic and nonenzymatic crosslinks is provided. To date, there is no report evaluating bone biopsy data of patients with IBD.
Glucocorticoid Use
It has been reported that patients with vertebral fractures who AQ have been treated with glucocorticoids (GCs) have a higher BMD but have a twofold risk of sustaining a fracture compared with patients who have vertebral fractures and postmenopausal osteoporosis [160]. Clinically, patients with GC-induced osteoporosis develop bone loss in the first few months of GC exposure and modest doses of GC tend to increase their risk of a fracture. Because a bone fracture may be imminent, even if their BMD is relatively high, deterioration of some other factors that contribute to bone quality, in addition to BMD, has been postulated as a likely contributing cause to GC-induced osteoporosis. A number of GC-mediated effects are responsible for bone fragility: (1) GC-induced impairments of osteoblast and osteocyte; (2) reduced intestinal absorption of calcium and increased renal elimination leading to a negative calcium balance that has been suggested to promote secondary hyperparathyroidism; and (3) glucocorticoids antagonize gonadal function and inhibit the osteoanabolic action of sex steroids [161, 162]. We demonstrated aberrant collagen crosslinking in bone and its relationship to bone strength in rats treated with prednisolone (10 mg kg−1 daily, intramuscularly [IM] for 4 weeks) [163]. Despite no reduction in BMD in this rat model, femoral bone strength markedly decreased. The total content of immature enzymatic divalent crosslinks (DHLNL + HLNL + LNL) was significantly decreased by GC administration (p = 0.0490), but the total content of mature pyridinium crosslinks (PYD + DPD) and AGE crosslinks were unaffected. The ratios of DHLNL/HLNL and PYD/DPDP, which indicate the extent of lysine hydroxylation, were not affected by GC treatment. Total concentration of enzymatic crosslinks (the sum of immature and mature crosslinks) were significantly lower in the GC-treated group than that in the control group (p = 0.042). In order to assess in more detail the relationship between the determinants of bone strength, such as BMD, femoral diameter, and collagen crosslinks, on the bone mechanical properties, the mechanical properties of interest were plotted against BMD, femoral diameter, or collagen crosslink parameters for the femoral midshaft, and a linear regression analysis was performed. The sum of enzymatic immature divalent and mature pyridinium crosslinks (p = 0.021, the immature crosslinks (p = 0.024), and the ratio of DHLNL/HLNL (p = 0.016) were significantly and positively correlated with the energy absorption. The concentration of mature pyridinium crosslinks showed a significant positive correlation to yield load (p = 0.006). BMD and femoral diameter did not correlate significantly to maximum load, stiffness, or energy absorption. To determine whether collagen crosslink parameters were independently related to bone strength after adjusting for the contribution made by BMD and the femoral diameter, we performed multiple regression analyses. Total enzymatic crosslinking and the ratio of DHLNL/HLNL contributed significantly to energy absorption independently of BMD and femoral diameter. The prediction model for bone strength improved the diagnostic sensitivity to 31.0 % for energy absorption, but maximum load and stiffness were less sensitive than energy absorption. Benson et al. [164] demonstrated that GCs produced a dose-dependent decrease in LOX activity in rat tissue, down to 78 % of the control, when administered over a 3-days period. TGF-β [47] and IGF-I [165] are other candidate molecules that may be downregulated by GCs because they act as positive regulators of LOX activity [166, 167]. Thus, GC use may reduce bone material properties by inhibiting regular enzymatic crosslinking.
Candidates for Treatment of Collagen Crosslinking
Deteriorating collagen crosslinks are replaced by the remodeling process. If bone remodeling is suppressed for a prolonged period, AGEs form in bone collagen in a time-dependent manner [91, 107, 108]. In a preclinical animal study, when >70 % suppression of bone remodeling continued for more than a year, excessive accumulation of AGEs occurred, resulting in microdamage formation [91, 107, 108]. Thus, to improve crosslink formation in bones, agents reducing oxidative stress and/or increasing lysyl oxidase activity without oversuppression of bone remodeling are required. Even when total enzymatic crosslink formation reaches its maximum in the primary mineralization process [6, 91], agents increasing lysyl oxidase activity may induce further enzymatic crosslinking in preexisting collagen. Some osteoporosis drugs are candidates for improving collagen enzymatic immature and mature crosslinking as well as AGEs formation. Selective estrogen receptor modulator (SERM), raloxifene, increased the actual concentrations of enzymatic immature and mature pyridinium crosslinks, while decreasing AGEs crosslink, PEN, after 16-weeks treatment. Since the treatment duration is only 16 weeks in this report, the marked increase in crosslink concentration cannot be explained by remodeling alone, which leads us to believe that there is also an increase in the crosslinks of preexisting collagen. Not all enzymatic crosslink precursors such as Lys and Hyl residues in collagen molecules are crosslinked. Therefore, if lysyl oxidase activity is increased by drugs or agents, the Lys and Hyl may form additional crosslinks in preexisting collagen fibers independent of bone remodeling. Furthermore, Gallant et al. [168] reported that ex vivo exposure of nonviable bone to raloxifene improves intrinsic toughness. These effects are cell independent and appear to be mediated by an increase in matrix-bound water. The hydroxyl groups (OH) on raloxifene were shown to be important in the increase of both water content and toughness. Thus, direct action to preexisting bone matrix in a cell-independent manner is also important in improving bone material property as well as maintaining physiological bone remodeling. In terms of enzymatic crosslink formation, candidates for increasing enzymatic crosslink formation in preexisting collagen fibers, such as active vitamin D3 [41] and its analogs, eldecalcitol [89], teriparatide [14], vitamin B6 [169], periostin [120–122], leucine-rich repeat proteins such as small proteoglycans [123], TGF-beta [37], CTGF [38] and IGF-1 [39], may be positive regulatory factors of lysyl oxidase. In contrast, a nonenzymatic AGEs crosslink, PEN, is well known to form even in preexisting collagen fibers as a result of oxidative stress. Therefore, the continuous reduction of oxidative stress by agents prevents further accumulation of PEN in preexisting bone collagen. Future studies are needed to investigate whether supplemental vitamin B6 such as pyridoxal [169] and pyridoxamine [170] can improve the abnormal crosslink formation. As pyridoxal is thought to act not only as a cofactor of lysyl oxidase but also as an inhibitor of AGEs formation, we propose that administration of pyridoxal or pyridoxal phosphate may be suitable for the amelioration of crosslink formation in diabetic WBN/Kob rats [169]. Another candidate inhibitor of AGEs is aminoguanidine, which is well documented to be potent in inhibiting AGEs formation [171]. Administration of aminoguanidine, however, would be expected to cause a further reduction in the levels of pyridoxal and pyridoxal phosphate [172], suggesting that long-term administration of aminoguanidine may have an adverse effect on lysyl oxidase activity and subsequent enzymatic crosslink formation. Aminoguanidine has now been withdrawn after clinical trials showed detrimental side effects. Thus, vitamin B6 may be more suitable as an inhibitor of AGEs in bones [173].
Further work is needed to clarify roles of collagen crosslink formation in bone. There is little information of biological interaction between collagen crosslinking and noncollagenous proteins such as osteocalcin, osteopontin, small proteoglycans, and periostin. Periostin regulates directly activation of lysyl oxidase [122] and plays an important role in the adaptation of the extracellular matrix architecture [121]. The other noncollagenous protein may have crucial roles in collagen crosslink formation and fibril formation.
Development of new agent reducing oxidative stress and/or increasing lysyl oxidase activity without oversuppression of bone remodeling is also needed as a further work.
Conclusions
Enzymatic crosslinking patterns regulated by Lys hydroxylation seem to directly affect the mineralization process under physiological conditions, whereas the total concentrations of enzymatic crosslinks affect bone strength at the material level. To increase bone strength, it is crucial to induce regular enzymatic crosslinking in collagen and inhibit AGE crosslinks. Enzymatic crosslink formation is regulated by the expression and activation of intracellular LHs and the extracellular actions of LOXs, whereas the amount of oxidative stress and glycation that occurs as a result of aging, hyperhomocysteinemia, and chronic diseases such as diabetes mellitus and CKD can increase the formation of AGE crosslinks (Fig. 1). The increasing number of reports in the literature addressing suitable treatments with the potential to decrease the risk of aberrant collagen crosslinking profiles may improve our ability to better personalize medications for patients at increased risk for bone fracture.
References
Buckwalter JA, Glimcher MJ, Cooper RR, Recker R (1996) Bone biology, I: structure, blood supply, cells, matrix, and mineralization. Instr Course Lect 45:371–386
Seeman E, Delmas PD (2006) Bone quality—the material and structural basis of bone strength and fragility. N Engl J Med 354:2250–2261
Manolagas SC (2010) From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev 31:266–300
Robins SP, Bailey AJ (1977) The chemistry of the collagen cross-links. Characterization of the products of reduction of skin, tendon and bone with sodium cyanoborohydride. Biochem J 163:339–346
Uzawa K, Grzesik WJ, Nishiura T, Kuznetsov SA, Robey PG, Brenner DA, Yamauchi M (1999) Differential expression of human lysyl hydroxylase genes, lysine hydroxylation, and cross-linking of type I collagen during osteoblastic differentiation in vitro. J Bone Miner Res 14:1272–1280
Saito M, Soshi S, Tanaka T, Fujii K (2004) Intensity-related differences in collagen post-translational modification in MC3T3-E1 osteoblasts after exposure to low and high intensity pulsed ultrasound. Bone 35:644–655
Bank RA, Robins SP, Wijmenga C, Breslau-Siderius LJ, Bardoel AF, van der Sluijs HA, Pruijs HE, TeKoppele JM (1999) Defective collagen crosslinking in bone, but not in ligament or cartilage, in Bruck syndrome: indications for a bone-specific telopeptide lysyl hydroxylase on chromosome 17. Proc Natl Acad Sci USA 96:1054–1058
Pornprasertsuk S, Duarte WR, Mochida Y, Yamauchi M (2005) Overexpression of lysyl hydroxylase-2b leads to defective collagen fibrillogenesis and matrix mineralization. J Bone Miner Res 20:81–87
Saito M, Marumo K, Fujii K, Ishioka N (1997) Single column high—performance liquid chromatographic—fluorescence detection of immature, mature and senescent cross-links of collagen. Anal Biochem 253:26–32
Saito M, Shiraishi A, Ito M, Sakai S, Hayakawa N, Mihara M, Marumo K (2010) Comparison of effects of alfacalcidol and alendronate on mechanical properties and bone collagen cross-links of callus in the fracture repair rat model. Bone 46:1170–1179
Eyre DR, Dickson IR, Ness KV (1988) Collagen cross-linking in human bone and articular cartilage. Age-related changes in the content of mature hydroxypyridinium residues. Biochem J 252:495–500
Knott L, Bailey AJ (1998) Collagen cross-links in mineralizing tissues: a review of their chemistry, function, and clinical relevance. Bone 22:181–187
Saito M, Fujii K, Soshi S, Tanaka T (2006) Reductions in degree of mineralization and enzymatic collagen cross-links and increases in glycation induced pentosidine in the femoral neck cortex in cases of femoral neck fracture. Osteoporos Int 17:986–995
Saito M, Marumo K, Kida Y, Uhsiku C, Kato S, Takao-Kawabata R, Kuroda T (2011) Changes in the contents of enzymatic immature, mature, and non-enzymatic senescent cross-links of collagen after once-weekly treatment with human parathyroid hormone (1–34) for 18 months contribute to improvement of bone strength in ovariectomized monkeys. Osteoporos Int 22:2373–2383
Marumo K, Saito M, Yamagishi M, Fujii K (2005) The “ligamentization” process in human anterior cruciate ligament reconstruction with autogenous patellar and hamstring tendons. Am J Sports Med 33:1166–1173
Yamauchi M, Katz EP (1993) The post-translational chemistry and molecular packing of mineralizing tendon collagens. Connect Tissue Res 29:81–98
Fujii K, Yamagishi T, Nagafuchi T, Tsuji M, Kuboki Y (1994) Biochemical properties of collagen from ligaments and periarticular tendons of the human knee. Knee Surg Sports Traumatol Arthrosc 2:229–233
Kato S, Saito M, Funasaki H, Marumo K (2013) Distinctive collagen maturation process in fibroblasts derived from rabbit anterior cruciate ligament, medial collateral ligament, and patellar tendon in vitro. Knee Surg Sports Traumatol Arthrosc. doi:10.1007/s00167-013-2773-8
Yamauchi M, Katz EP, Mechanic GL (1986) Intermolecular cross-linking and stereospecific molecular packing in type I collagen fibrils of the periodontal ligament. Biochemistry 25:4907–4913
Yamauchi M, Woodley DT, Mechanic GL (1988) Aging and cross-linking of skin collagen. Biochem Biophys Res Commun 152:898–903
Yamauchi M, Shiiba M (2008) Lysine hydroxylation and cross-linking of collagen. Methods Mol Biol 446:95–108
Yamauchi M, Sricholpech M (2012) Lysine post-translational modifications of collagen. Essays Biochem 52:113–133
Yeowell HN, Walker LC (1999) Tissue specificity of a new splice form of the human lysyl hydroxylase 2 gene. Matrix Biol 18:179–187
Yeowell HN, Walker LC (2000) Mutations in the lysyl hydroxylase 1 gene that result in enzyme deficiency and the clinical phenotype of Ehlers-Danlos syndrome type VI. Mol Genet Metab 71:212–224
Kuboki Y, Kudo A, Mizuno M, Kawamura M (1992) Time-dependent changes of collagen cross-links and their precursors in the culture of osteogenic cells. Calcif Tissue Int 50:473–480
Bank RA, Bayliss MT, Lafeber FP, Maroudas A, Tekoppele JM (1998) Ageing and zonal variation in post-translational modification of collagen in normal human articular cartilage. The age-related increase in non-enzymatic glycation affects biomechanical properties of cartilage. Biochem J 330:345–351
Rivera EM, Yamauchi M (1993) Site comparisons of dentine collagen cross-links from extracted human teeth. Arch Oral Biol 38:541–546
Miguez PA, Pereira PN, Atsawasuwan P, Yamauchi M (2004) Collagen cross-linking and ultimate tensile strength in dentin. J Dent Res 83:807–810
Hayashi M, Furuya Y, Minoshima K, Saito M, Marumo K, Nakashima S, Hongo C, Kim J, Ota T, Ebisu S (2012) Effects of heating on the mechanical and chemical properties of human dentin. Dent Mater 28:385–391
Saito M, Soshi S, Fujii K (2003) Effect of hyper- and microgravity on collagen post-translational controls of MC3T3-E1 osteoblasts. J Bone Miner Res 18:1695–1705
Gerstenfeld LC, Riva A, Hodgens K, Eyre DR, Landis WJ (1993) Post-translational control of collagen fibrillogenesis in mineralizing cultures of chick osteoblasts. J Bone Miner Res 8:1031–1043
Eyre DR, Paz A, Gallop PM (1984) Cross-linking in collagen and elastin. Annu Rrev Biochem 53:717–748
Bird TA, Levene CI (1982) Lysyl oxidase: evidence that pyridoxal phosphate is a co-factor. Biochem Biophys Res Commun 108:1172–1180
Wang SX, Mure M, Medzihradszky KF, Burlingame AL, Brown DE, Dooley DM, Smith AJ, Kagan HM, Klinman JP (1996) A crosslinked cofactor in lysyl oxidase: redox function for amino acid side chains. Science 273:1078–1084
Fujii K, Kajiwara T, Kurosu H (1979) Effect of vitamin B6 deficiency on the crosslink formation of collagen. FEBS Lett 97:193–195
Satio M, Fujii K, Mori Y, Marumo K (2006) Role of collagen enzymatic and glycation induced cross-links as a determinant of bone quality in spontaneously diabetic WBN/Kob rats. Osteoporos Int 17:1514–1523
Feres-Filho EJ, Choi YJ, Han X, Takala TE, Trackman PC (1995) Pre- and post-translational regulation of lysyl oxidase by transforming growth factor-beta 1 in osteoblastic MC3T3-E1 cells. J Biol Chem 270:30797–30803
Hong HH, Uzel MI, Duan C, Sheff MC, Trackman PC (1999) Regulation of lysyl oxidase, collagen, and connective tissue growth factor by TGF-beta1 and detection in human gingiva. Lab Invest 79:1655–1667
Reiser K, Summers P, Medrano JF, Rucker R, Last J, McDonald R (1996) Effects of elevated circulating IGF-1 on the extracellular matrix in high-growth C57BL/6J mice. Am J Physiol 271:R696–R703
Ozasa H, Tominaga T, Nishimura T, Takeda T (1981) Lysyl oxidase activity in the mouse uterine cervix is physiologically regulated by estrogen. Endocrinology 109:618–621
Nagaoka H, Mochida Y, Atsawasuwan P, Kaku M, Kondoh T, Yamauchi M (2008) 1,25(OH)2D3 regulates collagen quality in an osteoblastic cell culture system. Biochem Biophys Res Commun 377:674–678
Raposo B, Rodriguez C, Martinez-Gonzalez J, Badimon L (2004) High levels of homocysteine inhibit lysyl oxidase (LOX) and down regulate LOX expression in vascular endothelial cells. Atherosclerosis 177:1–8
Liu G, Nellaiappan K, Kagan HM (1997) Irreversible inhibition of lysyl oxidase by homocysteine thiolactone and its selenium and oxygen analogues. Implications for homocystinuria. J Biol Chem 272:32370–32377
Kang HA, Trelstad RL (1973) A collagen defect in homocystinuria. J Clin Invest 52:2571–2578
Saito M, Fujii K, Marumo K (2006) Degree of mineralization-related collagen crosslinking in the femoral neck cancellous bone in cases of hip fracture and controls. Calcif Tissue Int 79:160–168
Blouin S, Thaler HW, Korninger C, Schmid R, Hofstaetter JG, Zoehrer R, Phipps R, Klaushofer K, Roschger P, Paschalis EP (2009) Bone matrix quality and plasma homocysteine levels. Bone 44:959–964
Feres-Filho EJ, Menassa GB, Trackman PC (1996) Regulation of lysyl oxidase by basic fibroblast growth factor in osteoblastic MC3T3-E1 cells. J Biol Chem 271:6411–6416
Boak AM, Roy R, Berk J, Taylor L, Polgar P, Goldstein RH, Kagan HM (1994) Regulation of lysyl oxidase expression in lung fibroblasts by transforming growth factor-b1 and prostaglandin E2. Am J Respir Cell Mol Biol 11:751–755
Rodriguez C, Alcudia JF, Martinez-Gonzalez J, Raposo B, Navarro MA, Badimon L (2008) Lysyl oxidase (LOX) down-regulation by TNFalpha: a new mechanism underlying TNFalpha-induced endothelial dysfunction. Atherosclerosis 196:558–564
Turecek C, Fratzl-Zelman N, Rumpler M (2008) Collagen cross-linking influences osteoblastic differentiation. Calcif Tissue Int 82:392–400
Fernandes H, Dechering K, Van Someren E, Steeghs I, Apotheker M, Leusink A, Bank R, Janeczek K, Van Blitterswijk C, de Boer J (2009) The role of collagen crosslinking in differentiation of human mesenchymal stem cells and MC3T3-E1 cells. Tissue Eng Part A 15:3857–3867
Eyre DR, Paz A, Gallop PM (1984) Cross-linking in collagen and elastin. Annu Rev Biochem 53:717–748
Fujii K, Kuboki Y, Sasaki S (1976) Aging of human bone and cartilage collagen: changes in the reducible cross-links and their precursors. Gerontology 22:363–370
Bailey AJ, Shimokomaki MS (1971) Age related changes in the reducible cross-links of collagen. FEBS Lett 16:86–88
Robins SP, Shimokomaki M, Bailey AJ (1973) Age-related changes in the reducible components of intact bovine collagen fibres. Biochem J 131:771–780
Ogawa T, Ono T, Tsuda M (1982) A novel fluor in insoluble collagen: a crosslinking moiety in collagen molecule. Biochem Biophys Res Commun 107:1252–1257
Eyre DR, Oguchi H (1980) The hydroxypyridinium crosslinks of collagens: their measurement, properties and a proposed pathway of formation. Biochem Biophys Res Commun 92:403–410
Robins SP, Duncan A (1987) Pyridinium crosslinks of bone collagen and their location in peptides isolated from rat femur. Biochim Biophys Acta 914:233–239
Robins SP, Duncan A (1983) Cross-linking of collagen. Location of pyridinoline in bovine articular cartilage at two sites of the molecule. Biochem J 215:175–182
Brady JD, Robins SP (2001) Structural characterization of pyrrolic cross-links in collagen using a biotinylated Ehrlich’s reagent. J Biol Chem 276:18812–18818
Kuypers R, Tyler M, Kurth LB, Jenkins ID, Horgan DJ (1992) Identification of the loci of the collagen-associated Ehrlich chromogen in type I collagen confirms its role as a trivalent cross-link. Biochem J 283:129–136
Hanson DA, Eyre DR (1996) Molecular site specificity of pyridinoline and pyrrole cross-links in type I collagen of human bone. J Biol Chem 271:26508–26516
Banse X, Devogelaer JP, Lafosse A, Sims TJ, Grynpas M, Bailey AJ (1998) Cross-link profile of bone collagen correlates with structural organization of trabeculae. Bone 31:70–76
Wang X, Shen X, Li X, Agrawal CM (2002) Age-related changes in the collagen network and toughness of bone. Bone 31:1–7
Garnero P, Borel O, Gineyts E, Duboeuf F, Solberg H, Bouxsein ML, Christiansen C, Delmas PD (2006) Extracellular post-translational modifications of collagen are major determinants of biomechanical properties of fetal bovine cortical bone. Bone 38:300–309
Oxlund H, Mosekilde L, Ortoft G (1996) Reduced concentration of collagen reducible crosslinks in human trabecular bone with respect to age and osteoporosis. Bone 19:479–484
Saito M, Marumo K, Ushiku C, Kato S, Sakai S, Hayakawa N, Mihara M, Shiraishi A (2011) Effects of alfacalcidol on mechanical properties and collagen cross-links of the femoral diaphysis in glucocorticoid-treated rats. Calcif Tissue Int 88:314–324
Yamauchi M, Shiiba M (2008) Lysine hydroxylation and cross-linking of collagen. Methods Mol Biol 446:95–108
Robins SP (1982) Analysis of the crosslinking components in collagen and elastin. Methods Biochem Anal 28:329–379
Avery NC, Sims TJ, Bailey AJ (2009) Quantitative determination of collagen cross-links. Methods Mol Biol 522:103–121
Gineyts E, Borel O, Chapurlat R, Garnero P (2010) Quantification of immature and mature collagen crosslinks by liquid chromatography-electrospray ionization mass spectrometry in connective tissues. J Chromatogr B Analyt Technol Biomed Life Sci 878:1449–1454
Takahashi M, Hoshino H, Kushida K, Inoue T (1995) Direct measurement of crosslinks, pyridinoline, deoxypyridinoline, and pentosidine, in the hydrolysate of tissues using high-performance liquid chromatography. Anal Biochem 232:158–162
Viguet-Carrin S, Gineyts E, Bertholon C (2009) Simple and sensitive method for quantification of fluorescent enzymatic mature and senescent crosslinks of collagen in bone hydrolysate using single-column high performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci 877:1–7
Bank RA, Beekman B, Verzijl N, de Roos JA, Sakkee AN, TeKoppele JM (1997) Sensitive fluorimetric quantitation of pyridinium and pentosidine crosslinks in biological samples in a single high-performance liquid chromatographic run. J Chromatogr B Biomed Sci Appl 703:37–44
Banse X, Sims TJ, Bailey AJ (2002) Mechanical properties of adult vertebral cancellous bone: correlation with collagen intermolecular cross-links. J Bone Miner Res 17:1621–1628
Farlay D, Duclos ME, Gineyts E, Bertholon C, Viguet-Carrin S, Nallala J, Sockalingum GD, Bertrand D, Roger T, Hartmann DJ, Chapurlat R, Boivin G (2011) The ratio 1660/1690 cm−1 measured by infrared microspectroscopy is not specific of enzymatic collagen cross-links in bone tissue. PLoS One 6:e28736
Gourion-Arsiquaud S, Lukashova L, Power J, Loveridge N, Reeve J, Boskey AL (2013) Fourier transform infrared imaging of femoral neck bone: reduced heterogeneity of mineral-to-matrix and carbonate-to-phosphate and more variable crystallinity in treatment-naive fracture cases compared with fracture-free controls. J Bone Miner Res 28:150–161
Satio M, Marumo K (2010) Collagen cross-links as a determinant of bone quality: a possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporos Int 21:195–214
Monnier VM, Sell DR, Nagaraj RH, Miyata S, Grandhee S, Odetti P, Ibrahim SA (1992) Maillard reaction-mediated molecular damage to extracellular matrix and other tissue proteins in diabetes, aging, and uremia. Diabetes 41(Suppl 2):36–41
Robins SP, Bailey AJ (1972) Age-related changes in collagen: the identification of reducible lysine-carbohydrate condensation products. Biochem Biophys Res Commun 48:76–84
Nagai R, Shirakawa J, Fujiwara Y, Ohno R, Moroishi N, Sakata N, Nagai M (2014) Detection of AGEs as markers for carbohydrate metabolism and protein denaturation. J Clin Biochem Nutr 55:1–6
Monnier VM, Sell DR, Dai Z, Nemet I, Collard F, Zhang J (2008) The role of the amadori product in the complications of diabetes. Ann N Y Acad Sci 1126:81–88
Beisswenger PJ, Howell S, Mackenzie T, Corstjens H, Muizzuddin N, Matsui MS (2012) Two fluorescent wavelengths, 440(ex)/520(em) nm and 370(ex)/440(em) nm, reflect advanced glycation and oxidation end products in human skin without diabetes. Diabetes Technol Ther 14:285–292
Couppé C, Hansen P, Kongsgaard M, Kovanen V, Suetta C, Aagaard P, Kjaer M, Magnusson SP (1985) Mechanical properties and collagen cross-linking of the patellar tendon in old and young men. J Appl Physiol 107:880–886
Nakamura K, Nakazawa Y, Ienaga K (1997) Acid-stable fluorescent advanced glycation end products: vesperlysines A, B, and C are formed as crosslinked products in the Maillard reaction between lysine or proteins with glucose. Biochem Biophys Res Commun 232:227–230
Sims TJ, Rasmussen LM, Oxlund H, Bailey AJ (1996) The role of glycation cross-links in diabetic vascular stiffening. Diabetologia 39:946–951
Sell DR, Biemel KM, Reihl O, Lederer MO, Strauch CM, Monnier VM (2005) Glucosepane is a major protein cross-link of the senescent human extracellular matrix. Relationship with diabetes. J Biol Chem 280:12310–12315
Karim L, Tang SY, Sroga GE, Vashishth D (2013) Differences in non-enzymatic glycation and collagen cross-links between human cortical and cancellous bone. Osteoporos Int 24:2441–2447
Saito M, Grynpas MD, Burr DB, Allen MR, Smith SY, Doyle N, Amizuka N, Hasegawa T, Kida Y, Marumo K, Saito H (2014) Treatment with eldecalcitol positively affects mineralization, microdamage, and collagen crosslinks in primate bone. Bone 73:8–15. doi:10.1016/j.bone.2014.11.025
Willett TL, Sutty S, Gaspar A, Avery N, Grynpas M (2013) In vitro non-enzymatic ribation reduces post-yield strain accommodation in cortical bone. Bone 52:611–622
Saito M, Mori S, Mashiba T, Komatsubara S, Marumo K (2008) Collagen maturity, glycation induced-pentosidine, and mineralization are increased following 3-year treatment with incadronate in dogs. Osteoporos Int 19:1343–1354
Silva MJ, Brodt MD, Lynch MA, McKenzie JA, Tanouye KM, Nyman JS, Wang X (2009) Type 1 diabetes in young rats leads to progressive trabecular bone loss, cessation of cortical bone growth and diminished whole-bone strength and fatigue life. J Bone Miner Res 24:1618–1627
Viguet-Carrin S, Roux JP, Arlot ME, Merabet Z, Leeming DJ, Byrjalsen I, Delmas PD, Bouxsein ML (2006) Contribution of the advanced glycation end product pentosidine and of maturation of type I collagen to compressive biomechanical properties of human lumbar vertebrae. Bone 39:1073–1079
Biemel KM, Reihl O, Conrad J, Lederer MO (2001) Formation pathways for lysine-arginine cross-links derived from hexoses and pentoses by Maillard processes: unraveling the structure of a pentosidine precursor. J Biol Chem 276:23405–23412
Brennan M (1989) Changes in solubility, non-enzymatic glycation, and fluorescence of collagen in tail tendons from diabetic rats. J Biol Chem 264:20852–20947
Saito M (1999) Age-related changes in biochemical characteristics of collagen from human weight-bearing and non-weight-bearing bone. Tokyo Jikeikai Med J 114:327–337. http://sciencelinks.jp/j-east/article/200010/000020001000A0286989.php
Sanguineti R, Storace D, Monacelli F, Federici A, Odetti P (2008) Pentosidine effects on human osteoblasts in vitro. Ann N Y Acad Sci 1126:166–172
Ogawa N, Yamaguchi T, Yano T, Yamauchi M, Yamamoto M, Sugimoto T (2007) The combination of high glucose and advanced glycation end-products (AGEs) inhibits the mineralization of osteoblastic MC3T3-E1 cells through glucose-induced increase in the receptor for AGEs. Horm Metab Res 39:871–875
Mercer N, Ahmed H, Etcheverry SB, Vasta GR, Cortizo AM (2007) Regulation of advanced glycation end product (AGE) receptors and apoptosis by AGEs in osteoblast-like cells. Mol Cell Biochem 306:87–94
Cortizo AM, Lettieri MG, Barrio DA, Mercer N, Etcheverry SB, McCarthy AD (2003) Advanced glycation end-products (AGEs) induce concerted changes in the osteoblastic expression of their receptor RAGE and in the activation of extracellular signal-regulated kinases (ERK). Mol Cell Biochem 250:1–10
Khosravi R, Sodek KL, Faibish M, Trackman PC (2014) Collagen advanced glycation inhibits its Discoidin Domain Receptor 2 (DDR2)-mediated induction of lysyl oxidase in osteoblasts. Bone 58:33–41
Zhou Z, Han JY, Xi CX, Xie JX, Feng X, Wang CY, Mei L, Xiong WC (2008) HMGB1 regulates RANKL-induced osteoclastogenesis in a manner dependent on RAGE. J Bone Miner Res 23:1084–1096
Miyata T, Notoya K, Yoshida K, Horie K, Maeda K, Kurokawa K, Taketomi S (1997) Advanced glycation end products enhance osteoclast-induced bone resorption in cultured mouse unfractionated bone cells and in rats implanted subcutaneously with devitalized bone particles. J Am Soc Nephrol 8:260–270
Mitome J, Yamamoto H, Saito M, Yokoyama K, Marumo K, Hosoya T (2011) Non-enzymatic crosslinking pentosidine increase in bone collagen and are associated with disorders of bone mineralization in dialysis patients. Calcif Tissue Int 88:521–529
Valcourt U, Merle B, Gineyts E, Viguet-Carrin S, Delmas PD, Garnero P (2007) Non-enzymatic glycation of bone collagen modifies osteoclastic activity and differentiation. J Biol Chem 282:5691–5703
Okazaki R, Totsuka Y, Hamano K, Ajima M, Miura M, Hirota Y, Hata K, Fukumoto S, Matsumoto T (1997) Metabolic improvement of poorly controlled noninsulin-dependent diabetes mellitus decreases bone turnover. J Clin Endocrinol Metab 82:2915–2920
Tang SY, Allen MR, Phipps R, Burr DB, Vashishth D (2009) Changes in non-enzymatic glycation and its association with altered mechanical properties following 1-year treatment with risedronate or alendronate. Osteoporos Int 20:887–894
Allen MR, Gineyts E, Leeming DJ, Burr DB, Delmas PD (2008) Bisphosphonates alter trabecular bone collagen cross-linking and isomerization in beagle dog vertebra. Osteoporos Int 19:329–337
McCarthy AD, Etcheverry SB, Bruzzone L, Lettieri G, Barrio DA, Cortizo AM (2001) Non-enzymatic glycation of a type I collagen matrix: effect on osteoblastic development and oxidative stress. BMC Cell Biol 2:16
Nojiri H, Saita Y, Morikawa D, Kobayashi K, Tsuda C, Miyazaki T, Saito M, Marumo K, Yonezawa I, Kaneko K, Shirasawa T, Shimizu T (2011) Cytoplasmic superoxide causes bone fragility due to low turnover osteoporosis with impaired collagen cross-links. J Bone Miner Res 26:2682–2694
Derouiche F, Bôle-Feysot C, Naïmi D, Coëffier M (2014) Hyperhomocysteinemia-induced oxidative stress differentially alters proteasome composition and activities in heart and aorta. Biochem Biophys Res Commun 452:740–745
Saito M, Marumo K, Soshi S, Kida Y, Ushiku C, Shinohara A (2010) Raloxifene ameliorates detrimental enzymatic and non-enzymatic collagen cross-links and bone strength in rabbits with hyperhomocysteinemia. Osteoporos Int 21:655–666
Miyata T, de Strihou CVY, Kurokawa K, Baynes JW (1999) Alterations in nonenzymatic biochemistry in uremia: origin and significance of “carbonyl stress” in long-term uremic complications. Kidney Int 55:389–399
Kalninova J, Jakus V, Glejtkova M, Kuracka L, Sandorova E (2014) Impact of glycemic control on advanced glycation and inflammation in overweight and obese patients with type 2 diabetes mellitus. Bratisl Lek Listy 115:457–468
Okazaki K, Yamaguchi T, Tanaka K, Notsu M, Ogawa N, Yano S, Sugimoto T (2012) Advanced glycation end products (AGEs), but not high glucose, inhibit the osteoblastic differentiation of mouse stromal ST2 cells through the suppression of osterix expression, and inhibit cell growth and increasing cell apoptosis. Calcif Tissue Int 91:286–296
McNerny EM, Gong B, Morris MD, Kohn DH (2014) Bone fracture toughness and strength correlate with collagen cross-link maturity in a dose-controlled lathyrism mouse model. J Bone Miner Res. doi:10.1002/jbmr.2356
Oxlund H, Barckman M, Ortoft G, Andreassen TT (1995) Reduced concentrations of collagen cross-links are associated with reduced strength of bone. Bone 17:365S–371S
Opsahl W, Zeronian H, Ellison M, Lewis D, Rucker RB, Riggins RS (1982) Role of copper in collagen cross-linking and its influence on selected mechanical properties of chick bone and tendon. J Nutr 112:708–716
Knott L, Whitehead CC, Fleming RH, Bailey AJ (1995) Biochemical changes in the collagenous matrix of osteoporotic avian bone. Biochem J 310:1045–1051
Shimazaki M, Nakamura K, Kii I, Kashima T, Amizuka N, Li M, Saito M, Fukuda K, Nishiyama T, Kitajima S, Saga Y, Fukayama M, Sata M, Kudo A (2008) Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med 205:295–303
Kii I, Nishiyama T, Li M, Matsumoto K, Saito M, Amizuka N, Kudo A (2010) Incorporaiton of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J Biol Chem 285:2028–2039
Maruhashi T, Kii I, Saito M, Kudo A (2010) Interaction between periostin and BMP-1 promotes proteolytic activation of lysyl oxidase. J Biol Chem 85:13294–13303
Mochida Y, Parisuthiman D, Pornprasertsuk-Damrongsri S, Atsawasuwan P, Sricholpech M, Boskey AL, Yamauchi M (2009) Decorin modulates collagen matrix assembly and mineralization. Matrix Biol 28:44–52
Parisuthiman D, Mochida Y, Duarte WR, Yamauchi M (2005) Biglycan modulates osteoblast differentiation and matrix mineralization. J Bone Miner Res 20:1878–1886
Vashishth D, Gibson GJ, Khoury JI, Schaffler MB, Kimura J, Fyhrie DP (2001) Influence of nonenzymatic glycation on biomechanical properties of cortical bone. Bone 28:195–201
Tang SY, Zeenath U, Vashishth D (2007) Effects of non-enzymatic glycation on cancellous bone fragility. Bone 40:1144–1151
Nyman JS, Roy A, Acuna RL, Gayle HJ, Reyes MJ, Tyler JH, Dean DD, Wang X (2006) Age-related effect on the concentration of collagen crosslinks in human osteonal and interstitial bone tissue. Bone 39:1210–1217
Nyman JS, Roy A, Tyler JH, Acuna RL, Gayle HJ, Wang X (2007) Age-related factors affecting the postyield energy dissipation of human cortical bone. J Orthop Res 25:646–655
Zioupos P, Currey JD, Hamer AJ (1999) The role of collagen in the declining mechanical properties of aging human cortical bone. J Biomed Mater Res 45:108–116
Dequeker J, Merlevede W (1971) Collagen content and collagen extractability pattern of adult human trabecular bone according to age, sex and amount of bone mass. Biochim Biophys Acta 244:410–420
Bailey AJ, Sims TJ, Ebbesen EN (1999) Age-related changes in the biochemical properties of human cancellous bone collagen: relationship to bone strength. Calcif Tissue Int 65:203–210
Odetti P, Rossi S, Monacelli F, Poggi A, Cirnigliaro M, Federici M, Federici A (2005) Advanced glycation end products and bone loss during aging. Ann N Y Acad Sci 1043:710–717
van der Harst MR, DeGroot J, Kiers GH, Brama PA, van de Lest CH, van Weeren PR (2005) Biochemical analysis of the articular cartilage and subchondral and trabecular bone of the metacarpophalangeal joint of horses with early osteoarthritis. Am J Vet Res 66:1238–1246
van den Bos T, Speijer D, Bank RA, Brömme D, Everts V (2008) Differences in matrix composition between calvaria and long bone in mice suggest differences in biomechanical properties and resorption: special emphasis on collagen. Bone 43:459–468
Matsuura T, Tokutomi K, Sasaki M, Katafuchi M, Mizumachi E, Sato H (2014) Distinct characteristics of mandibular bone collagen relative to long bone collagen: relevance to clinical dentistry. Biomed Res Int. doi:10.1155/2014/769414
Isaksson H, Tolvanen V, Finnilä MA, Iivarinen J, Tuukkanen J, Seppänen K, Arokoski JP, Brama PA, Jurvelin JS (2009) Helminen HJ.Physical exercise improves properties of bone and its collagen network in growing and maturing mice. Calcif Tissue Int 85:247–256
Bailey AJ, Wotton SF, Sims TJ, Thompson PW (1993) Biochemical changes in the collagen of human osteoporotic bone matrix. Connect Tissue Res 29:119–132
Aoki C, Uto K, Honda K, Kato Y, Oda H (2013) Advanced glycation end products suppress lysyl oxidase and induce bone collagen degradation in a rat model of renal osteodystrophy. Lab Invest 93:1170–1183
Gamsjaeger S, Srivastava AK, Wergedal JE, Zwerina J, Klaushofer K, Paschalis EP, Tatakis DN (2014) Altered bone material properties in HLA-B27 rats include reduced mineral to matrix ratio and altered collagen cross-links. J Bone Miner Res 29:2382–2391
Guerra RC, Zuñiga-Muñoz A, Guarner Lans V, Díaz-Díaz E, TenaBetancourt CA, Pérez-Torres I (2014) Modulation of the activities of catalase, Cu–Zn, Mn superoxide dismutase, and glutathione peroxidase in adipocyte from ovariectomised female rats with metabolic syndrome. Int J Endocrinol. doi:10.1155/2014/175080
Ventura E, Durant R, Jaussent A, Picot MC, Morena M, Badiou S, Dupuy AM, Jeandel C, Cristol JP (2009) Homocysteine and inflammation as main determinants of oxidative stress in the elderly. Free Radic Biol Med 46:737–744
Shiraki M, Urano T, Kuroda T, Saito M, Tanaka S, Miyao-Koshizuka M, Inoue S (2008) The synergistic effect of bone mineral density and methylenetetrahydrofolate reductase (MTHFR) polymorphism (C677T) on fracture. J Bone Miner Metab 26:595–602
McLean RR, Jacques PF, Selhub J, Tucker KL, Samelson EJ, Broe KE, Hannan MT, Cupples LA, Kiel DP (2004) Homocysteine as a predictive factor for hip fracture in older persons. N Engl J Med 350:2042–2049
Yang J, Hu X, Zhang Q, Cao H, Wang J, Liu B (2012) Homocysteine level and risk of fracture: a meta-analysis and systematic review. Bone 51:376–382
Lubec B, Fang-Kircher S, Lubec T, Blom HJ, Boers GH (1996) Evidence for McKusick’s hypothesis of deficient collagen cross-linking in patients with homocystinuria. Biochim Biophys Acta 13:159–162
Herrmann M, Tami A, Wildemann B, Wolny M, Wagner A, Schorr H, Taban-Shomal O, Umanskaya N, Ross S, Garcia P, Hübner U, Herrmann W (2008) Hyperhomocysteinemia induces a tissue specific accumulation of homocysteine in bone by collagen binding and adversely affects bone. Bone 44:467–475
Leslie WD, Rubin MR, Schwarz AV, Kanis JA (2012) Type 2 diabetes and bone. J Bone Miner Res 27:2231–2237
Hammond MA, Gallant MA, Burr DB, Wallace JM (2014) Nanoscale changes in collagen are reflected in physical and mechanical properties of bone at the microscale in diabetic rats. Bone 60:26–32
Tomasek JJ, Meyers SW, Basinger JB, Green DT, Shew RL (1994) Diabetic and age-related enhancement of collagen-linked fluorescence in cortical bones of rats. Life Sci 55:855–861
Ebesunun MO, Obajobi EO (2012) Elevated plasma homocysteine in type2 diabetes mellitus :a risk factor for cardiovascular diseases. Pan Afr Med J 12:48
Baynes JW (1991) Role of oxidative stress in development of complications in diabetes. Diabetes 40:405–412
Miller PD (2014) Bone disease in CKD: a focus on osteoporosis diagnosis and management. Am J Kidney Dis 64:290–304
Kazama JJ, Iwasaki Y, Fukagawa M (2013) Uremic osteoporosis. Kidney Int Suppl (2011) 3:446–450
LowryMB Lotinun S, Leontovich AA, Zhang M, Maran A, Shogren KL, Palama BK, Marley K, Iwaniec UT, Turner RT (2008) Osteitis fibrosa is, mediated by platelet-derived growth factor-A via a phosphoinositide, 3-kinase-dependent signaling pathway in a rat model for chronic, hyperparathyroidism. Endocrinology 149:5735–5746
Muntner P, Hamm LL, Kusek JW, Chen J, Whelton PK, He J (2004) The prevalence of nontraditional risk factors for coronary heart disease in patients with chronic kidney disease. Ann Intern Med 140:9–17
Kaji H, Yamauchi M, Yamaguchi T, Shigematsu T, Sugimoto T (2010) Mild renal dysfunction is a risk factor for a decrease in bone mineral density and vertebral fractures in Japanese postmenopausal women. J Clin Endocrinol Metab 95:4635–4642
LaCroix AZ, Lee JS, Wu L, Cauley JA, Shlipak MG, Ott SM, Robbins J, Curb JD, Leboff M, Bauer DC, Jackson RD, Kooperberg CL, Cummings SR (2008) Cystatin-C, renal function, and incidence of hip fracture in postmenopausal women. J Am Geriatr Soc 56:1434–1441
Oussalah A, Guéant JL, Peyrin-Biroulet L (2011) Meta-analysis: hyperhomocysteinaemia in inflammatory bowel diseases. Aliment Pharmacol Ther 34:1173–1184
Krajcovicova A, Hlavaty T, Killinger Z, Miznerova E, Toth J, Letkovsky J, Nevidanska M, Cierny D, Koller T, Zelinkova Z, Huorka M, Payer J (2014) Combination therapy with an immunomodulator and anti-TNF α agent improves bone mineral density in IBD patients. J Crohns Colitis. doi:10.1016/j.crohns.2014.08.004
van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C (2003) Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 48:3224–3229
Canalis E (2005) Mechanisms of glucocorticoid action in bone. Curr Osteoporos Rep 3:98–102
Yao W, Cheng Z, Pham A, Busse C, Zimmermann EA, Ritchie RO, Lane NE (2008) Glucocorticoid-induced bone loss in mice can be reversed by the actions of parathyroid hormone and risedronate on different pathways for bone formation and mineralization. Arthritis Rheum 58:3485–3497
Saito M, Marumo K, Ushiku C, Kato S, Sakai S, Hayakawa N, Mihara M, Shiraishi A (2011) Effects of alfacalcidol on mechanical properties and collagen cross-links of the femoral diaphysis in glucocorticoid-treated rats. Calcif Tissue Int 88:314–324
Benson SC, LuValle PA (1998) Inhibition of lysyl oxidase and prolyl hydroxylase activity in glucocorticoid treated rats. Biochem Biophys Res Commun 99:557–562
Centrella M, McCarthy TL, Canalis E (1991) Glucocorticoid regulation of transforming growth factor beta 1 activity and binding in osteoblast-enriched cultures from fetal rat bone. Mol Cell Biol 11:4490–4496
Delany AM, Durant D, Canalis E (2001) Glucocorticoid suppression of IGF I transcription in osteoblasts. Mol Endocrinol 15:1781–1789
Reiser K, Summers P, Medrano JF, Rucker R, Last J, McDonald R (1996) Effects of elevated circulating IGF-1 on the extracellular matrix in high-growth C57BL/6J mice. Am J Physiol 271:R696–703
Gallant MA, Brown DM, Hammond M, Wallace JM, Du J, Deymier-Black AC, Almer JD, Stock SR, Allen MR, Burr DB (2014) Bone cell-independent benefits of raloxifene on the skeleton: a novel mechanism for improving bone material properties. Bone 61:191–200
Saito M, Fujii K, Soshi S (2005) Effects of vitamin B6 and Vitamin K2 on bone mechanical properties and collagen cross-links in spontaneously diabetic WBN/Kob rats. J Bone Miner Res 20:S286
Ahmad S, Shahab U, Baig MH, Khan MS, Khan MS, Srivastava AK, Saeed M (2013) Inhibitory effect of metformin and pyridoxamine in the formation of early, intermediate and advanced glycation end-products. PLoS One 8:e72128
Hammes HP, Martin S, Federlin K, Geisen K, Brownlee M (1991) Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc Natl Acad Sci USA 88:11555–11558
Khatami M, Suldan Z, David I, Li W, Rockey JH (1988) Inhibitory effects of pyridoxal phosphate, ascorbate and aminoguanidine on nonenzymatic glycosylation. Life Sci 43:1725–1731
Voziyan PA, Hudson BG (2005) Pyridoxamine: the many virtues of a maillard reaction inhibitor. Ann N Y Acad Sci 1043:807–816
Conflict of interest
Mitsuru Saito and Keishi Marumo have no conflict of interest.
Human and Animal Rights and Informed Consent
All research, discussed in this review has been subject to ethics approval and, informed consent has been obtained in animal and human studies.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Saito, M., Marumo, K. Effects of Collagen Crosslinking on Bone Material Properties in Health and Disease. Calcif Tissue Int 97, 242–261 (2015). https://doi.org/10.1007/s00223-015-9985-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-015-9985-5