
Abstract
Clinical and experimental studies have reported that phosphate overload plays a central role in the pathogenesis of vascular calcification in chronic kidney disease. However, it remains undetermined whether phosphate induces cellular senescence during vascular calcification. We established a modified uremic rat model induced by a diet containing 0.3 % adenine that showed more slowly progressive kidney failure, more robust vascular calcification, and longer survival than the conventional model (0.75 % adenine). To determine the effect of phosphate on senescence of vascular smooth muscle cells (VSMCs) and the protective effect of phosphate binders, rats were divided into four groups: (1) normal control rats; (2) rats fed with the modified adenine-based diet (CKD); (3) CKD rats treated with 6 % lanthanum carbonate (CKD-LaC); and (4) CKD rats treated with 6 % calcium carbonate (CKD-CaC). After 8 weeks, CKD rats showed circumferential arterial medial calcification, which was inhibited in CKD-LaC and CKD-CaC rats. CKD rats showed increased protein expression of senescence-associated β-galactosidase, bone-related proteins, p16 and p21, and increased oxidative stress levels in the calcified area, which were inhibited by both phosphate binders. However, serum levels of oxidative stress and inflammatory markers, serum fibroblast growth factor 23, and aortic calcium content in CKD-CaC rats were higher than those in CKD-LaC rats. In conclusion, phosphate induces cellular senescence of VSMCs in the modified uremic rat model, and phosphate binders can prevent both cellular senescence and calcification of VSMCs via phosphate unloading. Our modified adenine-based uremic rat model is useful for evaluating uremia-related complications, including vascular calcification.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cellular senescence, also called stress-induced premature senescence, is a state of irreversible cell-cycle arrest of mitotic cells [1] and leads to a reduced capacity to respond to various stimuli. Senescent cells exhibit a flattened and enlarged morphology and express a different series of genes such as p16, p21, p53, and retinoblastoma protein [2]. Recent studies have reported that cellular senescence is involved in various organ dysfunctions in chronic kidney disease (CKD) [3, 4]. Cellular senescence has also been shown to be related to vascular calcification (VC) that contributes to the high cardiovascular mortality in CKD: in vitro studies have reported that replicative senescence of vascular smooth muscle cells (VSMCs) promotes VC [5]. Although phosphate (Pi) is one the most powerful inducers of VC [6], it remains undetermined whether Pi overload-related cellular senescence of VSMC is involved in the VC associated with CKD.
Pi binders are useful to counteract Pi overload in CKD. Clinical studies have already confirmed that Ca-containing Pi binders promote VC more than non-Ca-containing Pi binders [7, 8]. Experimental studies have shown that Ca overload accelerates VC by several known mechanisms [1]. However, it remains unknown whether calcium (Ca)-containing Pi binders and non-Ca-containing Pi binders have differential effects on cellular senescence of VSMC, oxidative stress, and inflammation in CKD.
Developing animal models that exhibit extensive and robust VC is another important issue for research in vascular biology [9]. Various types of animal models with VC have been proposed [10]. The adenine-fed renal failure model is one of the most frequently used rat models for its relatively easy induction [11]. Although adenine-fed uremic rat models develop a series of renal failure-related phenotypes including arterial medial calcification, the original model using 0.75 % adenine has several vital limitations: relatively low prevalence and degree of VC, severe and rapid malnutrition, and high fatality in 4–6 weeks [12–14]. Accordingly, a modified adenine-fed uremic rat model with a high probability of developing extensive arterial medial calcification and a longer survival rate is required.
In the preset study, we established a new uremic rat model that develops robust and extensive arterial medial calcification with a higher probability than the conventional (0.75 %) adenine-fed uremic rat model by feeding a modified adenine-based diet. In addition, we investigated the role of Pi overload in CKD focusing on cellular senescence of VSMC and VC, and compared the therapeutic potential of Pi binders (lanthanum carbonate, LaC, and calcium carbonate, CaC) for Pi-related changes in this new uremic rat model.
Materials and Methods
Animal Care and Study Protocols
Male Sprague–Dawley rats (10-week old) were purchased from Kyudo Co. Ltd (Saga, Japan) and fed a standard diet for 7 days before being used in each protocol. All rats had free access to food and water. Both the standard diet and synthetic diet were purchased from Oriental Yeast Co., Ltd (Tokyo, Japan).
In protocol 1 (survival study), the survival rate over 8 weeks (until day 56) was compared between the conventional and new uremic rat models fed adenine-containing diets; (1) CKD-HA, uremic rats fed a conventional high adenine-based diet (0.75 % adenine, 1.0 % Ca, 1.2 % Pi, 19 % grain-based protein); and (2) CKD-LA, uremic rats fed a modified low adenine-based diet (0.3 % adenine, 1.0 % Ca, 1.2 % Pi, 20 % lactose, 19 % casein-based protein) (Supplemental Fig. 1a).
In protocol 2 (model comparison study), to compare the biochemical parameters and VC between CKD-HA and CKD-LA, rats were raised for 2, 4, and 6 weeks in the CKD-HA group, and for 2, 4, 6, and 8 weeks in the CKD-LA group (n = 8 rats for each time point in each group; 56 total rats). Blood, 24-h urine, and aorta were obtained at each time period (Supplemental Fig. 1b).
In protocol 3 (treatment study), 32 rats were randomly divided into four groups (n = 8 for each group) and raised under each specific diet as follows for 8 weeks: (1) CNT, rats fed standard diet; (2) CKD, rats fed modified adenine-based diet; (3) CKD-LaC, CKD rats treated with 6 % LaC, and (4) CKD-CaC, CKD rats treated with 6 % CaC (Supplemental Fig. 1c). The content of each diet is shown in Supplemental Table 1. In the present study, we used LaC as a non-Ca-containing Pi binder, because sevelamer chloride, another non-Ca-containing Pi binder, was shown to have pleiotropic effects including anti-inflammatory action that may ameliorate the progression of VC [15, 16].
1 day before being killed, rats were housed in metabolic cages for 24 h and urine was collected for each protocol. Urine was centrifuged at 1500×g for 15 min and the supernatant stored at −30 °C. Rats were killed under sevoflurane anesthesia, and blood and aorta were collected. The abdominal aorta was immersed in formalin for histological analysis; the remainder was stored at −80 °C. Serum and urinary levels of albumin, urea nitrogen, creatinine (Cr), Ca, Pi, and magnesium were measured using an automated analyzer (Auto Analyzer HITACHI 7020; Hitachi High-Technologies Corporation, Tokyo, Japan). The following biochemical parameters were determined by commercially available rat ELISA kits: serum intact fibroblast 23 (Kainos Laboratories Inc., Tokyo, Japan), urinary8-hydroxy-2’-deoxyguanosine (8-OHdG; JaICA, Shizuoka, Japan), and serum tumor necrosis factor-alpha (TNF-α; R&D Systems, Minneapolis, MN, USA). All kits were used according to the manufacturers’ instructions, and their qualities were within analytical levels.
Examination of Arterial Calcification and Renal Fibrosis
Four-micrometer sections from paraffin-embedded aorta were deparaffinized and processed for von Kossa staining. Semi-quantitative determination of the degree and prevalence of VC was performed using the four-level scale developed from previous studies [17]: no calcification (calcified area was not detected), mild calcification (calcified area was 1–33 % of the aortic ring), moderate calcification (calcified area was 34–66 % of the aortic ring), severe calcification (calcified area was 67–100 % of the aortic ring).
To quantitatively evaluate the degree of VC, frozen aortic tissue was weighed and hydrolyzed in 1 mL of 6 mol/L hydrochloric acid for 24 h. The Ca and Pi content of the supernatant was determined using commercially available kits (Calcium E-test and Phospha C-test; Wako, Osaka, Japan) and normalized by wet tissue weight (μg/mg wet tissue weight).
Two-micrometer sections from paraffin-embedded kidney were deparaffinized and processed for Periodic-Acid-Schiff staining and Sirius-Red staining using the standard method for evaluating renal structure and fibrosis. Representative histological images were captured using light microscopy on an Eclipse E800 microscope (Nikon, Tokyo, Japan).
Immunohistochemistry
Immunohistochemistry was performed as previously described [18]. Briefly, 4-µm sections from paraffin-embedded aorta were deparaffinized, rehydrated, and prepared for antigen retrieval. Antigen retrieval was performed by microwave for 15 min in citrate buffer (pH 6) for p16, p21, senescence-associated β-galactosidase (SA-β-gal), runt-related gene 2 (Runx2), and osteocalcin. For antigen retrieval for 8-OHdG, sections were treated with RNase solution at 37 °C for 1 h, proteinase K solution (10 μg/mL) for 15 min, hydrochloric acid (4 mol/L) for 20 min, and Trizma base (50 mM) for 5 min at room temperature. p16, p21, and SA-β-gal were used as markers for cellular senescence, Runx2 and osteocalcin as makers for osteoblastic transdifferentiation, and 8-OHdG as the marker for oxidative stress [18, 19]. After inactivation of intrinsic peroxidase by incubation in 0.3 % hydrogen peroxidase, sections were treated with 5 % skim milk for 30 min at room temperature and incubated in a humidified chamber for 1 h at 37 °C with the following primary antibodies: mouse monoclonal anti-β-galactosidase antibody (1:200; Promega Corp., Madison, WI, USA), mouse monoclonal anti-p16 antibody (1:100; sc-1661; Santa Cruz Biotechnologies, Santa Cruz, CA, USA), mouse monoclonal anti-p21 antibody (1:50; sc-6246, Santa Cruz Biotechnologies), rabbit polyclonal anti-Runx2 antibody (1:50; sc-10758, Santa Cruz Biotechnologies), goat polyclonal anti-osteocalcin antibody (1:25; sc-18319, Santa Cruz Biotechnologies), and mouse monoclonal anti-8-OHdG antibody (1:200, Japan Institute for the Control of Aging, Nikken Seil Co. Ltd., Shizuoka, Japan). After washing three times in PBS/Tween-20, sections were incubated with horseradish peroxidase (HRP)-coupled secondary antibody (Nichirei Corporation, Tokyo, Japan) for 30 min at room temperature. HRP was visualized by reaction with 3,3′-diaminobenzidine tetrahydrochloride and hydrogen peroxide.
Real-Time PCR
Total RNA was extracted from rat tissue frozen in liquid nitrogen by the guanidinium thiocyanate phenol–chloroform method, according to the manufacturer’s instructions using TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA), and used to prepare complementary DNA by reverse transcription using a PrimeScript™ RT reagent kit (Perfect Real Time; Takara Bio Inc., Otsu, Japan). Real-time quantitative PCR was performed using SYBR Premix Ex Taq™ (Takara Bio Inc.), Applied Biosystems 7500 Real-time PCR systems (Applied Biosystems, CA, USA), and the following primers purchased from Takara Bio Inc.: rat glyceraldehyde 3-phosphate dehydrogenase (GAPDH), RA015380; rat TNF-α, RA043092; rat alkaline phosphatase, RA041418; rat Runx2, RA067967; rat nicotinamide adenine dinucleotide phosphate oxidase 4 (Nox4), RA064409; rat p16, RA047733; and rat p21, RA063054. The cycling conditions were 30 s at 95 °C, followed by 40 cycles of 5 s at 95 °C for denaturation and 40 s of annealing at 60 °C. The specificity of the PCR products was confirmed by analysis of the melting curves and additionally by agarose gel electrophoresis. All measurements were performed in duplicate, and mRNA fold changes were calculated using the 2−ΔΔCt method using GAPDH as an internal reference.
Statistical Analyses
Statistical analyses were performed using JMP version 10.0 (SAS Institute, Tokyo, Japan). Data are presented as mean ± SD unless otherwise specified. Kaplan–Meier method and log-rank test were used to compare the survival rate between CKD-HA and CKD-LA. Differences between the two groups were analyzed using one-way ANOVA followed by Dunnett’s test for serial, parametric data. Differences among groups were analyzed by Cochran-Armitage tests for ordinal data for protocol 2 and by one-way ANOVA followed by Tukey’s multiple tests for protocol 3. A two-tailed p value of <0.05 was considered statistically significant.
Results
Modified Uremic Rat Had Longer Survival, Slower Progression of Uremia, and Less Malnutrition than the Conventional Model (Protocol 1 and Protocol 2)
Figure 1 shows the survival curve described by the Kaplan–Meier method in both the groups. Only two of 16 rats survived after 8 weeks in CKD-HA group, while 12 out of 16 rats survived after 8 weeks in CKD-LA group; the survival rate in CKD LA group was significantly longer than that in CKD-HA group (log-rank test, P < 0.05).

Survival curve in two adenine-fed rats (protocol 1, survival study). CKD-HA: uremic rats fed a conventional high adenine diet (adenine 0.75 %, Ca 1.0 %, Pi 1.2 %, 19 % grain-based protein); and CKD-LA: uremic rats fed a modified low adenine diet (adenine 0.3 %, Ca 1.0 %, Pi 1.2 %, 20 % lactose, 19 % casein-based protein). Ca calcium, CKD chronic kidney disease, Pi phosphate
Next, we compared biochemical parameters and vascular pathology of CKD-LA rats at week 8 with those of CKD-HA rats at week 6. Tables 1 and 2 summarize the physical and biochemical parameters obtained from urine and serum every 2 weeks in both groups. Figure 2 shows representative photomicrographs of kidney specimens stained with Periodic-Acid-Schiff and Sirius-Red method obtained every 2 weeks in both the groups. Both CKD-HA rats and CKD-LA rats showed progressive increases in blood urea nitrogen, serum creatinine levels, and urine volume and progressive decreases in body weight, food intake, and serum albumin levels, followed by hyperphosphaturia and hyperphosphatemia. However, the progression of these parameters was slower in CKD-LA than in CKD-HA. Periodic-Acid-Schiff staining and Sirius-Red staining of the kidney showed that fibrotic areas, dilated tubules, and deposition of adenine crystals in the renal cortex increased in both the groups in a time-dependent manner (Fig. 2). The progression of renal injury was also slower in CKD-LA than in CKD-HA.

Kidney histology in two adenine-fed rats (protocol 2, model comparison study). Representative photomicrographs of kidney specimens stained with a Periodic-Acid-Schiff and b Sirius-Red at each week in two rat models. CKD-HA; uremic rats fed a conventional high adenine diet (adenine 0.75 %, Ca 1.0 %, Pi 1.2 %, 19 % grain-based protein); and CKD-LA: uremic rats fed a modified low adenine diet (adenine 0.3 %, Ca 1.0 %, Pi 1.2 %, 20 % lactose, 19 % casein-based protein). Ca calcium, CKD chronic kidney disease, Pi phosphate
Serum levels of urea and creatinine in CKD-LA rats at week 8 were almost the same as those in CKD-HA at week 6. Histologically, renal injury in CKD-LA rats at week 8 was similar to that in CKD-HA rats at week 6. Serum Pi levels and urinary Pi excretion in CKD-LA rats at week 8 were higher than those in CKD-HA rats at week 6. Serum albumin level and body weight in CKD-LA rats at week 8 were also higher than in CKD-HA rats at week 6.
Modified Adenine-Based Uremic Rats Induced More Robust and Extensive Arterial Medial Calcification than Conventional Adenine-Based Rats (Protocol 2)
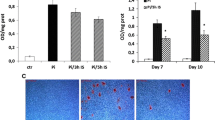
Figure 3a and b shows semi-quantitative analysis of aorta stained using the von Kossa method revealing the prevalence and degree of VC at each time point in the two groups. CKD-HA rats developed VC at week 4 and week 6, and CKD-LA rats developed VC at week 6 and week 8 (Fig. 3b). The degree and prevalence of arterial medial calcification in CKD-LA rats at week 8 was significantly more severe than in CKD-HA rats at week 6 with less variation. The Ca and Pi content in CKD-LA rats at week 8 was greater than those in CKD-HA rats at week 6 (Fig. 3c). Arterial medial calcification in CKD-LA rats at week 8 spread from the ascending aorta to the bilateral femoral arteries (data not shown).

Prevalence and extent of VC in two adenine-fed rats (protocol 2, model comparison study). a Representative photomicrographs of abdominal aorta stained using the von Kossa method and semi-quantification of the degree of VC. No calcification: calcified area is absent; mild calcification: calcified area covers 1–33 % of the aortic ring; moderate calcification: calcified area covers 34–66 % of the aortic ring; and severe calcification: calcified area covers 67–100 % of the aortic ring (original magnification, ×40) b Prevalence and degree of VC at every 2 weeks for both the groups. c Ca and Pi content in the aorta at each time period in each group. CKD: uremic rats fed a conventional high adenine diet (adenine 0.75 %, Ca 1.0 %, Pi 1.2 %, 19 % grain-based protein) (CKD-HA); and CKD-LA: uremic rats fed a modified low adenine diet (adenine 0.3 %, Ca 1.0 %, Pi 1.2 %, 20 % lactose, 19 % casein-based protein), Ca calcium, CKD chronic kidney disease, Pi phosphate, VC vascular calcification. Data are expressed as mean ± SEM. A two-tailed P < 0.05 was considered statistically significant. a P < 0.05 vs. CKD-HA at week 2, b P < 0.05 vs. CKD-LA at week 2
Phosphate Binders Prevented Phosphate-Induced Mineral Derangement (Protocol 3)
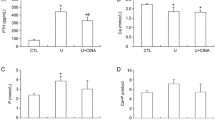
Serum and urinary biochemical parameters in each group are shown in Table 3. Serum creatinine and urea nitrogen was significantly higher in CKD, CKD-LaC, and CKD-CaC rats than that in CNT rats (P < 0.05), while no significant difference was observed among the other three CKD groups. Serum Pi, intact FGF23 levels, and urinary Pi excretion were significantly higher in CKD rats than in CNT rats (P < 0.05); treatment with LaC or CaC significantly decreased these parameters (P < 0.05). The urinary Ca level in CKD-CaC rats was significantly higher than those in the other three groups (P < 0.05). The levels of urinary protein were comparable among the four groups.
Pi Binders Ameliorated Pi-Induced Aortic Calcification in Uremia
As shown in Fig. 4, both Ca and Pi contents in the aorta of CKD rats were significantly higher than in CNT rats, while treatment with LaC or CaC suppressed both Ca and Pi in the aorta induced by high-Pi loading. However, Ca and Pi contents in the aorta in CKD-CaC rats were significantly greater than those in CKD-LaC rats.

Effects of phosphate overload on biochemical parameters and VC in chronic kidney disease (protocol 3, treatment study). Quantification of Ca content (a) and Pi content (b) in aorta. CNT: normal rats fed a standard diet, CKD uremic rats fed a modified adenine-containing diet, CKD-LaC CKD: rats treated with 6 % lanthanum carbonate, CKD-CaC CKD rats treated with 6 % calcium carbonate. Data are expressed as mean ±SEM. One-way ANOVA followed by Tukey’s post hoc tests was performed. Ca calcium, CKD chronic kidney disease, Pi phosphate, VC vascular calcification. Data are expressed as mean ± SEM. A two-tailed P < 0.05 was considered statistically significant. a P < 0.05 vs. CNT, b P < 0.05 vs. CKD
Pi Binders Prevented Pi-Induced Osteoinductive Signaling and Senescence of VSMCs
Figure 5 shows the relative mRNA expression in aortas from each group. The mRNA expressions of Runx2, alkaline phosphatase, Nox4, TNF-α, p16, and p21 in CKD rats were increased compared with that in CNT rats. Treatment with LaC and CaC equally abolished all these Pi-related changes. There were no significant differences in relative mRNA expression of Runx2, alkaline phosphatase, Nox4, TNF-α, p16, and p21 between CKD-LaC and CKD-CaC rats, although mRNA expression of osteoblastic markers (Runx2 and alkaline phosphatase) tended to be higher in CKD-CaC than in CKD-LaC rats.

Effects of phosphate overload on the phenotype of vascular smooth muscle cells (protocol 3, treatment study). The mRNA expression of a Runx2, b ALP, c TNF-α, d Nox4, e p16, and f p21 in the rat aorta. The mRNA expression was corrected to the level of GAPDH. CNT: normal rats fed a standard diet; CKD: uremic rats fed a modified adenine-containing diet; CKD-LaC: CKD rats treated with 6 % lanthanum carbonate; CKD-CaC: CKD rats treated with 6 % calcium carbonate. ALP alkaline phosphatase, BMP-2 bone morphogenetic protein-2, GAPDH glyceraldehyde 3-phosphate dehydrogenase, Nox4 nicotinamide adenine dinucleotide phosphate oxidase 4, Runx2 runt-related gene 2, TNF-α tumor necrosis factor-alpha. Data are expressed as mean ±SEM. One-way ANOVA followed by Tukey’s post hoc tests was performed. Data are expressed as mean ± SEM. A two-tailed P < 0.05 was considered statistically significant. a P < 0.05 vs. CNT, b P < 0.05 vs. CKD
Pi binders Prevented Pi-Induced Oxidative Stress, Cellular Senescence, and Calcification of VSMCs
To determine whether senescence-related proteins were associated with VC induced by Pi overload, we performed immunohistochemistry for Runx2, osteocalcin, SA-β-gal, p16, p21, and 8-OHdG in the abdominal aortas from each group (Fig. 6). No CNT rats were positive for Runx2, osteocalcin, SA-β-gal, p16, and p21. Both Runx2 and osteocalcin were positively stained in and around the calcified area of the CKD aorta. 8-OHdG was accentuated in both the calcified area and non-calcified area in CKD rats. SA-β-gal, p16, and p21 were also positively stained in and around the calcified area of the aorta in CKD rats. Localizations of SA-β-gal, p16, and p21 were closely associated with that of Runx2, osteocalcin, and 8-OHdG in CKD. Treatment with Pi binders suppressed the expressions of p16, p21, and SA-β-gal (cellular senescence-related proteins), Runx2 and osteocalcin (osteoblastic lineage proteins), and 8-OHdG, indicating that senescence-associated proteins, osteoblastic transdifferentiation, and oxidative stress were induced by Pi overload in CKD.

Histological analysis of the effects of Pi overload on cellular senescence of vascular smooth muscle in aorta (protocol 3, treatment study). Von Kossa staining and immunohistochemistry for Runx2, osteocalcin, SA-β-gal, p16, p21, and 8-OHdG of abdominal aorta for each group (original magnification, ×200). CNT: normal rats fed a standard diet; CKD: uremic rats fed a modified adenine-containing diet; CKD-LaC: CKD rats treated with 6 % lanthanum carbonate; and CKD-CaC: CKD rats treated with 6 % calcium carbonate. Ca calcium, CKD chronic kidney disease, 8-OHdG 8-hydroxy-2’-deoxyguanosine, Pi phosphate, Runx2 runt-related gene 2, SA-β-gal senescence-associated β-galactosidase
Pi Binders had Differential Effects on Systemic Inflammation and Oxidative Stress
To compare the effects of the two Pi binders on systemic inflammation and oxidative stress, we measured serum TNF-α levels and the urinary 8-OHdG/Cr ratio in CKD-LaC and CKD-CaC rats. Both serum and urinary markers in CKD-CaC rats were significantly higher than those in CKD-LaC rats (Fig. 7).

Different effects of two phosphate binders on oxidative stress and inflammation markers (protocol 3, treatment study). a Serum TNF-α level and b urinary 8-OHdG/Cr ratio. Serum TNF-α was used as a marker of inflammation, and urinary 8-OHdG/Cr ratio was used as the marker of systemic oxidative stress level. CKD-CaC: uremic rats treated with 6 % calcium carbonate; CKD-LaC: uremic rats treated with 6 % lanthanum carbonate. Cr creatinine, 8-OHdG 8-hydroxy-2’-deoxyguanosine, TNF-α tumor necrosis factor-alpha. Data are expressed as mean ± SEM. A two-tailed P < 0.05 was considered statistically significant. a P < 0.05 versus CKD-LaC
Discussion
Recent studies have reported that cellular senescence plays an important role in the pathogenesis of organ dysfunction in CKD [2–5]. However, Pi loading-induced cellular senescence and its effect on VC in VSMC has not been fully investigated in CKD. The present study demonstrated several novel findings. First, a new adenine-based uremic rat model showed slowly progressive renal failure, more extensive and robust arterial medial calcification, and longer survival compared with the conventional, adenine-based uremic rat model. Second, Pi overload-induced cellular senescence of VSMC and VC in the modified adenine-based uremic rat model. Third, CaC and LaC treatment inhibited senescence and transdifferentiation of VSMCs into osteoblastic lineage cells and almost completely prevented VC. Fourth, CKD rats treated by CaC had greater aortic Ca deposition and increased systemic inflammation and oxidative stress than CKD rats treated by LaC.
It has been a challenge to create a uremic rat model that consistently develops extensive and robust arterial medial calcification without genetic manipulation or a low protein diet [9–14]. In the present study, to overcome the limitations of the conventional adenine rat model, we established a new synthetic diet that modified the conventional adenine-based diet in three ways and compared the modified and conventional adenine-based uremic rat models. First, we used a 0.3 % adenine-based diet to slow progression of CKD and to avoid rapid malnutrition, because a 0.75 % adenine-based diet induces severe and rapid progression of renal failure and malnutrition, leading to high mortality in 4–6 weeks [12–14]. Second, we selected a casein-based diet as the protein source because a casein-based diet can promote Ca and Pi absorption from the gastrointestinal tract [20]. Third, we added 20 % lactose to the diet to further enhance Ca and Pi absorption from the gastrointestinal tract [21]. Both conventional and modified adenine-fed rats developed similar levels of serum creatinine, but the progression of uremia was relatively slower and the serum Pi level was higher in the new model than in the conventional model. The new model survived for 8 weeks and developed robust VC. This modified synthetic diet provided a rat model with extensive and robust arterial medial calcification and a high probability of developing VC after 8 weeks, without using a low protein diet, under a relatively long lifespan. From the animal care viewpoint and in accordance with the “three Rs” (reduction, refinement, and replacement), this model is also useful, because a rat model with longer survival and robust calcification can reduce the number of rats used for the study of VC. Collectively, the features of the present modified rat model are suitable for researchers to investigate the complex mechanisms of arterial medial calcification in uremia.
The impact of cellular senescence on Pi-induced VC is an important subject. In vitro studies have reported that senescence of VSMCs is involved in phenotypic changes of VSMCs into osteoblastic lineage cells, contributing to the increase in calcification [5, 22]. Furthermore, cellular senescence of VSMCs is also involved in arterial intimal calcification in atherosclerotic plaques [23]. A recent experimental study reported that indoxyl sulfate, a uremic toxin, increased protein expression of senescence-associated proteins and VC in the aorta of a 5/6 nephrectomized rat model, which were prevented by administration of AST120, an absorber of indol in the gastrointestinal tract [24]. In the present study, senescence-related proteins p16 and p21 and SA-β-gal increased in and around the calcified area. Both Pi binders prevented the expression of senescence-associated markers in the aorta by inhibiting Pi overload. Recently, Takemura et al. reported in their in vitro study that Pi loading increased SA-β-gal and p21 expression in VSMCs and calcification, which were prevented by resveratrol, an inducer of sirt1 [25]. They also reported that down-regulation of p21 by siRNA decreased calcification, suggesting that p21 plays a critical role in the pathogenesis of cellular senescence of VSMCs and in VC. These results suggest that senescence-associated proteins, including p16 and p21, which are up-regulated by Pi overload, are important in the pathology of VC, and regulation of cell senescence may be a promising target for the prevention of Pi-induced VC.
The mechanistic role of Pi overload in the senescence of VSMCs should be elucidated. Recent experimental studies have shown that oxidative stress is involved in transdifferentiation of VSMCs and results in VC, and that anti-oxidative treatment prevents VC. Pi overload induces reactive oxygen species in mitochondria via NADPH oxidase [19, 26]. In the present study, both oxidative stress and senescence markers were co-localized at the calcified area and were ameliorated by both Pi binders. Because oxidative stress is a strong inducer of VSMC senescence [27], these results collectively indicate that Pi-induced oxidative stress might promote cellular senescence of VSMCs, resulting in progression of VC.
Pi overload is an important and potent inducer of VC in CKD, and activates multiple steps of VC [6]. In the present study, treatment with two different Pi binders significantly decreased urinary Pi excretion and serum Pi levels in uremic rats, leading to the prevention of VC, although uremic levels were comparable among CKD, CKD-LaC, and CKD-CaC. Furthermore, Pi binders blunted the increased expression of Runx2 and osteocalcin observed in the aorta of CKD rats. These results are consistent with previous studies that reported a high-Pi diet induces phenotypic changes of VSMCs into osteoblastic lineage cells and results in VC in uremic rats; these effects are prevented by adequate Pi binder use [28, 29]. Consequently, the present study showed that this newly developed rat model with a low adenine-based diet is useful to determine whether certain interventions can prevent VC.
Clinical studies have shown that Ca-based Pi binders promote VC more than non-Ca-based Pi binders in CKD patients [7, 8]. Recent experimental studies have shown that Ca loading promotes VC by activating various pathways [6] and promotes the formation of calciprotein particles, aggregates of Ca, Pi, α-fetuin, and other proteins [30]. Because calciprotein particles are shown to accelerate inflammation in macrophages, lymphocytes, and VSMCs [31–33], it has become important to avoid their formation. In the present study, aortic Ca content, systemic oxidative stress level, and inflammatory response in CKD-CaC rats were higher than in CKD-LaC rats. These results were consistent with the previous studies [34, 35]. However, there were no significant differences between two different Pi binders in the phenotypic change of VSMCs determined by real-time PCR and immunohistochemistry, although mRNA of bone-related markers in the aorta tented to be higher in CKD-CaC than in CKD-LaC. One of the explanations is that systemic oxidative stress and inflammation decreased soluble inhibitors of VC-like fetuin in CKD-CaC rats, because soluble inhibitors are negatively regulated by inflammation [30]. In addition, the serum FGF23 level in CKD-CaC rats was higher than that in CKD-LaC rats, although the serum Pi level and urinary Pi excretion level were comparable between the two groups. Because recent studies showed that dietary Ca loading induces FGF23 production and FGF23 affects VSMCs in the absence of klotho [36–38], it is possible that Ca overload in CKD-CaC can promote VC independent of osteogenic transdifferentiation of VSMCs. Hence, further studies are needed to clarify the precise mechanisms of how a Ca-based Pi binder exacerbates VC.
There are several limitations in the present study. First, we selected LaC, not sevelamer hydrochloride, as a non-Ca-containing Pi binder. Clinically, sevelamer is widely used and avoids the Ca-loading seen with CaC. Because sevelamer is shown to have anti-inflammatory effects [39–41], we may have observed off-target effects on the serum TNF-α level, urinary 8-OHdG level, and aortic Ca content if we had used sevelamer in the present study. Second, the ability of 6 % LaC to lower urinary Pi level was similar to that of 6 % CaC. Clinically, LaC is shown to be more effective than CaC in binding intestinal Pi [42–44], but in the present study, LaC and CaC were comparable in reducing urinary Pi excretion, indicating that their Pi-binding capacities were almost equivalent. Animal studies have also shown that the same dose of LaC is as effective as CaC [45]. These results indicate that Pi-binding capacity can vary depending on the situation where a given Pi binder is used. Thus, further studies are needed to clarify the difference between CaC and LaC in the Pi-binding capacity and their differential effects on VC and inflammation within the uremic milieu.
In conclusion, Pi overload-induced cellular senescence of VSMCs and arterial medial calcification in uremic rats was inhibited by Pi binder treatment in a novel rat model that showed extensive and robust arterial medial calcification. These results suggest that cellular senescence can play a role in the pathogenesis of Pi overload-related, arterial medial calcification in CKD, and that Pi binders can counteract the cellular senescence of VSMCs, leading to amelioration of VC. Furthermore, our new rat model enables researchers to investigate uremia-related sequela, including VC, because this modified uremic rat model has a longer survival period and develops more consistent and robust phenotypes.
References
Campisi J, d’Adda di Fagagna F (2007) Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8:729–740
Ben-Porath I, Weinberg RA (2005) The signals and pathways activating cellular senescence. Int J Biochem Cell Biol 37:961–976
Yang H, Fogo AB (2010) Cell senescence in the aging kidney. J Am Soc Nephrol 21:1436–1439
Kooman JP, Broers NJ, Usvyat L, Thijssen S, van der Sande FM, Cornelis T, Levin NW, Leunissen KM, Kotanko P (2013) Out of control: accelerated aging in uremia. Nephrol Dial Transplant 28:48–54
Nakano-Kurimoto R, Ikeda K, Uraoka M, Nakagawa Y, Yutaka K, Koide M, Takahashi T, Matoba S, Yamada H, Okigaki M, Matsubara H (2009) Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am J Physiol Heart Circ Physiol 297:H1673–H1684
Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM (2011) Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res 109:697–711
Qunibi W, Moustafa M, Muenz LR, He DY, Kessler PD, Diaz-Buxo JA, Budoff M, CARE-2 Investigators (2008) A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis 51:952–965
Di Iorio B, Bellasi A, Russo D, INDEPENDENT Study Investigators (2012) Mortality in kidney disease patients treated with phosphate binders: a randomized study. Clin J Am Soc Nephrol 7:487–493
Shobeiri N, Adams MA, Holden RM (2010) Vascular calcification in animal models of CKD: a review. Am J Nephrol 31:471–481
Atkinson J (2008) Age-related medial elastocalcinosis in arteries: mechanisms, animal models, and physiological consequences. J Appl Physiol 105:1643–1651
Yokozawa T, Zheng PD, Oura H, Koizumi F (1986) Animal model of adenine-induced chronic renal failure in rats. Nephron 44:230–234
Katsumata K, Kusano K, Hirata M, Tsunemi K, Nagano N, Burke SK, Fukushima N (2003) Sevelamer hydrochloride prevents ectopic calcification and renal osteodystrophy in chronic renal failure rats. Kidney Int 64:441–450
Tamagaki K, Yuan Q, Ohkawa H, Imazeki I, Moriguchi Y, Imai N, Sasaki S, Takeda K, Fukagawa M (2006) Severe hyperparathyroidism with bone abnormalities and metastatic calcification in rats with adenine-induced uraemia. Nephrol Dial Transplant 21:651–659
Price PA, Roublick AM, Williamson MK (2006) Artery calcification in uremic rats is increased by a low protein diet and prevented by treatment with ibandronate. Kidney Int 70:1577–1583
Aoshima Y, Mizobuchi M, Ogata H, Kumata C, Nakazawa A, Kondo F, Ono N, Koiwa F, Kinugasa E, Akizawa T (2012) Vitamin D receptor activators inhibit vascular smooth muscle cell mineralization induced by phosphate and TNF-α. Nephrol Dial Transplant 27:1800–1806
Zhao G, Xu MJ, Zhao MM, Dai XY, Kong W, Wilson GM, Guan Y, Wang CY, Wang X (2012) Activation of nuclear factor-kappa B accelerates vascular calcification by inhibiting ankylosis protein homolog expression. Kidney Int 82:34–44
Yu C, Chen B, Zhao T, Wang R, Akhtar J, Wang H, Zhang H (2013) High phosphate feeding induced arterial medial calcification in uremic rats: roles of Lanthanum carbonate on protecting vasculature. Life Sci 93:646–653
Yamada S, Taniguchi M, Tokumoto M, Toyonaga J, Fujisaki K, Suehiro T, Noguchi H, Iida M, Tsuruya K, Kitazono T (2012) The antioxidant tempol ameliorates arterial medial calcification in uremic rats: important role of oxidative stress in the pathogenesis of vascular calcification in chronic kidney disease. J Bone Miner Res 27:474–485
Gruber HE, Ingram JA, Norton HJ, Hanley EN Jr (2007) Senescence in cells of the aging and degenerating intervertebral disc: immunolocalization of senescence-associated beta-galactosidase in human and sand rat discs. Spine 32:321–327
Moe SM, Chen NX, Seifert MF, Sinders RM, Duan D, Chen X, Liang Y, Radcliff JS, White KE, Gattone VH 2nd (2009) A rat model of chronic kidney disease-mineral bone disorder. Kidney Int 75:176–184
Kawata T, Nagano N, Obi M, Miyata S, Koyama C, Kobayashi N, Wakita S, Wada M (2008) Cinacalcet suppresses calcification of the aorta and heart in uremic rats. Kidney Int 74:1270–1277
Burton DG, Matsubara H, Ikeda K (2010) Pathophysiology of vascular calcification: pivotal role of cellular senescence in vascular smooth muscle cells. Exp Gerontol 45:819–824
Burton DG, Giles PJ, Sheerin AN, Smith SK, Lawton JJ, Ostler EL, Rhys-Williams W, Kipling D, Faragher RG (2009) Microarray analysis of senescent vascular smooth muscle cells: a link to atherosclerosis and vascular calcification. Exp Gerontol 44:659–665
Adijiang A, Higuchi Y, Nishijima F, Shimizu H, Niwa T (2010) Indoxyl sulfate, a uremic toxin, promotes cell senescence in aorta of hypertensive rats. Biochem Biophys Res Commun 399:637–641
Takemura A, Iijima K, Ota H, Son BK, Ito Y, Ogawa S, Eto M, Akishita M, Ouchi Y (2011) Sirtuin A retards hyperphosphatemia-induced calcification of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 31:2054–2062
Zhao MM, Xu MJ, Cai Y, Zhao G, Guan Y, Kong W, Tang C, Wang W (2011) Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int 79:1071–1079
Zhou RH, Vendrov AE, Tchivilev I, Niu XL, Molnar KC, Rojas M, Carter JD, Tong H, Stouffer GA, Madamanchi NR, Runge MS (2012) Mitochondrial oxidative stress in aortic stiffening with age: the role of smooth muscle cell function. Arterioscler Thromb Vasc Biol 32:745–755
Neven E, Dams G, Postnov A, Chen B, De Clerck N, De Broe ME, D’Haese PC, Persy V (2009) Adequate phosphate binding with lanthanum carbonate attenuates arterial calcification in chronic kidney disease. Nephrol Dial Transplant 24:1790–1799
De Schutter TM, Behets GJ, Geryl H, Peter ME, Steppan S, Gundlach K, Passlick-Deetjen J, D’Haese PC, Neven E (2013) Effect of a magnesium-based phosphate binder on medial calcification in a rat model of uremia. Kidney Int 83:1109–1117
Jahnen-Dechent W, Heiss A, Schäfer C, Ketteler M (2011) Fetuin-A regulation of calcified matrix metabolism. Circ Res 108:1494–1509
Pazár B, Ea HK, Narayan S, Kolly L, Bagnoud N, Chobaz V, Roger T, Lioté F, So A, Busso N (2011) Basic calcium phosphate crystals induce monocyte/macrophage IL-1β secretion through the NLRP3 inflammasome in vitro. J Immunol 186:2495–2502
New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, Libby P, Shanahan CM, Croce K, Aikawa E (2013) Macrophage-derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res 113:72–77
Shroff RC, McNair R, Skepper JN, Figg N, Schurgers LJ, Deanfield J, Rees L, Shanahan CM (2010) Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 21:103–112
Cozzolino M, Staniforth ME, Liapis H, Finch J, Burke SK, Dusso AS, Slatopolsky E (2003) Sevelamer hydrochloride attenuates kidney and cardiovascular calcifications in long-term experimental uremia. Kidney Int 64:1653–1661
Moe SM, Chen NX, Newman CL, Gattone VH 2nd, Organ JM, Chen X, Allen MR (2014) A Comparison of calcium to zoledronic acid for improvement of cortical bone in an animal model of CKD. J Bone Miner Res 29:902–910
Jimbo R, Kawakami-Mori F, Mu S, Hirohama D, Majtan B, Shimizu Y, Yatomi Y, Fukumoto S, Fujita T, Shimosawa T (2014) Fibroblast growth factor 23 accelerates phosphate-induced vascular calcification in the absence of Klotho deficiency. Kidney Int 85:1103–1111
Six I, Okazaki H, Gross P, Cagnard J, Boudot C, Maizel J, Drueke TB, Massy ZA (2014) Direct, acute effects of Klotho and FGF23 on vascular smooth muscle and endothelium. PLoS One 9:e93423
Rodriguez-Ortiz ME, Lopez I, Muñoz-Castañeda JR, Martinez-Moreno JM, Ramírez AP, Pineda C, Canalejo A, Jaeger P, Aguilera-Tejero E, Rodriguez M, Felsenfeld A, Almaden Y (2012) Calcium deficiency reduces circulating levels of FGF23. J Am Soc Nephrol 23:1190–1197
Vlassara H, Uribarri J, Cai W, Goodman S, Pyzik R, Post J, Grosjean F, Woodward M, Striker GE (2012) Clin J Am Soc Nephrol 7:934–942
Sun PP, Perianayagam MC, Jaber BL (2009) Endotoxin-binding affinity of sevelamer: a potential novel anti-inflammatory mechanism. Kidney Int Suppl 114:S20–S25
Hauser AB, Azevedo IR, Gonçalves S, Stinghen A, Aita C, Pecoits-Filho R (2010) Sevelamer carbonate reduces inflammation and endotoxemia in an animal model of uremia. Blood Purif 30:153–158
Hutchison AJ, Maes B, Vanwalleghem J, Asmus G, Mohamed E, Schmieder R, Backs W, Jamar R, Vosskuhler A (2005) Efficacy, tolerability, and safety of lanthanum carbonate in hyperphosphatemia: a 6-month, randomized, comparative trial versus calcium carbonate. Nephron Clin Pract 100:c8–c19
Shigematsu T, Lanthanum Carbonate Group (2008) Multicenter prospective randomized, double-blind comparative study between lanthanum carbonate and calcium carbonate as phosphate binders in Japanese hemodialysis patients with hyperphosphatemia. Clin Nephrol 70:404–410
Daugirdas JT, Finn WF, Emmett M, Chertow GM (2011) The phosphate binder equivalent dose. Semin Dial 24:41–49
Takashima T, Sanai T, Miyazono M, Fukuda M, Kishi T, Nonaka Y, Yoshizaki M, Sato S, Ikeda Y (2014) A comparison of the long-term effects of lanthanum carbonate and calcium carbonate on the course of chronic renal failure in rats with adriamycin-induced nephropathy. PLoS One 9:e97859
Conflict of interest
Shunsuke Yamada, Narihito Tatsumoto, Masanori Tokumoto, Hideko Noguchi, Hiroaki Ooboshi, Takanari Kitazono, and Kazuhiko Tsuruya have nothing to disclose.
Human and Animal Rights and Informed Consent
All experimental protocols were reviewed and approved by the Committee on Ethics of Animal Experimentation at Kyushu University Faculty of Medicine (A25-073-1).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yamada, S., Tatsumoto, N., Tokumoto, M. et al. Phosphate Binders Prevent Phosphate-Induced Cellular Senescence of Vascular Smooth Muscle Cells and Vascular Calcification in a Modified, Adenine-Based Uremic Rat Model. Calcif Tissue Int 96, 347–358 (2015). https://doi.org/10.1007/s00223-014-9929-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-014-9929-5