
Abstract
Osteogenesis imperfecta (OI) is a hereditary disease characterized by low bone mass, increased bone fragility, short stature, and skeletal deformities. This study focuses on OI type I, the mildest form of the disease. Bisphosphonates represent the prevailing standard of care in patients with OI. Teriparatide (TPD) is a PTH analog with bone-anabolic actions which has been approved for osteoporosis treatment. Thirteen postmenopausal women with type I OI who had been on treatment with neridronate for at least 2 years and who incurred new vertebral fracture during treatment were treated with TPD for 18 months. Bone mineral density (BMD) increased significantly over 18 months up to 3.5 % at the lumbar spine (p = 0.001), while no significant changes were noted in hip BMD. Serum markers of bone formation and of bone resorption increased significantly during the treatment. The Wnt inhibitors serum dickkopf-1 (DKK1) and sclerostin were also measured. A nonsignificant increase was seen in serum sclerostin levels, while serum DKK1 rose gradually and significantly during TPD treatment. In patients affected by type I OI, TPD treatment is associated with a remarkable response in markers of bone formation. This suggests a normal osteoblastic response to TPD. However, the observed increases in BMD were somewhat lower than those in postmenopausal or senile osteoporosis treated with TPD for the same lag time. Our results open the possibility to develop TPD for the treatment of adult type I OI, but particularly for the lack of a control group, a properly designed controlled study is warranted.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteogenesis imperfecta (OI) is a hereditary disease, mostly autosomal and dominant due to collagen defects, associated with mutations of the genes coding for the chains of collagen type 1 gene (COL1A1 or COL1A2). The disorder is characterized by low bone mass, increased bone fragility, short stature, and skeletal deformities. The most important defect in adults with OI is the inability to acquire an adequate thickness of the cortices of long bone and to achieve or maintain normal trabecular density [1]. A variety of OI types have been described, with considerable differences in severity [2]. OI type I is the mildest form of the disease and has a triad of features: recurrent fractures, blue sclera, and hearing loss [2]. Fractures often begin with ambulation, but the risk decreases after puberty and rises again after the fourth to fifth decades of life [2]. Skeletal deformities are moderate, and the stature is close to normal. Patients with type I OI phenotype have a null COL1A1 allele with consequent type I collagen haploinsufficiency. Thus, type I OI is a clinically and biochemically homogenous group and the only dominant OI form in which structurally abnormal collagen is not present in the mature bone tissue [2].
Bisphosphonates are antiresorptive compounds that are widely used in patients with OI, and they represent the currently prevailing standard of care for moderate to severe forms of the disease, especially in children [3]. The only bisphosphonate registered so far for the treatment of OI is neridronate, which was studied in controlled trials both in children [4, 5] and in adults [6], with evidence of a significant 64 % decrease in fracture number. In a recent Cochrane review [7] to assess the effectiveness and safety of bisphosphonates in people with OI, only two trials in adult populations were identified. In the first study, 2 years of treatment with neridronate [6] was associated with 6.9 and 5.8 % mean increases in spine and hip bone mineral density (BMD), respectively. In the study by Chevrel et al. [8] with alendronate given for 4 years, spine BMD rose by 10 % and hip BMD by 3.3 %.
The rationale for using bisphosphonates in OI is based on the observation that bone turnover markers are often found to be slightly increased [9]. However, the hallmark of the pathophysiology of type I OI remains an inadequate osteoblast synthesis of collagen type I associated with inadequate bone formation.
Teriparatide (TPD, recombinant human PTH 1–34) is a PTH analog with bone-anabolic actions, approved for osteoporosis treatment both in postmenopausal women and in men. Its administration is associated with a twofold increase in markers of bone formation such as N-propeptide of type I collagen (P1NP), followed by an increase also of markers of bone resorption [10]. Owing to its bone-anabolic activity TPD might be effective also in OI, but it remains to be understood whether the functionally impaired osteoblasts of type I OI respond to TPD treatment .
The identification of the Wnt/ß-catenin signaling pathway as a major promoter of bone formation has led to considerable interest in potential crosstalk between this pathway and osteoclastic bone resorption [11]. The Wnt pathway influences bone formation through its effects on osteoblast number, maturation, and progenitor differentiation. In turn, these actions are opposed by various intracellular and secreted factors [11]. The secreted Wnt antagonists sclerostin and dickkopf-1 (DKK1) block Wnt signaling by binding to Wnt coreceptors, such as low-density lipoprotein receptor-related protein 5 (LRP5) and LRP6, thus inhibiting the canonical Wnt/ß-catenin signaling pathway [11, 12]. Sclerostin is primarily, though not exclusively [13], expressed by mature osteocytes, where it appears to regulate bone mass in response to mechanical loading [14]. DKK1 expression is widespread in embryonic mice, while in adults it is almost exclusively confined to osteoblasts and maturing osteocytes [15]. Recently, we showed that DKK1 and sclerostin are also involved in the bone metabolic response to long-term treatment with teriparatide [16], bisphosphonates [17], and denosumab [18].
Here, we show the results of a pilot clinical trial with TPD in postmenopausal women with type I OI previously treated with neridronate, and we explore the Wnt functionality in these patients.
Materials and Methods
The study population included 13 postmenopausal women affected by type I OI. The disease was clinically diagnosed by the presence of a family history of OI, blue sclerae, and previous low-trauma fractures in childhood. A genetic confirmation was available for eight patients. In three other patients a genetic diagnosis was obtained from one of their affected children.
All patients had been on treatment with neridronate (100 mg infused intravenously for 30 min every 3 months) [6] for at least 2 years, and no other osteoporosis medications had been taken before. All patients incurred a new vertebral fracture during neridronate treatment. Thus, all patients had at least one vertebral fracture at the thoracic spine (eight subjects) and/or at the lumbar spine (five women). Three patients had a history of bilateral hip fractures, eight patients a previous Colles fracture, and four a previous fracture of the tibia or fibula. The time elapsed from the last fracture to initiation of TPD ranged 10–18 months.
All patients had their dietary calcium intake regularly evaluated and maintained above 1,000 mg/day through diet or additional supplementation. Vitamin D3 supplements (≥25,000 U monthly) were given if the serum 25(OH) vitamin D levels fell below 20 ng/mL [19].
In all patients neridronate treatment was discontinued and they were fully reassessed before commencing TPD treatment within 1 month. The pens were distributed directly by our center every 2 months, and the patients returned the used pens.
Blood was collected at baseline and every 6 months until the 18 month, when TPD administration was discontinued. Serum samples were stored at −50 °C until they were assayed for P1NP, bone alkaline phosphatase (bAP), C-terminal telopeptide of type I collagen (CTX), DKK1, and sclerostin in our laboratory.
Bone turnover markers were measured by the IDS-ISYS Multi-Discipline automated analyzer (Immunodiagnostic System, Boldon, UK) based on chemiluminescence technology. The coefficients of variation (CV, intra-assay) measured in our laboratory were 4 % for P1NP, 2.0 % for bAP, and 3 % for CTX. Serum DKK1 and sclerostin were measured by ELISA (Biomedica Medizinprodukte, Vienna, Austria) with a sensitivity of 0.38 and 2.6 pmol/L and intra-assay CV of 7 and 5 %, respectively. The interassays were assessed on four separate occasions in four serum samples, and the CV was 8.2 and 6.9 % for DKK1 and sclerostin, respectively.
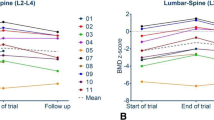
BMD was measured in all patients by dual-energy X-ray absorptiometry (DXA, QDR Delphi; Hologic, Waltham, MA) at the lumbar spine (L1–L4) and at the total proximal hip. Spine DXA measurements were collected for all patients, but one deformed vertebral body was excluded for analysis in five patients. An appropriate hip evaluation could not be obtained in three patients for previous bilateral hip fractures. The CV of the procedure was 1.0 % for spine and 1.2 % for hip BMD.
All statistical procedures were carried out using a computer program (SPSS version 13.0; SPSS Inc., Chicago, IL). Data are expressed as mean ± standard deviation (SD). Comparisons between baseline and follow-up values at each time point were performed by paired t test. Significance was set at p < 0.05.
The study was approved by the institutional review board of the Medical School of Verona. Written informed consent was obtained from all the women.
Results
The mean age of the study population was 59.5 ± 6.6 years (range 52–72), and the duration of previous neridronate treatment ranged 5–9 years (mean 7).
The mean values for each of the biochemical parameters for all time points are listed in Table 1, while the percent changes in biochemical parameters and in BMD are listed in Table 2. All data were normally distributed. All patients fully complied with treatment for 18 months. Mild nausea was reported by seven patients immediately after the injection, but this never led to treatment discontinuation. BMD at the lumbar spine increased significantly throughout the treatment, and at the end of the 18 month treatment period vertebral bone mass increased by 3.5 % (p = 0.001), while no significant changes were noted in hip BMD (Table 2).
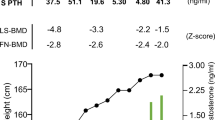
Values for all biochemical parameters were detectable. P1NP, bAP, and serum CTX rose significantly during the treatment with an apparent peak value at 12 months (Table 2). A nonsignificant increase was observed in serum sclerostin levels, while serum DKK1 rose gradually and significantly versus the baseline value, as early as the 6 month time point (Table 2).
The percentage changes in DKK1 were positively correlated with the percentage changes in P1NP (r = 0.42, p < 0.01) and bAP (r = 0.48, p < 0.01) (results not shown). None of the patients reported new fractures during the treatment period.
Discussion
In this study we have shown for the first time that postmenopausal women with type I OI respond to TPD treatment as in postmenopausal osteoporosis in terms of changes in bone turnover markers.
Mean baseline bone turnover markers were in the lower part of the normal range, as expected for their previous long-term therapy with neridronate. We have previously shown [9] that in OI adult patients bone turnover markers are clearly increased, and this provided a strong rationale for the use of an antiresorbing agent in OI. Only serum CTX levels were comparable to those found in healthy controls, and we attributed this finding to a relative lack of pyridinoline crosslinks at the C-terminal portion of collagen type I in OI [9], envisaging a possible inadequate response to anabolic agents. However, the two- and four-fold increases in bAP and P1NP, respectively, suggest an excellent osteoblastic response to TPD. With our results it remains to be established whether the tremendous increase we observed in P1NP should be attributed to increased cell number or also to cellular stimulation of collagen synthesis.
In this study we also measured the changes of two antagonists of the Wnt pathway: serum DKK1 and sclerostin. This evaluation of the WNT pathway was considered important in order to explain a possible lack of response to TPD. Some of the effects on bone turnover of bone active agents used for the treatment of osteoporosis seem to be related to changes in the WNT/β-catenin signaling [20]. Bisphosphonates directly suppress osteoclastic activity, and this suppression of bone resorption is typically associated with a later decrease also of bone formation. Recently, we showed that chronic treatment with a bisphosphonate or denosumab is associated in osteoporotic women with gradual increases in serum levels of sclerostin, which mirrors the decline in bone formation markers [17, 18]. Even more important in the context of the present study was our observation that the weaning of the positive bone-anabolic effect of TPD in postmenopausal osteoporosis after the first few months of treatment is related to an increase in DKK1 [16].
All patients were ambulatory and with almost normal physical activity and skeletal load. Baseline DKK1 and sclerostin were at the lower part of the normal range and significantly lower than those found in an age- and sex-matched group derived from a parallel study (results not shown). This cannot be explained by the previous neridronate therapy since we showed that long-term treatment with neridronate is rather associated with higher serum levels of sclerostin and unchanged DKK1 levels [17]. The results of the present study might suggest that both Wnt inhibitors are somewhat decreased in OI, suggesting a sort of feedback between inadequate bone formation and a compensatory mechanism leading to increased activation of the Wnt pathway. Larger numbers of observations are warranted in order to explore this hypothesis.
During TPD treatment a significant increase of serum DKK1 was observed, confirming the results we observed in osteoporotic subjects after the first year of treatment [16]. Here, the effect of TPD treatment on DKK1 levels seems to be slightly more precocious, becoming statistically significant as early as after 6 months of therapy. This increase is positively correlated with elevation of bone formation markers, namely, bAP and P1NP, but not with the increase in a bone resorption marker such as CTX. This suggests that chronic stimulation of osteoblasts by a PTH analogue might trigger a homeostatic reaction, with oversecretion of DKK1 and consequent damping of the TPD effects.
The BMD changes were considerably lower than those reported in postmenopausal osteoporosis after 18 months of therapy with TPD: +3.5 versus +9–10 % at the spine reported in the pivotal trial in postmenopausal osteoporosis [21]. It has been reported that the BMD increase induced by TPD treatment might be somewhat less robust for patients previously treated with bisphosphonates [22], but the changes observed in postmenopausal osteoporosis previously treated with bisphosphonates [23] are still considerably higher than those seen here. This point remains to be clarified, even though we cannot exclude that our results might have been influenced by the small number of patients included in the study. Together with its uncontrolled nature, this remains the main limitation of the study. However, it should be pointed out that a trial with TPD in growing individuals was not considered at the time acceptable by our regulatory authorities for the oncological risk and that before commencing this study TPD could not be offered as a first-line therapy in postmenopausal type I OI.
In conclusion, we have observed that treatment of type I OI with TPD is associated with a remarkable response in P1NP and other markers of bone turnover. This suggests a normal bone response in this disease characterized by an inadequate osteoblast production of collagen type 1. The observed increases in BMD are somewhat lower than those seen in postmenopausal or senile osteoporosis. A larger number of treatment-naive patients are needed before drawing any conclusions, but the results of this pilot study open the possibility to develop TPD for the treatment of adult type I OI. Bisphosphonates remain the first-line treatment in OI, but the results of this study open a new option in those patients with type I OI in whom the positive effects of bisphosphonate treatment appear to be clinically suboptimal. A major limitation of our study is its uncontrolled nature. For the lack of a control group, a properly designed controlled study remains warranted.
References
Gatti D, Colapietro F, Fracassi E, Sartori E, Antoniazzi F, Braga V, Rossini M, Adami S (2003) The volumetric bone density and cortical thickness in adult patients affected by osteogenesis imperfecta. J Clin Densitom 6:173–177
Forlino A, Cabral WA, Barnes AM, Marini JC (2011) New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol 7:540–557
Cheung MS, Glorieux FH (2008) Osteogenesis imperfecta: update on presentation and management. Rev Endocr Metab Disord 9:153–160
Gatti D, Antoniazzi F, Prizzi R, Braga V, Rossini M, Tatò L, Viapiana O, Adami S (2005) Intravenous neridronate in children with osteogenesis imperfecta: a randomized controlled study. J Bone Miner Res 20:758–763
Antoniazzi F, Zamboni G, Lauriola S, Donadi L, Adami S, Tatò L (2006) Early bisphosphonate treatment in infants with severe osteogenesis imperfecta. J Pediatr 149:174–179
Adami S, Gatti D, Colapietro F, Fracassi E, Braga V, Rossini M, Tatò L (2003) Intravenous neridronate in adults with osteogenesis imperfecta. J Bone Miner Res 18:126–130
Phillipi CA, Remmington T, Steiner RD (2008) Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev 4:CD005088
Chevrel G, Schott AM, Fontanges E, Charrin JE, Lina-Granade G, Duboeuf F, Garnero P, Arlot M, Raynal C, Meunier PJ (2006) Effects of oral alendronate on BMD in adult patients with osteogenesis imperfecta: a 3-year randomized placebo-controlled trial. J Bone Miner Res 21:300–306
Braga V, Gatti D, Rossini M, Colapietro F, Battaglia E, Viapiana O, Adami S (2004) Bone turnover markers in patients with osteogenesis imperfecta. Bone 34:1013–1016
Girotra M, Rubin MR, Bilezikian JP (2006) The use of parathyroid hormone in the treatment of osteoporosis. Rev Endocr Metab Disord 7:113–121
Baron R, Rawadi G (2007) Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology 148:2635–2643
Ott SM (2005) Sclerostin and Wnt signaling—the pathway to bone strength. J Clin Endocrinol Metab 90:6741–6743
Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, Skonier JE, Zhao L, Sabo PJ, Fu Y, Alisch RS, Gillett L, Colbert T, Tacconi P, Galas D, Hamersma H, Beighton P, Mulligan J (2001) Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot–containing protein. Am J Hum Genet 68:577–589
Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido TM, Harris SE, Turner CH (2008) Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem 283:5866–5875
Li J, Sarosi I, Cattley RC, Pretorius J, Asuncion F, Grisanti M, Morony S, Adamu S, Geng Z, Qiu W, Kostenuik P, Lacey DL, Simonet WS, Bolon B, Qian X, Shalhoub V, Ominsky MS, Zhu Ke H, Li X, Richards WG (2006) Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone 39:754–766
Gatti D, Viapiana O, Idolazzi L, Fracassi E, Rossini M, Adami S (2011) The waning of teriparatide effect on bone formation markers in postmenopausal osteoporosis is associated with increasing serum levels of DKK1. J Clin Endocrinol Metab 96:1555–1559
Gatti D, Viapiana O, Adami S, Idolazzi L, Fracassi E, Rossini M (2012) Bisphosphonate treatment of postmenopausal osteoporosis is associated with a dose dependent increase in serum sclerostin. Bone 50:739–742
Gatti D, Viapiana O, Fracassi E, Idolazzi L, Dartizio C, Povino MR, Adami S, Rossini M (2012) Sclerostin and DKK1 in postmenopausal osteoporosis treated with denosumab. J Bone Miner Res 27:2259–2263
Ross AC, Taylor CL, Yaktine AL, Del Valle HB (eds) (2010) Institute of Medicine Committee to review dietary reference intakes for vitamin D and calcium dietary reference intakes for calcium and vitamin D. National Academics Press, Washington
Rossini M, Gatti D, Adami S (2013) Involvement of wnt/β-catenin signaling in the treatment of osteoporosis Calcif Tissue Int (in press)
Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, Wang O, Mitlak BH (2001) Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 344:1434–1441
Boonen S, Marin F, Obermayer-Pietsch B, Simões ME, Barker C, Glass EV, Hadji P, Lyritis G, Oertel H, Nickelsen T, McCloskey EV, EUROFORS Investigators (2008) Effects of previous antiresorptive therapy on the bone mineral density response to two years of teriparatide treatment in postmenopausal women with osteoporosis. J Clin Endocrinol Metab 93:852–860
Finkelstein JS, Wyland JJ, Lee H, Neer RM (2010) Effects of teriparatide, alendronate, or both in women with postmenopausal osteoporosis. J Clin Endocrinol Metab 95:1838–1845
Acknowledgments
We thank Caterina Fraccarollo and Cristina Bosco for the ELISAs.
Conflict of interest
S. Adami has consultant/advisory role to Amgen and Merck. All other authors have stated that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gatti, D., Rossini, M., Viapiana, O. et al. Teriparatide Treatment in Adult Patients with Osteogenesis Imperfecta Type I. Calcif Tissue Int 93, 448–452 (2013). https://doi.org/10.1007/s00223-013-9770-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-013-9770-2