
Abstract
Human deciduous teeth have been proposed as a promising source of mesenchymal stem cells for application in bone and dental tissue engineering. We established cultures of mesenchymal stem cells from the pulp of human deciduous teeth (deciduous teeth stem cells, DTSCs) and analyzed their morphologic, growth, immunophenotypic, and osteo/odontogenic differentiation characteristics using different isolation methods and culturing environments. We compared the biologic behavior of DTSCs isolated either by enzymatic dissociation (DTSCs-ED) or by direct outgrowth from pulp tissue explants (DTSCs-OG). We found that different isolation methods give rise to different populations/lineages of cells with respect to their phenotypic and differentiation characteristics. DTSCs-ED cultures comprised heterogeneous cell populations, whereas DTSCs-OG comprised more homogenous spindle-shaped cells. We have characterized DTSCs as STRO-1+/CD146+/CD34+/CD45− cells. However, the percentage of STRO-1+ and CD34+ cells was higher in DTSCs-ED (STRO-1, 17.01 ± 5.04%; CD34, 19.79 ± 4.66%) compared to DTSCs-OG cultures (STRO-1, 5.18 ± 2.39%; CD34, 9.94 ± 3.41%), probably as a result of a higher release of stem/progenitor cells from the perivascular niche during enzymatic dissociation. DTSCs isolated using either method displayed an active potential for cellular migration and biomineralization, giving rise to 3D mineralized structures when challenged with dexamethasone, monopotassium phosphate, and β-glycerophosphate. These cellular aggregates progressively expressed differentiation markers of functional odontoblasts, including dentin sialophosphoprotein, bone sialoprotein, osteocalcin, and alkaline phosphatase, having the characteristics of osteodentin. However, in DTSCs-ED, the mineralization rate and the amount of mineralized matrix produced was higher compared to DTSCs-OG cultures. Therefore, DTSCs-ED cells display enhanced biomineralization potential, which might be of advantage for application in clinical therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Human exfoliated deciduous teeth have been proposed as a promising source of multipotent mesenchymal stem cells (MSCs) to be used in various clinical applications, including dental tissue engineering [1], repair of bone defects [2], and treatment of neural tissue injury and degenerative diseases [1, 3, 4]. Their clinical utility is attributed to their simple and convenient isolation, lack of ethical controversy, and negligible immunogenicity, thus permitting allogenic transplantation without the use of immunosuppressive agents [3, 5, 6]. Therefore, these cells are considered a valuable source for banking and long-term cryopreservation in several countries worldwide [7–9].
Deciduous teeth differ significantly from permanent teeth with respect to their developmental processes, tissue structure and function. Earlier studies have shown that stem cells derived from human exfoliated deciduous teeth (SHED) represent a more immature cell population compared to stem cells originating from the pulp of adult teeth (dental pulp stem cells, DPSCs). SHED are characterized by a higher proliferation rate, increased cell population doublings and high osteoinductive capacity in vivo compared to DPSCs [1, 10]. However, unlike DPSCs, SHED failed to reconstitute a complete dentin–pulp–like complex when transplanted in immunocompromised mice in vivo [1]. Such data suggest that SHED are distinctly different form adult DPSCs in terms of odontogenic differentiation and osteogenic induction.
Before the development of a protocol for dental tissue engineering using multipotent MSCs of dental origin, it is crucial to investigate certain aspects of cellular behavior under different conditions. Previous studies have shown that the isolation method and the culture conditions can give rise to different populations or lineages of pulp cells under in vitro passage [11]. Pulp cells have been isolated either by the outgrowth method, i.e., cells outgrow from pulp tissue explants [12–14], or by enzymatic digestion with collagenase/dispase [15, 16]. Although both methods seem to give rise to cultures containing odontoblast progenitor cells when used for the isolation of MSCs from adult teeth (DPSCs) [11], to our knowledge, there is no report analyzing the odontogenic potential of MSCs derived from deciduous teeth using these two methods.
Therefore, the aim of this study was the comparative analysis of the morphologic, growth, immunophenotypic, and in vitro osteo/odontogenic differentiation characteristics of MSCs derived from the dental pulp of human deciduous teeth (deciduous teeth stem cells, DTSCs), using two well-established isolation methods (outgrowth vs. enzymatic dissociation). The data presented in this study provide evidence toward understanding the underlying mechanisms of DTSC-induced biomineralization and reparative dentin formation, thus improving current dental tissue engineering protocols via stem cell-based approaches.
Materials and Methods
Cell Culture
The human DTSCs cultures established in this study were derived from the dental pulp of human extracted deciduous teeth of children aged 1–8 years (n = 10). Teeth were extracted for malposition in the dental arch or for orthodontic purposes. The collection of the samples was performed according to the guidelines of the institutional review board and the parents of all donors signed informed consent.
Cell cultures were established either by direct cell outgrowth from pulp tissue explants (OG method) [12–14] or by enzymatic dissociation (ED method) [15, 16]; the cell lines were designated DTSCs-OG and DTSCs-ED, respectively. Briefly, teeth were cleaned and cut around the cementum-enamel junction with sterilized dental fissure burs to reveal the pulp chamber. For the ED method, the pulp tissue was retrieved, minced and then digested in a solution of 3 mg/ml collagenase type I (Gibco/Invitrogen, Karlsruhe, Germany) and 4 mg/ml dispase (Roche Diagnostics, Mannheim, Germany) for 1 h at 37°C. Single-cell suspensions were obtained by passing the cells through a 70-μm strainer (BD Biosciences, Heidelberg, Germany). Cells were seeded at a density of 104/cm2 with α modification of Eagle medium (α-MEM, Gibco/Invitrogen), supplemented with 15% fetal bovine serum (FBS; Lonza, Verviers, Belgium), 100 μM l-ascorbic acid phosphate, 2 mM l-glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin (Biochrom AG, Berlin, Germany), and 0.25 mg/ml amphotericin B (Promo Cell, Heidelberg, Germany) and incubated at 37°C in 5% CO2.
For the OG method, pulp tissue was minced into 1–2-mm fragments, which were placed in 25-cm2 culture flasks with Dulbecco modified Eagle medium (DMEM) (Biochrom) supplemented with 10% FBS and antibiotics as above and incubated at 37°C in 10% CO2. The outgrown cells at confluence were transferred to a larger 75-cm2 flask (passage 1), and then continuously passed for further experiments. Cells from passages 2–4 were used for all experiments.
In addition, for cell growth analysis, both DTSCs-ED and DTSCs-OG cells were seeded at a density of 2 × 105 cells/well in six-well plates. The cell number was assessed every 24 h with a hemocytometer after collecting the cells of the corresponding wells with trypsinization (6 replicates for each time point of 24, 48, 72, and 96 h respectively), and the corresponding growth curves were calculated.
Flow Cytometric Surface Marker Expression Analysis
DTSCs-ED and DTSCs-OG cells at passage 2 were collected by treatment with 0.25% trypsin/0.25 mM EDTA (ethylenediaminetetraacetic acid; Biochrom AG). For each analysis, 106 cells per tube were first Fc-blocked with 1 μg of human IgG for 10 min at room temperature (RT) and subsequently stained by incubation with fluorescein isothiocyanate–conjugated mouse anti-human STRO-1 (Santa Cruz Biotechnology, Santa Cruz, CA), phycoerythrin-conjugated mouse anti-human CD146 (BD Biosciences), allophycocyanin-conjugated mouse anti-human CD34 (BD Biosciences), and phycoerythrin-conjugated mouse anti-human CD45 (BD Biosciences) at various combinations. After 20 min of incubation at ambient temperature in the dark, cells were washed with 2 ml fluorescence-activated cell sorter (FACS) wash solution (PBS without Ca2+ and Mg2+ (PBS(–)) + 1% bovine serum albumin + 0.1% NaN3) and centrifuged for 5 min at 230×g. Supernatant was removed; cells were resuspended in 200 μl FACS wash solution and analyzed with a BD LSR II Flow Cytometer (BD Biosciences). A total of 100,000 events were acquired for each sample. Dead cells were stained with DAPI (4′,6-diamidino-2-phenylindole) (Sigma-Aldrich, Taufkirchen, Germany) and excluded from further analysis. Data were analyzed by Summit 5.1 software (Beckman Coulter). All experiments were repeated at least three times.
Odontogenic/Osteogenic Differentiation In Vitro
For the induction of odontogenic/osteogenic differentiation, cells from DTSCs-ED and DTSCs-OG cultures were seeded in six-well plates at a density of 2 × 105/well. After 3–4 days, confluent monolayers were induced for osteo/odontogenic differentiation by exposure to α-MEM complete culture medium (CCM) supplemented with 0.01 μM dexamethasone disodium phosphate, 1.8 mM monopotassium phosphate (KH2PO4), and 5 mM β-glycerophosphate (all from Sigma-Aldrich). The cells were cultured for a total of 3 weeks; the differentiation medium was changed every 3–4 days. Cultures exposed to normal CCM without the additional supplements for the same period of time were used as negative control (uninduced control). At the end of each week, both induced and uninduced cultures were evaluated for mineralization by Alizarin Red S (AR-S) staining and processed for immunocytochemical analysis of alkaline phosphatase (ALP) activity.
AR-S Mineralization Assay
For the assessment of in vitro mineralization, cell cultures were washed twice (PBS(–)) (Gibco-Invitrogen, Carlsbad, CA) and fixed with 10% neutral buffered formalin (NBF) (Sigma-Aldrich) for 1 h at RT. Cultures were then stained with 1% AR-S (Sigma-Aldrich) (pH 4.2) for 20 min at RT, followed by rinsing three times with deionized water (dH2O) to reduce nonspecific staining. Mineralized nodules were visualized and photographed with an inverted microscope (Olympus Optical, Japan) equipped with a digital camera (Olympus E-410, Olympus Optical, Japan), and the amount of mineralized tissue per well was estimated by two independent observers. Subsequently, quantification of the total mineralized tissue produced per well was performed by extracting the AR-S from the stained sites by adding 2 ml of cetylpyridinium chloride buffer (10% w/v) in 10 mM Na2HPO4 (pH 7) for 2 h at 37°C. Subsequently, 200 μl aliquots were transferred to a 96-well plate, and the optical density (OD550nm) of the solution was measured with a microplate reader (Spectra Max 250, MWG Biotech). Mineralized tissue formation was represented as OD per microgram of total cellular protein, determined in parallel experiments by Bradford Protein assay [17]. Experiments were performed in triplicate wells and repeated at least three times.
Histochemical Detection of ALP Activity
Cells in six-well plates were washed twice with PBS(−) and fixed with 10% NBF, as described in the AR-S protocol. ALP activity was visualized by incubating the cells for 2 h at 37°C with 0.1 mg/ml naphthol-AS-MX phosphate in N,N-dimethylformamide and 0.6 mg/ml Fast Blue BB Salt in 0.2 M Tris (hydroxymethyl)-aminomethane buffer (pH 8.9) (all from Sigma-Aldrich). The cells were rinsed with dH2O and evaluated for ALP activity under an inverted microscope (Olympus Optical, Japan). Experiments were performed in triplicate wells and repeated at least three times.
Immunocytochemical Detection of Dentin Sialophosphoprotein Expression
Cell cultures were processed for immunocytochemical detection of dentin sialophosphoprotein (DSPP) expression 14 days after induction of osteo/odontogenic differentiation. Cells were washed with PBS(−) and fixed with 10% NBF for 30 min at RT. Cells were first incubated with 1.5% blocking serum in PBS to avoid nonspecific staining and then with mouse anti-human dentin sialoprotein (DSP) (LFMb-21) primary antibody (dilution 1:100, Santa Cruz) for 1 h at RT. Then cells were incubated with goat anti-mouse secondary antibody (dilution 1:200; Santa Cruz) for 1 h at RT and processed for enzymatic immunohistochemical staining with a broad-spectrum immunoperoxidase ABC kit (Santa Cruz) according to the manufacturer’s protocol. Finally, cells were counterstained with hematoxylin and examined under an inverted microscope.
Semiquantitative Reverse Transcription–Polymerase Chain Reaction Analysis
Total RNA was extracted from cells with NucleoSpin RNA II kit (Macherey-Nagel, Düren, Germany) at days 0, 9, and 15 after induction of differentiation and treated with RNase-free DNase I (Sigma) to remove contaminant DNA. Reverse transcription–polymerase chain reaction (RT-PCR) reactions were performed in a one-step procedure using 0.5 μg total RNA as starting template and the Robus T I RT-PCR kit (F-580L, Finnzymes). Briefly, RNA template was diluted in a 25-μl PCR reaction of 1 × PCR reaction buffer containing 1.5 mM MgCl2/200 mM each of dNTP/0.04 U/μl of DyNAzyme EXT DNA Polymerase)/0.1 U/μl of AMV reverse transcriptase (RT) and 10 pmol of each human-specific primer sets: bone sialoprotein (BSP) (sense: 5′-ATGGAGAGGACGCCACGCCT-3′, antisense: 5′-GGTGCCCTTGCCCTGCCTTC-3′), osteocalcin (OCN) (sense: 5′-GACTGTGACGAGTTGGCTGA-3′, antisense: 5′-AAGAGGAAAGAAGGGTGCCT-3′), DSPP (sense: 5′-GGG ACACAGGAAAAGCAGAA-3′, antisense: 5′-TGCTCCATTCCCACTAGGAC-3′ and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (sense: 5′-GAAGGTGAAGGTCGGAGT-3′, antisense: 5′-GAAGATGGTGATGGGATTTC-3′) as housekeeping gene. The primers were designed using Primer-Blast software from NCBI nucleotide sequence database (http://www.ncbi.nlm.nih.gov/BLAST). The reactions were performed in a PCR thermal cycler (Bio-Rad iCycler, Munich, Germany) at 50°C for 30 min for cDNA synthesis, 94°C for 2 min for one cycle, and then 94°C/(45 s), 56°C/(60 s), 72°C/(60 s) for 30 cycles, with a final 10-min extension at 72°C. Reactions without RT were used as negative controls in all cases. After amplification, 5 μl of each reaction was analyzed by 1.5% w/v agarose gel electrophoresis and visualized by ethidium bromide staining.
Statistical Analysis
All assays were performed on at least three independent experiments, and the results were expressed as mean ± SD. A paired t-test analysis was used to determine the differences in the amount of mineralized matrix produced by DTSCs-ED and DTSCs-OG cultures at various time points after induction of osteo/odontogenic differentiation (significance assumed for P < 0.05).
Results
Morphological and Growth Characteristics of DTSCs Cultures
Significant morphological differences in DTSCs isolated using different methods were observed. DTSCs-ED cultures were apparently heterogenous (Fig. 1a). Most cells were spindle shaped or fibroblast-like, but there were also some cuboidal or large polygonal cells. On the other hand, DTSCs-OG cultures were morphologically more homogeneous. Most cells exhibited a typical fibroblastic-like shape with long cytoplasmic processes and were arranged in an orderly fashion, with a tendency to align themselves in parallel lines (Fig. 1b). Despite the phenotypic differences observed, the growth rates of both types of DTSCs cultures were similar, as shown by the corresponding growth curves (Fig. 1c), and remained consistent during the next 10 passages (data not shown).

Morphologic characteristics of DTSCs cultures established from the dental pulp of deciduous teeth of children aged 1–8 years old, using either the ED or OG method. a In DTSCs-ED cultures, cells were polymorphic in shape, mostly fibroblast-like, whereas some appeared cuboidal or polygonal (scale bars = 25 μm). b In DTSCs-OG cultures, cells were more homogenous, with typical fibroblastic shape and long cytoplasmic processes, tending to align themselves in parallel lines. c Typical growth curves of DTSCs-ED and DTSCs-OG cells at passage 4
Immunophenotypic Characterization
Immunofluorescent analysis revealed significant differences in the surface epitope profiles of DTSCs isolated using different methods (Table 1, Fig. 2a, b). DTSCs-ED cultures showed a higher expression of STRO-1 (in 17.01 ± 5.04% of the cell population) and CD34 (19.79 ± 4.66%) compared to DTSCs-OG cultures (STRO-1, 5.18 ± 2.39%; CD34, 9.94 ± 3.41%). However, in both types of DTSCs cultures, most of the cell population expressed the perivascular marker CD146 (DTSCs-ED, 99.36 ± 0.76%; DTSCs-OG, 91.84 ± 4.30%). In contrast, DTSCs did not express the leukocyte precursor marker CD45 (Table 1, Fig. 2a, b), which suggests the stromal origin of these cells and the absence of hematopoietic precursor contamination.

Single-parameter histograms showing the expression of STRO-1, CD146, CD34, and CD45 in a DTSCs-ED and b DTSCs-OG cultures (red line isotype control, green line marker of interest). Both types of DTSCs cultures were positive for STRO-1, CD146, and CD34 but negative for CD45. A higher expression of STRO-1 and CD34 could be observed in DTSCs-ED cultures in most cases. Results from one representative experiment for each type of DTSCs are shown
Mineralized Tissue Formation by DTSCs During Osteogenic/odontogenic Differentiation
During the first 7–10 days after induction of osteo/odontogenic differentiation by selected medium containing dexamethasone as well as inorganic and organic phosphates, cells at confluence (Fig. 3a) started to migrate in an oriented manner and to aggregate, producing dense cellular 3D structures (Fig. 3b, c). In DTSCs-OG cultures, cells moved from the periphery toward the center of the well, giving rise to elongated 3D organized structures (Fig. 3b). In DTSCs-ED cultures, cellular movement led mainly to the formation of multiple cellular aggregates that grew up to build rounded 3D structures or colony-like clusters (Fig. 3c). Subsequently, these organized structures were the first to be mineralized, as shown by the calcium-specific AR-S staining (Fig. 3d–i).

(a–c) Representative phase-contrast microscopy photographs of DTSCs-OG (a, b) and DTSCs-ED (c) cultures 1 week after induction of differentiation. Cells in adherent monolayers (a) started migrating from the periphery toward the center of well and formed organized elongated 3D structures (b) or aggregates of high density (c). (a) Center of the plate; (b, c) periphery of plate. (d–f) AR-S staining of DTSCs-OG cultures at 1 week (d), 2 weeks (e), and 3 weeks (f) after induction of differentiation. The mineralization process initiated inside the organized structures but also with randomly distributed mineralized nodules all over the monolayer. At 3 weeks after induction, almost 50–60% of the monolayer was covered by mineralized matrix (f). In DTSCs-ED cultures at 1 week (g), 2 weeks (h), and 3 weeks (i) after induction, the mineralization process progressed at a higher rate, and at 3 weeks, mature mineralized tissue (brown color) was covering the entire monolayer (i). In uninduced DTSCs-OG (j, k) and DTSCs-ED (l) cultures, organized structures could be also observed, but the mineralization was restricted to a few spontaneous nodules (scale bars = 25 μm)
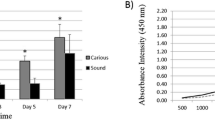
During the first week, multiple single mineralized nodules (250 ± 28/well) of small size could be observed in DTSCs-ED cultures throughout the whole adherent monolayer (Fig. 3g), whereas in DTSCs-OG cultures, a smaller number (13 ± 5/well) of larger mineralized nodules could be recorded (Fig. 3d). Subsequently, mineralization gradually increased, covering almost 100% of the adherent monolayer 3 weeks after induction of differentiation in DTSCs-ED cultures (Fig. 3i, Table 2), while the formation of mineralized matrix did not exceed 50–60% of the adherent monolayer in DTSCs-OG cultures at the same time (Fig. 3f, Table 2). These observations were also confirmed by spectrophotometric quantification of the mineralized tissue formation using the cetylpyridinium chloride extraction method (Fig. 4). In fact, the accumulated amount of mineralized matrix produced by DTSCs-ED cells was statistically significantly higher at 2 and 3 weeks after induction of differentiation compared to DTSCs-OG cells (P < 0.01).

AR-S quantification for evaluating mineralized tissue formation in DTSCs-ED and DTSCs-OG cultures induced for differentiation. Data are shown as mean OD per μg of total protein ± SD (n = 6). Asterisk indicates statistically significant differences in mineralized tissue deposition between DTSCs-ED and DTSCs-OG cultures at each time point (7, 14, and 21 days) (P < 0.05)
It is also important to note that cell migration and aggregation was also observed in DTSCs-uninduced cultures of both types treated with normal α-MEM medium (CCM) without any osteo/odontogenic supplements during the same 3-week period (Fig. 3j–l). However, mineralization in this case was limited, being restricted to some scattered mineralized nodules formed spontaneously (Table 2). This was due to the absence of the source of inorganic (KH2PO4) and organic (β-glycerophosphate) phosphates in the culture medium.
Expression of Differentiation Markers by DTSCs Cells During Osteogenic/Odontogenic Differentiation
DTSCs-ED- and DTSCs-OG-induced cultures progressively expressed several differentiation markers that are normally present during reparative dentinogenesis, including BSP, DSPP, and OCN, as shown by RT-PCR analysis (Fig. 5). Expression of these markers increased in a parallel way with the deposition of mineralized matrix from baseline levels (day 0) up to day 15 after induction of differentiation. There was also a relatively earlier expression (day 9) of these markers, especially of DSPP, in DTSCs-ED compared to DTSCs-OG cultures. In addition, immunocytochemical analysis for DSPP expression in DTSCs-induced cultures revealed a strong expression of this protein inside the organized 3D structures, as well as in cells forming such structures (Fig. 6a, b, d, e). DSPP-positive cells were lining the structures vertically and were finally entrapped within the newly formed reparative dentin (Fig. 6b, e). In DTSCs-OG cultures, an obvious elongation and polarization of the dentinogenic cells could be observed (Fig. 6b), whereas in DTSCs-ED cultures, there was a more random distribution of DSPP-positive cells, which in their majority retained their normal spindle-shaped phenotype (Fig. 6e). On the other hand, in uninduced DTSCs-ED (Fig. 6f) and DTSCs-OG (Fig. 6c) cultures, the expression of DSPP remained minimal, even after 2 weeks in culture.

Representative agarose gels containing RT-PCR products from (a) DTSCs-ED and (b) DTSCs-OG cell cultures at 0, 9, and 15 days after induction of differentiation. Baseline expression of these genes gradually increased with prolonged exposure to differentiation culture conditions. DSPP showed a relatively earlier expression in DTSCs-ED compared to DTSCs-OG cultures. BSP, bone sialoprotein (product: 322 bp); OCN, osteocalcin (product: 137 bp); DSPP, dentin sialophosphoprotein (product: 422 bp); GAPDH, glyceraldehyde-3-phosphate dehydrogenase (product: 226 bp)

Immunohistochemical staining for dentin sialophosphoprotein (DSPP). Pronounced expression of DSPP was observed in DTSCs-OG (a, b) and DTSCs-ED (d, e) cultures 14 days after induction of osteo/odontogenic differentiation. DSPP was mainly expressed in organized structures and in cells forming these structures (b, e). Uninduced DTSCs-OG (c) and DTSCs-ED (f) cultures displayed a very low expression for DSPP (scale bars = 25 μm)
DTSCs-induced cultures also showed a strong ALP activity present in 75–100% of the total cell population. ALP is a widely used marker, mainly expressed in differentiated cells producing mineralized matrix [18]. This enzymatic activity was observed as early as 1 week after induction of differentiation in DTSCs cultures obtained by both methods (Fig. 7a–h) and remained at high levels after 3 weeks, although the extensive mineral depositions at that time technically prevented to some extent the penetration of the ALP substrate (Fig. 7d, h). On the contrary, the ALP activity was very low (<25%) in DTSCs uninduced (control) cultures (Fig. 7a, e).

Histochemical staining showing ALP activity in DTSCs-ED (a–d) and DTSCs-OG (e–h) cultures. ALP was strongly expressed (75–100% of the cell population) in both types of cultures 1 week (b, f) after induction of osteo/odontogenic differentiation. ALP expression remained stable during the following 3 weeks, although at week 3, the extensive mineral depositions technically prevented, to some extent, the penetration of the ALP substrate (d, h). Uninduced DTSCs-ED (a) and DTSCs-OG (e) cell cultures displayed a much lower (<25%) ALP activity (scale bars = 25 μm)
Discussion
This study aimed to analyze the biologic characteristics of multipotent MSCs derived from the pulp of human deciduous teeth (DTSCs) and to elucidate the patterns of their differentiation toward osteo/odontogenic lineages under different isolation conditions and culturing environments. Two well-established isolation methods were applied: the OG method [13] and the ED method [15]. The first has been widely used by many investigators to study various aspects of pulp biology [12, 14, 19–22]. These studies have shown that primary cell cultures of human pulp cells established via the OG method can be induced to differentiate in vitro into cells of odontoblastic phenotype, characterized by polarized cell bodies and accumulation of mineralized nodules [12, 14]. In most recent studies, on the other hand, the ED method has been proposed as the method of choice for the isolation of dental tissue–derived MSCs of various origins [1, 15, 23, 24]. This method has been found to give rise to pulp cells able to differentiate into osteo/odontoblastic lineages in vitro and to produce dentin–pulp complexes in vivo [15].
In a study by Huang et al. [11], both methods were comparatively used for the isolation of DPSCs from adult teeth (third molars) and were found to give rise to populations or lineages of cells with different proliferation rates (higher for the ED method) and gene expression patterns. We add to this knowledge by showing in this study that MSCs derived from the pulp of deciduous teeth (DTSCs) using these two methods represent different cell populations with respect to their morphologic, immunophenotypic, and osteo/odontogenic differentiation characteristics.
From a morphological point of view, we have found that DTSCs-ED cultures display a significant heterogeneity comprising different cell sizes and morphologies even within the same colonies (Fig. 1a). This finding was not surprising because the ED method allows different types of cells to be released during tissue dissociation [15, 25]. Fibroblast-like cells, as well as immature stem/progenitor cells, endothelial cells, and pericytes released from the perivascular niche [26], are those that attach on the culture disc, whereas blood cells and debris are discarded when the medium is changed. The OG method, on the other hand, allows for a more uniform migration of fibroblast-like cells (Fig. 1b), whereas nonmigrating cells disintegrate within the tissue explants [11]. Therefore, it is highly possible that only a part of the whole population of pulp cells capable of attaching and dividing is represented in the DTSCs-OG cultures. Despite this observation, no difference could be detected in the growth rates of these two types of cultures, as shown by the corresponding growth curves (Fig. 1c).
The existence of different cell populations also provides a possible explanation for the differences observed in the immunophenotypic profiles of these two types of cultures (Table 1, Fig. 2a, b). The higher expression of STRO-1 and CD34 observed in DTSCs-ED compared to DTSCs-OG cultures most probably reflects the different numbers of stem/progenitor cells released by these two methods. In a study by Miura et al. [1], almost 9% of the total pulp cell population derived from exfoliated deciduous teeth isolated via the ED method was reported to be positive for STRO-1. The significant diversity among different studies in surface receptor expression can be attributed to several factors, including passage number, state of confluence, composition of the media and incubation environments, and the experimental procedures used [27, 28]. To these factors we should also add the interindividual variations among different donors and the characteristics of teeth used (e.g., stage of root resorption and exfoliated vs. extracted deciduous teeth).
It must be also emphasized that in this study, most of the cell population in both types of DTSCs-established cultures was found to express the perivascular marker CD146 (ED method, 99.36 ± 0.76%; OG method, 91.84 ± 4.30%) (Table 1, Fig. 2a, b), which confirms the perivascular origin of these cells [29, 30]. Shi and Gronthos [29] demonstrated that the expression of CD146 in the dental pulp of adult teeth is restricted to blood vessel walls but is absent in the surrounding fibrous tissue, odontoblast layer, and perineurium of the nerve, indicating that stem cells are localized in the perivascular region of the pulp tissue. These authors found that STRO-1+/CD146+ cells also expressed the pericyte marker 3G5 and for this reason they assumed that they are pericytes. The high degree of expression of CD146 found in our study for DTSCs is in accordance with this hypothesis.
Another interesting finding in our study was the expression of CD34 antigen in DTSCs cultures established by both methods (Table 1, Fig. 2a, b). CD34 is an epitope commonly expressed on human hematopoietic stem cells but also some primitive stromal stem cells from bone marrow [31]. This finding is not in agreement with previous studies [1, 25, 32] and also contrasts with a general definition of MSCs recently proposed by the Mesenchymal and Tissue Stem Cell Committee of the International Society for Cellular Therapy, which includes cells negative for CD34, CD45, CD14, CD11a, etc. [27, 33]. In our study, however, CD34+ cells lacked expression of the hematopoietic and inflammatory cell marker CD45 [34, 35], which suggests a stromal origin of DTSCs cells and an absence of hematopoietic precursor contamination. This finding is in agreement with a previous study by Laino et al. [36], who identified a c-kit (CD117)+/STRO-1+/CD34+/CD45− population inside the pulp of adult teeth. The controversy concerning CD34 expression among studies can be attributed to the different experimental conditions, mainly the high serum concentration usually used in most MSCs culture protocols, which has been found responsible for the rapid loss of the CD34 surface epitope during expansion culture [34].
The present study also showed that DTSCs developed by both methods displayed an active migratory and contractile potential, producing dense cellular 3D formulations 7–10 days after reaching confluence, irrespective of exposure to differentiation-inductive conditions (i.e., both in induced and uninduced cultures) (Fig. 3). In DTSCs-OG cultures, cell migration occurred mainly from the periphery toward the center of the culture plate, leading to characteristic elongated 3D organized structures (Fig. 3b), whereas cells in DTSCs-ED cultures aggregated more randomly, producing rounded 3D structures or colony-like clusters (Fig. 3c). These observations suggest that this phenomenon of cellular migration and organization is an inherent phenotypic characteristic of dental MSCs populations during expansion culture that occurs after prolonged confluence. It is likely to be triggered by the high adhesion forces developed among cells after prolonged culture that make the monolayer contractile, behaving more like a tissue. Active migration was also reported for other premature cell populations of mesenchymal origin in previous in vitro studies [37]. However, further investigation is needed to elucidate the molecular signals that orchestrate the different patterns of cellular migration and organization observed in different types of dental MSCs cultures.
In addition, in cultures exposed to mineralization-inductive conditions, by exposure to media containing dexamethasone, KH2PO4, and β-glycerophosphate, these cellular aggregates became the centers where an extensive matrix mineralization occurred (Fig. 3d–i). All of these supplements have been reported to play a significant role in the enhancement of extracellular mineralized matrix formation [38–40]. The mineralization rate was lower in DTSCs-OG (covering 50–60% of the monolayer at 3 weeks) compared to the DTSCs-ED cultures (100% at 3 weeks) (Figs. 3, 4; Table 2). This finding could be mainly attributed to the different populations of stem\progenitor cells released by these two methods, as already mentioned. In addition, another contributing factor could be the lower FBS concentration in the medium commonly used for the expansion of cells using the OG method (DMEM + 10% FBS) compared to the ED method (α-MEM + 15% FBS). In this regard, it is supported that FBS plays an essential role in MSCs proliferation and differentiation [31, 41]. Further investigation using identical media and serum concentrations could clarify the possible role of FBS concentration.
Mineralization of induced DTSCs cultures was accompanied by an increasing expression of both osteogenic and odontogenic differentiation markers, including ALP, BSP, OCN, and DSPP (Figs. 5–7). Their expression increased in an analogous way with cellular progression along the odontoblastic lineage (Fig. 5). Moreover, DTSCs-ED displayed a relatively earlier expression of these markers, especially DSPP, compared to DTSCs-OG cells, which is in accordance with the earlier and more extensive mineralized tissue formation in DTSCs-ED cultures. BSP, OCN, and DSPP are glycoproteins that share common expression in bones and teeth [42–44]. DSPP has also been detected in bone, but at a level of 1/400 compared to that of dentin [42]. Even though there are no specific odontoblastic markers, BSP, DSPP, ALP, osteocalcin, and osteonectin have been mainly used as indicators of odontoblastic differentiation [15, 45, 46]. In addition, the expression of DSPP in functional odontoblasts in early stages of odontogenesis is consistent with the hypothesis that it plays an active role in the mineralization of dentin matrix [47] and can therefore be considered a rather representative marker of odontoblastic differentiation. In our study, DSPP was strongly expressed inside the organized structures as well as in migrating cells forming these structures, which confirms their odontoblastic phenotype (Fig. 6). This finding is also in accordance with in vivo data, suggesting that dental pulp cells adjacent to reparative dentin stain more intensely for DSP and dentin phosphoroprotein (DPP) compared to undifferentiated cells within the dental pulp tissue, indicating an up-regulation of these proteins during reparative dentinogenesis [48]. The elongated and polarized phenotype of DTSCs-OG cells is more consistent with that of functional odontoblasts producing reparative dentin with tubular structure, whereas the fibroblast-like shape of DTSCs-ED cells is mostly linked to a rather nonspecific response leading to the production of an atubular osteotypic matrix or osteodentin [49]. In our study, the mineralized matrix produced by both types of cells seemed to lack any specific tubular structure. However, previous work has demonstrated that the matrix produced in vitro by pulp cultures established by the OG method lacked any tubuli, but its organic and mineral composition was similar to human dentin. This was confirmed by the analogy between the Fourier-transformed infrared spectroscopy–mass spectrometry spectra obtained in vitro and those of dentin in vivo [13]. On the other hand, no information exists concerning the characteristics of mineralized matrix produced by dental MSCs cultures established by ED. Therefore, further characterization of this matrix with respect to its ultrastructure and crystallinity would provide more information with respect to the nature of the tissue produced, which would be of significance for future tissue engineering applications.
It is clear from the data presented in this study that the two isolation methods indeed yield different populations of cells. The ED method seems to release a higher amount of stem/progenitor cells, which migrate more randomly after induction of odontogenic differentiation, producing quickly mineralized tissue. Despite the higher mineralization rate and the positivity of the matrix produced for DSPP, the DTSCs-ED cells retained their spindle-shaped morphology without showing any specific elongation and polarization. The OG method, on the other hand, gives rise to rather homogenous cultures of fibroblast-like cells with a lower initial number of stem/progenitor cells and a lower mineralization rate after induction. However, these cells seem to acquire the phenotypic characteristics of functional odontoblasts, presenting a typical cell elongation and polarization of the cell bodies vertical to the dentin-like matrix produced. The isolation of pulp cells with a potential to differentiate into functional odontoblasts is a critical step toward pulp tissue regeneration and new dentin formation. Therefore, even though the ED method seems to give rise to pulp cells of higher mineralization potential, additional investigation is required to demonstrate which of these two methods is superior for the purpose of functional dentin/pulp tissue regeneration.
In conclusion, the findings presented in this study support that different isolation methods and/or culturing environments give rise to population or lineages of DTSCs with different morphologic, immunophenotypic, and osteo/odontogenic differentiation characteristics. Therefore, this study sets the basis for further multiparameter exploration to identify other differences in pulp cells isolated by different methods and under various culture conditions. These include changes occurring during passage in lineage commitment/differentiation, epigenetic changes during expansion culture, and identification of the subpopulations of cells that are most capable of differentiation toward functional odontoblasts producing reparative dentin. The different cell origins and cell populations (e.g., mesenchymal, endothelial, pericytes) released by different methods can be a problem or limitation but also a further challenge for study. Despite the observed differences, the high potential of DTSCs to differentiate into mineralizing cells under the conditions presented in this study highlights the potential application of such an approach for future dental tissue or bone engineering strategies.
References
Miura M, Gronthos S, Zhao M, Lu B, Fisher LW, Robey PG, Shi S (2003) SHED: stem cells from human exfoliated deciduous teeth. Proc Natl Acad Sci USA 100:5807–5812
Zheng Y, Liu Y, Zhang CM, Zhang HY, Li WH, Shi S, Le AD, Wang SL (2009) Stem cells from deciduous tooth repair mandibular defect in swine. J Dent Res 88:249–254
Huang GTJ, Gronthos S, Shi S (2009) Mesenchymal stem cells derived from dental tissues vs those from other sources: their biology and role in regenerative medicine. J Dent Res 88:792–806
Morsczeck C, Völlner F, Saugspier M, Brandl C, Reichert TE, Driemel O, Schmalz G (2010) Comparison of human dental follicle cells (DFCs) and stem cells from human exfoliated deciduous teeth (SHED) after neural differentiation in vitro. Clin Oral Investig 14:433–440
Pierdomenico L, Bonsi L, Calvitti M, Rondelli D, Arpinati M, Chirumbolo G, Becchetti E, Marchionni C, Alviano F, Fossati V, Staffolani N, Franchina M, Grossi A, Bagnara GP (2005) Multipotent mesenchymal stem cells with immunosuppressive activity can be easily isolated from dental pulp. Transplantation 80:836–842
Noel D, Djouad F, Bouffi C, Mrugala D, Jorgensen C (2007) Multipotent mesenchymal stem cells and immune tolerance. Leuk Lymphoma 48:1283–1289
Arora V, Arora P, Munshi AK (2009) Banking stem cells from human exfoliated deciduous teeth (SHED): saving for the future. J Clin Pediatr Dent 33:289–294
Petrovic V, Stefanovic V (2009) Dental tissue—new source for stem cells. Sci World J 14:1167–1177
Sloan AJ, Waddington RJ (2009) Dental pulp stem cells: what, where, how? Int J Paediatr Dent 19:61–70
Shi S, Bartold PM, Miura M, Seo BM, Robey PG, Gronthos S (2005) The efficacy of mesenchymal stem cells to regenerate and repair dental structures. Orthod Craniofac Res 8:191–199
Huang GT, Sonoyama W, Chen J, Park SH (2006) In vitro characterization of human dental pulp cells: various isolation methods and culturing environments. Cell Tissue Res 324:225–236
Tsukamoto Y, Fukutani S, Shin-Ike T, Kubota T, Sato S, Suzuki Y, Mori M (1992) Mineralized nodule formation by cultures of human dental pulp-derived fibroblasts. Arch Oral Biol 37:1045–1055
About I, Bottero MJ, Denato P, de Camps J, Franquin JC, Mitsiadis TA (2000) Human dentin production in vitro. Exp Cell Res 258:33–41
Couble ML, Farges JC, Bleicher F, Perrat-Mabillon B, Boudeulle M, Magloire H (2000) Odontoblast differentiation of human dental pulp cells in explants cultures. Calcif Tissue Int 66:129–138
Gronthos S, Mankani M, Brahim J, Robey PG, Shi S (2000) Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci USA 97:13625–13630
Onishi T, Kinoshita S, Shintani S, Sobue S, Ooshima T (1999) Stimulation of proliferation and differentiation of dog dental pulp cells in serum-free culture medium by insulin-like growth factor. Arch Oral Biol 44:361–371
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Yang X, Walboomers XF, van den Beucken JJ, Bian Z, Fan M, Jansen JA (2009) Hard tissue formation of STRO-1 selected rat dental pulp stem cells in vivo. Tissue Eng Part A 15:367–375
Park SH, Hsiao GY, Huang GT (2004) Role of substance P and calcitonin gene–related peptide in the regulation of inteleukin-8 and monocytes chemotactic protein-1 expression in human dental pulp. Int Endod J 37:185–192
Nakashima M, Nagasawa H, Yamada Y, Reddi AH (1994) Regulatory role of transforming growth factor-beta, bone morphogenetic protein-2, and protein-4 on gene expression of extracellular matrix proteins and differentiation of dental pulp cells. Dev Biol 162:18–28
Nakao K, Itoh M, Tomita Y, Tomooka Y, Tsuji T (2004) FGF-2 potently induces both proliferation and DSP expression in collagen type I gel cultures of adult incisor immature pulp cells. Biochem Biophys Res Commun 325:1052–1059
About I, Mitsiadis TA (2001) Molecular aspects of tooth pathogenesis and repair: in vivo and in vitro models. Adv Dent Res 15:59–62
Seo BM, Miura M, Gronthos S, Bartold PM, Batouli S, Brahim J, Young M, Robey PG, Wang CY, Shi S (2004) Investigation of multipotent postnatal stem cells from human periodontal ligament. Lancet 364:149–155
Sonoyama W, Liu Y, Fang D, Yamaza T, Seo BM, Zhang C, Liu H, Gronthos S, Wang CY, Shi S, Wang S (2006) Mesenchymal stem cell-mediated functional tooth regeneration in Swine. PLoS One 20:1:e79
Gronthos S, Brahim J, Li W, Fisher LW, Cherman N, Boyde A, DenBesten P, Robey PG, Shi S (2002) Stem cell properties of human dental pulp stem cells. J Dent Res 81:531–535
Ellerström C, Hyllner J, Strehl R (2010) Single cell enzymatic dissociation of human embryonic stem cells: a straightforward, robust, and standardized culture method. Methods Mol Biol 584:121–134
Karp LM, Leng Teo GS (2009) Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell 4:206–216
Javazon EH, Begs KJ, Flake AW (2004) Mesenchymal stem cells: paradoxes of passaging. Exp Hematol 32:414–425
Shi S, Gronthos S (2003) Perivascular niche of postnatal mesenchymal stem cells in human bone marrow and dental pulp. J Bone Miner Res 18:696–704
Spath L, Rotilio V, Alessandrini M, Gambara G, De Angelis L, Mancini M, Mitsiadis TA, Vivarelli E, Naro F, Filippini A, Papaccio G (2010) Explant-derived human dental pulp stem cells enhance differentiation and proliferation potentials. J Cell Mol Med 14(6B):1635–1644
García-Pacheco JM, Oliver C, Kimatrai M, Blanco FJ, Olivares EG (2001) Human decidual stromal cells express CD34 and STRO-1 and are related to bone marrow stromal precursors. Mol Hum Reprod 7:1151–1157
Koyama N, Okubo Y, Nakao K, Bessho K (2009) Evaluation of pluripotency in human dental pulp cells. J Oral Maxillofac Surg 67:501–506
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317
Simmons PJ, Torok-Storb B (1991) CD34 expression by stromal precursors in normal human adult bone marrow. Blood 78:2848–2853
Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ, Harris S, Wiedemann LM, Mishina Y, Li L (2003) Identification of the haematopoietic stem cell niche and control of the stem cell size. Nature 425:836–840
Laino G, Graziano A, D’ Acquino R, Pirozzi G, Lanza V, Valiante S, De Rosa A, Naro F, Vivarelli E, Papaccio G (2006) An approachable human adult stem cell source for hard-tissue engineering. J Cell Physiol 206:693–701
Sandulache VC, Perekh A, Dohar JE, Hebda PA (2007) Fetal dermal fibroblasts retain a hyperactive migratory and contractile phenotype under 2- and 3-demensional constraints compared to normal adult fibroblasts. Tissue Eng 13:2791–2801
Bellows CG, Aubin JE, Heersche JN (1987) Physiological concentrations of glucocorticoids stimulate formation of bone nodules from isolated rat calvaria cells in vitro. Endocrinology 121:1985–1992
Bellows CG, Aubin JE, Heersche JNM (1991) Initiation and progression of mineralization of bone nodules formed in vitro: the role of alkaline phosphatase and organic phosphate. Bone Miner 14:27–40
Tanaka H, Murphy CL, Murphy C, Kimura M, Kawai S, Polak JM (2004) Chondrogenic differentiation of murine embryonic stem cells: effects of culture conditions and dexamethasone. J Cell Biochem 93:454–462
Brink HE, Satlling SS, Nicoll SB (2005) Influence of serum on adult and fetal dermal fibroblast migration adhesion and collagen expression. In Vitro Cell Dev Biol Animal 41:252–257
Qin C, Brunn JC, Cadena E, Ridall A, Tsujigiwa H, Nagatsuka H, Nagai N, Butler WT (2002) The expression of dentin sialophosphoprotein gene in bone. J Dent Res 81:392–394
Fisher LW, Fedarko NS (2003) Six genes expressed in bones and teeth encode the current members of the SIBLING family of proteins. Connect Tissue Res 44(suppl 1):33–40
Viereck V, Siggelkow H, Tauber S, Raddatz D, Schutze N, Hüfner M (2002) Differential regulation of cfba1/Runx2 and osteocalcin gene expression by vitamin D3, dexamethasone and local growth factors in primary human osteoblasts. J Cell Biochem 86:348–356
Wei X, Ling J, Wu L, Liu L, Xiao Y (2007) Expression of mineralization markers in dental pulp cells. J Endod 33:703–708
Liu H, Gronthos S, Shi S (2006) Dental pulp stem cells. Methods Enzymol 419:99–113
D’ Souza RN, Cavender A, Sunavala G, Alvarez J, Ohshima T, Kulkarni AB, MacDougall M (1997) Gene expression pattern of murine dentin matrix protein 1 (Dmp 1) and dentin sialophosphoprotein (DSPP) suggest distinct developmental functions in vivo. J Bone Miner Res 12:2040–2049
Lee L, Liu J, Clarkson BH, Lin CP, Godovikova V, Ritchie HH (2006) Dentin–pulp complex responses to carious lesions. Caries Res 40:256–264
Tziafas D, Smith AJ, Lesot H (2000) Designing new treatment strategies in vital pulp therapy. J Dent 28:77–92
Acknowledgment
This study was supported by a grant from DAAD (German Academic Exchange Service).
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors have stated that they have no conflict of interest.
Rights and permissions
About this article
Cite this article
Bakopoulou, A., Leyhausen, G., Volk, J. et al. Assessment of the Impact of Two Different Isolation Methods on the Osteo/Odontogenic Differentiation Potential of Human Dental Stem Cells Derived from Deciduous Teeth. Calcif Tissue Int 88, 130–141 (2011). https://doi.org/10.1007/s00223-010-9438-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-010-9438-0