
Abstract
The effects of low-magnitude, high-frequency (LMHF) mechanical stimulation on osteoblastic cells are poorly understood. We have developed a system that generates very small (15–40 με), high-frequency (400 Hz, sine) deformations on osteoblast cultures (MC3T3-E1). We investigated the effects of these LMHF stimulations mainly on extracellular matrix (ECM) synthesis. The functional properties of this ECM after decellularization were evaluated on C3H10T1/2 mesenchymal stem cells (MSCs). LMHF stimulations were applied 20 min once daily for 1, 3, or 7 days in MC3T3-E1 culture (1, 3, or 7 dLMHF). Cell number and viability were not affected after 3 or 7 dLMHF. Osteoblast response to LMHF was assessed by an increase in nitric oxide secretion, alteration of the cytoskeleton, and focal contacts. mRNA expression for fibronectin, osteopontin, bone sialoprotein, and type I collagen in LMHF cultures were 1.8-, 1.6-, 1.5-, and 1.7-fold higher than controls, respectively (P < 0.05). In terms of protein, osteopontin levels were increased after 3 dLMHF and ECM organization was altered as shown by fibronectin topology after 7 dLMHF. After decellularization, 7 dLMHF-ECM or control ECM was reseeded with MSCs. Seven dLMHF-ECM improved early events such as cell attachment (2 h) and focal contact adhesion (6 h) and, later (16 h), modified MSC morphological parameters. After 5 days in multipotential medium, gene-expression changes indicated that 7 dLMHF-ECM promoted the expression of osteoblast markers at the expense of adipogenic marker. LMHF stimulations of osteoblasts are therefore efficient and sufficient to generate osteogenic matrix.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Cyclic mechanical loading is able to increase bone formation by stimulating osteogenesis [1]. Several mechanical parameters, such as strain rate, number of cycles, and resting periods, have been shown to influence bone cell response to loading [2]. Strain rate can be further subdivided into strain magnitude and loading frequency. Fritton et al. [3] showed that, during normal activities, low-magnitude (<100 με) bone deformations occur more often than higher strains (>1,000 με), suggesting that low-magnitude mechanical stimuli could be used to promote adaptive bone formation. Recent in vivo studies have focused on low-magnitude mechanical signals because of their safety profile. Some teams [4, 5] working on animals have demonstrated the osteogenic effects of short-duration, low-magnitude mechanical stimuli applied at high frequencies The loss of strain amplitude appears to be compensated for by application of higher-frequency loading [6]. The results of recent clinical studies have shown the value of these mechanical regimens for frail patients unable to perform regular physical exercise [7, 8]. However, exploration of low-magnitude high-frequency (LMHF) mechanical signals proved to be more difficult than expected. Although a few teams have regularly published positive results, other recent studies have not always demonstrated benefits at all bone sites [9]. Moreover, conditions vary so widely between studies (frequency ranges, acceleration, and exposure time) that it is difficult to derive a general and consistent pattern. In vitro data are missing, which could highlight the role of LMHF loading on matrix synthesis. These studies have only focused on changes of secondary messengers [10, 11].
The extracellular matrix (ECM) plays an important role in maintaining and mediating bone function. It is also a source of factors supporting bone cell differentiation as several studies suggest that the 3D matrix structure and organization can influence the phenotypic behavior of cells [12–14]. ECM is therefore considered to be an essential interface to functionalize cellularized bone substitute, providing osteoinductive properties. From this perspective, it appeared of interest to examine the osteogenic potential of very LMHF mechanical stimulation on cellular response in terms of quantitative and qualitative modifications of ECM.
To explore the osteoblast response to mechanical signals, the first part of the present investigation consisted of developing an original device able to generate controlled very LMHF stimulations of cell cultures. This study investigates whether osteoblasts would consistently respond to the applied strain and was designed to determine to what extent ECM production and organization can be modified by these LMHF stimulations. Osteoblast responsiveness (MC3T3-E1) to LMHF was tested by examining typical mechanotransducers (nitric oxide, cytoskeleton, and focal contacts). ECM was also analyzed at the mRNA and protein levels, and image analysis was performed to examine ECM organization.
The final objective of this study was to assess the ECM properties produced by these LMHF osteoblasts (MC3T3-E1). For this purpose, decellularized matrices were used as substrate for mesenchymal stem cells (MSCs) capable of osteodifferentiation (C3H10-T1/2). Cell behavior was evaluated in terms of MSC–matrix interactions and MSC commitment. Short-term cell adhesion and subsequent cell differentiation were analyzed by determining typical markers of osteoblastic and adipogenic differentiation, such as alkaline phosphatase, Runx2, type I collagen, fibronectin (FN), and PPARγ2.
Materials and Methods
Cell Culture
Mouse osteoblastic MC3T3-E1 cells, subclone 4 (MC-4), were maintained in T75 flasks, in alpha modified Eagle medium (αMEM) supplemented with 10% FBS, 2 mM l-glutamine, and antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin) in a humidified atmosphere of 5% CO2 at 37°C. The mouse pluripotent MSC line C3H10T1/2 (clone-8, American Type Culture Collection; LGC Promochem, Molsheim, France) was maintained in complete αMEM supplemented with 10% FBS and 2 mM l-glutamine, and antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin) in a humidified atmosphere of 5% CO2 at 37°C and passaged before confluence. In these experiments, cells were used between passages 10 and 15.
LMHF Stimulation
For mechanical stimulation experiments, MC3T3-E1 cells were trypsinized with 1× trypsin-EDTA and plated at 8,000 cells/cm2 in 100-mm Petri dishes (Falcon, Lincoln Park, NJ), with 8 ml of culture medium supplemented with 50 μg/ml ascorbate phosphate (Sigma-Aldrich, St. Louis, MO).
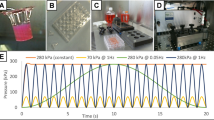
Starting 24 h after seeding, MC3T3-E1 cells were subjected to LMHF loading induced by a buzzer (FARNELL 1193664 Ø = 50 mm) fixed under the Petri dish with a thin (12 μm) doubled-sided adhesive tape. Petri dishes were supported by three soft plots to avoid interfering with buzzer movements. The sinusoidal input signal was controlled by a generator and amplified by a tension amplifier (Fig. 1a). Signal frequency was chosen to match the resonance frequency of the Petri dish. For determination of resonance frequency, the experimental dish was excited by a computer-controlled gain phase analyzer (HP 4194A; Hewlett-Packard, Palo Alto, CA). This gain phase analyzer applied a frequency sweep from 0 to 1,000 Hz and simultaneously recorded the gain. The excitation signals from the gain phase analyzer were amplified by a power amplifier (2100RF; ENI, Rochester NY) in order to maximize the excitation absolute value. Vibrations of the Langevin transducer were measured with a laser-vibrometer (Polytec, Irvine, CA). The reflect signals were processed by the electronic unit (OFV302) and fed to the test channel of the gain phase analyzer. The power amplifier output was also fed to the reference channel of the gain phase analyzer to measure gain. The maximum displacement (μm) of the center of the Petri dish corresponded to the resonance frequency (400 Hz) (Fig. 1b).

Production and characterization of micromechanical stimulation. a Diagram of the mechanical stimulator with four Petri dishes in parallel. A generator connected to an amplifier produces a sinusoidal tension (±30 V) for each buzzer fixed under the Petri dish. b Determination of Petri dish resonance frequency. Petri dishes were filled with 8 ml of culture medium. Displacement of the center of the Petri dish was measured for frequencies ranging 0–1,000 Hz. The two curves represent measurements on two different Petri dishes. c Strain gauge repartition: Three gauges were positioned along the radius inside the strained surface: at the center, at a point midway between the center and the end support, and at the edge of the Petri dish. Surface deformation: The strained surface was represented at t1 and t2, showing alternatively compression and tension. Strain gauge measurements: The graph represents the measurements of the three strain gauges during 0.01 second at resonance frequency. Strain modeling: This represents the tangential strain along the radius (r) at fixed time (t1). Mechanical interfaces are represented by lines a and b, which correspond, respectively, to the piezoelectric material and total surface of the buzzer. r0 indicates the strain nodal circle. Area 1 corresponds to strains from 15 to 25 με and area 2 corresponds to strains from 25 to 40 με. (Color figure online)
To quantitatively verify the load-induced strain fields (με), strain gauges (Kyowa, Tokyo, Japan) were glued (Mbond 200 adhesive; Vishay Micro-Measurements, Shelton, CT) inside the Petri dish. Three strain gauges were placed on the cultured surface along the radius: at the center, at a point midway between the center and the end support, and at the edge of the Petri dish culture (Fig. 1c). These gauges were aligned tangentially to the radius in order to measure the Petri dish tangential deformation expressed as microstrain (με). The strains on the Petri dish surface were precisely (±1 με) recorded with a dynamic datalogger (Resist4000; Microlight, www.milight.com), which measured signals from strain gauges with three simultaneous channels at a maximum rate of 4,500 measurements/second/channel. The datalogger is equipped with a serial link for transfer of data to a personal computer (PC) entirely compatible with Excel software (Microsoft, Redmond, WA). Osteoblast cultures were stimulated at 400 Hz with a sinusoidal signal, and the recorded strains were situated between ±15 and ±40 με, as shown in Fig. 1c.
The tangential LMHF strain (με) on the inner surface of the polystyrene substrates was qualitatively characterized by modeling (Fig. 1c). Modeling was performed by the Element Finite technique using ANSYS commercial software (ANSYS, Canonsburg, PA). The behavior of the functional mode is shown in Fig. 1c and is identified as the fundamental bending mode of the dish. According to the composite plate theory, the radial component of the tangential strain direction exhibited variations at each mechanical interface (a and b). Moreover, the presence of a strain nodal circle (r0) indicated that the strain field was divided into two areas of opposite sign (i.e., an extensive area for r < r0 and a compressive area for r > r0).
LMHF stimulation was applied 20 min once daily for 1, 3, or 7 days on osteoblast cultures (1, 3, or 7 dLMHF). During mechanical stimulation, cultures were maintained in a humidified atmosphere of 95% air and 5% CO2 at 37°C.
Cell Growth and Viability
Cell density was quantified after DAPI staining (Chemicon, Temecula, CA) at a working concentration of 1 μg/mL by counting the nuclei at days 0, 1, 2, 3, and 7. Staining was examined with a Leitz (Wetzlar, Germany) DMRB fluorescence microscope. Images were acquired with a Roper Scientific (Trenton, NJ) CoolSnapfx camera using Meta Imaging series 4.6.6 software (Universal Imaging, Ypsilanti, MI).
Cell activity was studied on the same dishes after 3 and 7 days of culture. Cultures were washed twice with culture medium without phenol red, and Alamar blue (AB) solution was added to this medium, resulting in a final concentration of 10%. After incubation for 90 min at 37°C, the medium was collected and the absorbance was determined at 570 and 600 nm with a spectrophotometer (940; Kontron, Munich, Germany). The percentage of AB reduction (%AB reduction) was calculated according to the manufacturer’s protocol. The number of viable cells correlates with the magnitude of dye reduction and is expressed as a percentage of control.
Nitric Oxide Measurements
Nitric oxide (NO) was measured as nitrite (NO2 −) accumulated in the MC3T3-conditioned culture medium. An aliquot (200 μl) of culture medium was collected after 3, 5, or 7 strain episodes. NO2 − was assayed using Griess reagent (1% sulfanilamide, 0.1% naphthylethylene-diamine-dihydrochloride). The reference curve was prepared by serial dilutions of NO2 − standard solution (0.1 M sodium NO2 −) using the culture medium as diluent. Absorbance was measured at 540 nm.
RNA Extraction and RT-PCR
RNA extraction and RT-PCR were performed on MC3T3-E1 cells to quantify matrix mRNA expression and on C3H10T1/2 cells to analyze cell differentiation. MC3T3-E1 cells were harvested 12 h after the last strain sequence, and C3H10T1/2 cells were harvested after 2 days of preculture and 5 days in permissive culture.
Total RNA was isolated using the RNeasy Plus Mini Kit (Qiagen, Valencia, CA). Purified RNA was dissolved in RNase-free water, and its concentration was determined at 260 nm. RNA quality was checked on a 2% agarose gel with 1 μg/ml ethidium bromide. Samples were stored at −80°C until use. cDNA was synthesized from 2 μg of total RNA with the first-strand cDNA synthesis kit for RT-PCR (AMV; Roche, Indianapolis, IN). For quantitative real-time PCR, 8 μl of cDNA mixture diluted to 1:20 in water was subjected to real-time PCR using SYBR green I dye (Lightcycler Faststart DNA Master SYBR Green I; Roche, Penzberg, Germany). Reactions were performed in 20 μl PCR mixture containing 4 μl 5× Master Mix (dNTP mixture with dUTP, MgCl2, SYBR Green I, Taq DNA polymerase, and reaction buffer) and 2 μl of 10 μM primers. Primer sequences of mouse FN, type I collagen, bone sialoprotein (BSP), osteopontin (OPN), alkaline phosphatase (ALP), Runx2, and peroxisome proliferator-activated receptor gamma 2 (PPARγ2) are listed in Table 1. Cyclophilin was used as control. Amplified products were examined for size estimation on 2% agarose gel with 1 μg/ml ethidium bromide and DNA molecular weight marker. Values are expressed as percent of control after normalization to cyclophilin values.
OPN Immunoassay
Twenty-four hours after the end of mechanical treatments, ELISAs were performed to measure OPN in cell lysate and culture medium using the DuoSet mouse Osteopontin ELISA Development System (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. Cells were collected in lysis buffer composed of PBS with 0.5% Nonidet 40 and 1% protease inhibition cocktail (PIC; Sigma, St. Quentin Fallavier, France). PIC was also added to the culture medium (1%). Each sample was run in duplicate in the assays. Data were corrected for fresh culture medium or fresh lysis buffer values and normalized to total protein content of the cell lysate measured with the BCA Protein Assay Kit (Interchim Pierce, Monluçon, France).
Fluorescence Labeling
For FN immunostaining, cell layers were washed in PBS, fixed with 4% paraformaldehyde in PBS for 20 min at room temperature, washed, and incubated for 2 h with 1/100 diluted rabbit anti-mouse-FN (Sigma). After washing, cells were incubated for 1 h with 1/250 diluted Alexa Fluor488-tagged secondary antibody (Invitrogen, Carlsbad, CA).
For actin labeling, cell layers were fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 in PBS (3 min), and incubated for 45 min at 37°C with a rhodamine–phalloidin (Interchim) stock solution (1/50 dilution).
For vinculin or PPARγ2 immunostaining, cells were fixed and permeabilized with 70% methanol, washed, and incubated for 2 h with primary antibodies. After washing, cells were incubated for 1 h with Alexa Fluor488.
Preparations were mounted in Fluokeep (Argene, North Massapequa, NY), examined by fluorescence microscopy (DMRB; Leica, Bensheim, Germany), and imaged by a camera (CoolSnapfx) using Meta Imaging series 4.6.6. software. Images were acquired in the region of interest corresponding to area 1 (Fig. 1c).
DNA Measurement
The Invitrogen PicoGreen DNA quantitation kit was used to measure double-stranded DNA concentrations in solution. All reagents (dsDNA reagent, TE buffer: 200 mM Tris–HCl, 20 mM EDTA [pH 7.5], and lambda DNA standard) were obtained from the kit, and the assay was performed as described in the protocol. Data were corrected for cell-free values. Samples were placed in a black 96-well plate and excited at 485 nm. Emission was measured at 538 nm using a fluorescent plate reader (Fluoroscan Ascent; Thermo Labsystems, Worcester, MA).
Culture of C3H10 T 1/2 on Cell-Free Matrices
Matrices were prepared from MC3T3-E1 cultures after 7 days of LMHF and the corresponding unstrained cultures. The procedure for preparing cell-free matrix was modified from a published protocol [15]. Cultures were washed once with PBS and twice with wash buffer I (100 mM Na2HPO4 [pH 9.6], 2 mM MgCl2, 2 mM EDTA). Each dish was incubated at 37°C for 15 min in the presence of 8 ml lysis buffer (8 mM Na2HPO4 [pH 9.6], 1% NP-40); then, lysis buffer was replaced with 8 ml of fresh lysis buffer for 30–60 min. Matrices were washed four times with 8 ml H2O. Matrices were used immediately or could be stored at 4°C in PBS containing 100 units/ml penicillin and 100 μg/ml streptomycin until needed. Decellularization was controlled by nuclear DAPI staining. FN was immunostained to observe matrix topology and quantified by image analysis, as described below. Nondecellularized matrices were observed in parallel.
C3H10T1/2 cells were plated at a density of 5,000 cells/cm2 on tissue culture polystyrene (TCPS) or decellularized matrices and maintained for 2 days in standard medium (preculture). Permissive medium (PM) was composed of αMEM supplemented with 10% FCS (PromoCell, Heidelberg, Germany), 50 μg/ml ascorbic acid, 10−6 M β-glycerophosphate, 10−8 M all trans-retinoic acid, 10−8 M dexamethasone, 1% insulin, and 5 × 10−5 M 3-isobutyl-1-methylxanthine. Culture medium was changed every 2 days. On day 2, cultures were switched to the PM for both osteoblast and adipocyte differentiation.
Cell Attachment
Calcein AM (Invitrogen) was used to label viable cells 2 h postseeding on cell-free matrix. Cells were washed with PBS and incubated for 20 min at 37°C with 4 ml of 10 μg/ml calcein. In live cells, nonfluorescent calcein AM is converted to a green fluorescent calcein after acetoxymethyl ester hydrolysis by intracellular esterases. Cell attachment was controlled by fluorescent cell count using ImageJ software (http://rsb.info.nih.gov/ij).
Image Analysis
Image analysis was performed with ImageJ software.
FN network analyses were performed after background removal. To selectively detect linear structures in images, we used a Hessian filter based on edge detection. After threshold, the resulting image revealed closed lacunae. To determine areas of lacunae, the parameter “mean area” was selected in the program of the particles analyzer. Focal contact area and number were determined according to Usson et al. [16]. After image binarization, morphological parameters including cell area and cell perimeter were automatically measured by the computer program. Shape factors were generated from these data. The shape factor 4 pi (area/perimeter2) assesses the deviation from circularity: A circle has a value of 1, and values approaching 0.0 indicate an increasingly elongated polygon. Feret diameter indicates the longest distance between any two points along the cell.
Statistical Analysis
Data were analyzed with STATISTICA6 software (StatSoft, Tulsa, OK). For MC3T3-E1 control and LMHF cultures, comparisons were performed with the Mann–Whitney test. For C3H10T1/2 cultures on different substrates (TCPS, control matrix, or LMHF matrix) group comparisons were performed with Kruskal–Wallis one-way analysis of variance. When P values were <0.05, post hoc comparisons among individual samples were performed with Mann–Whitney test with adjusted α value.
Results
Production and Characterization of LMHF Strain
Due to the rigidity of Petri dishes, the resonance frequency had to be used for signal amplification. The resonance frequency of 100-mm Petri dishes filled with 8 mL of culture medium was measured at 400 Hz, a frequency which produces a maximum vertical displacement (16 μm) of the center of the Petri dish (Fig. 1b). Modeling of tangential strain along the radius of the Petri dish showed compressive or extensive area on both sides of the nodal circle at a fixed time (Fig. 1c). Three strain gauges, positioned at various places on the inner surface of the polystyrene substrates, as shown in Fig. 1c, were used to measure strain magnitudes, which were consistent with the models. As illustrated, the signal waveform induced maximal strains of approximately ±15 με at the center, ±25 με at the mid-radius, and ±40 με at the periphery. Area 1 (15–25 με) can be estimated to represent 75% of the inside surface of the Petri dish and area 2 (25–40 με) can be estimated to represent 25%.
MC3T3 E1 Cells are Sensitive to LMHF Stimulation
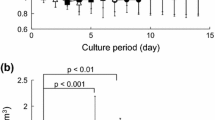
The cell densities measured in control or mechanically stimulated cultures at days 1, 2, 3, and 7 are shown in Fig. 2a. No significant difference was observed between controls and strained cultures regardless of the time point tested. The cell population increased from about 8,900 cells/cm2 at day 1 to 45,700 cells/cm2 at day 3 and 75,400 cells/cm2 at day 7 (Fig. 2a) in both conditions. No significant difference was observed in the viability of cells cultured between control and LMHF cultures after 3 or 7 days of culture (Fig. 2b). NO release, a parameter of bone cell response to load, was assessed. A modest but significant increase in the NO accumulated in the culture medium was measured after 3 dLMHF and maintained after 5 and 7 days of LMHF (Fig. 2c). Cytoskeletal organization was assessed by actin labeling 15 min after the third session of LMHF (3 dLMHF). Control cells showed an abundance of straight aligned F-actin fibrils. In contrast, LMHF cells exhibited curled stress fibers, demonstrating that cells sense LMHF and respond by reorganization of the cytoskeleton (Fig. 2d). The number of focal contacts immunolabeled with vinculin antibodies was similar between the two conditions (data not shown), whereas the mean focal contact area was 40% higher in the LMHF group than in controls (Fig. 2d).

MC3T3 E1 cells are sensitive to micromechanical stimulation. a Density of MC3T3 cells unstrained or in strain culture (1, 2, 3, or 7 dLMHF). Unstrained (empty bars) and strained (gray bars) cultures were grown in parallel. Quantification of cell nuclei. n = 3, 10 fields, mean ± SEM. b Viability of unstrained or strain culture cells (3 or 7 dLMHF). Unstrained (empty bars) and strained (gray bars) cultures were grown in parallel. * P < 0.05 vs. control, n = 4, mean ± SEM. c NO accumulation in culture medium of unstrained (filled diamond) or strain culture (filled square) (3, 5, or 7 dLMHF). Unstrained and strained cultures were grown in parallel. * P < 0.05 vs. control, n = 6, mean ± SEM. d Actin and vinculin immunostaining on unstrained culture (ctrl) or 3 dLMHF culture and quantification of immunolabeled vinculin area per cell. n = 3, 10 fields per dish, mean ± SEM
Effects of LMHF on Matrix Gene Expression and Matrix Protein Production
A single 20-min strain sequence was sufficient to increase mRNA expression of FN (1.8-fold), OPN (1.7-fold), and BSP (1.5-fold) (Fig. 3a). Fractionated application of LMHF was also tested: When stimulation was divided into 10 2-min sessions over 20 h, a less marked increase of FN and BSP mRNA was also observed (data not shown). One daily 20-min LMHF sequence was therefore used for the following experiments. The increase in FN mRNA was maintained after 3 and 7 dLMHF, whereas OPN mRNA and BSP mRNA expression appeared to be biphasic, reaching control levels after 3 dLMHF and reincreasing after 7 dLMHF (Fig. 3a). A late response for type I collagen expression was noted with a 1.7-fold increase after 7 dLMHF (Fig. 3a). OPN protein at day 1 increased in the soluble fraction and at day 3 was already integrated in the matrix, in line with the increased mRNA expression measured after the first LMHF sequence (Fig. 3a).

Effects of LMHF on matrix gene expression and matrix protein production in MC3T3-E1 cultures. a Matrix protein mRNA levels in 1 dLMHF MC3T3-E1 cells. Cultures were strained for 1, 3, or 7 dLMHF; and analysis was performed 12 h poststrain. Unstrained (empty bars) and strained (gray bars) cultures were grown in parallel. Values were normalized to cyclophilin mRNA level and expressed as a ratio to controls (Ctrl). * P < 0.05 vs. control, n = 4, mean ± SEM. b OPN protein was measured in lysate and in culture medium after 1 or 3 dLMHF. Unstrained (empty bars) and strained (gray bars) cultures were grown in parallel. Results are normalized on total cellular protein content and expressed as a ratio to controls (Ctrl). * P < 0.05 vs. control, n = 4, mean ± SEM
LMHF Strain Alters Matrix Organization
FN immunolabeling was performed in order to visualize the matrix network after 3 and 7 dLMHF of MC3T3 cultures, showing initiation of matrix organization after 3 dLMHF (data not shown). This pattern, characterized by an increase in lacunar structures of the matrix, further progressed after 7 dLMHF (Fig. 4a). Following decellularization treatment, no nuclear material was observed in the matrix, as visualized by DAPI staining (Fig. 4a), demonstrating complete efficiency of the decellularization process. FN imaging showed that the characteristic pattern with larger lacunae was maintained.

Effects of LMHF stimulation on matrix organization. a Immunolabeled FN network and DAPI staining on control (Ctrl) and strain cultures (7 dLMHF) for matrix with cells or cell-free matrix. n = 3, 20 fields per dish. b Mean of matrix lacunar areas in cell-free matrices of control (empty bars) and 7 dLMHF (gray bars) cultures, mean ± SEM. Lacunar areas were separated into two size categories: small (40–5,000 pixel2) and large (5,000–25,000 pixel2) lacunae
Quantitative analysis of the FN pattern was performed on the matrices after decellularization. Measurement of lacunar area (Fig. 4b) revealed that the mean area of the largest (>5,000 pixel2) lacunae was twofold increased in 7 dLMHF cultures (Fig. 4b), whereas the smallest lacunae (<5,000 pixel2) had similar mean areas in both matrices. These analyses showed that the matrix pattern was reorganized during mechanical stimulation.
Short-Term Behavior of MSCs Grown on Matrix Produced Under LMHF
To evaluate the osteogenic properties of ECM, C3H10T1/2 MSCs were plated on acellular ECM from control and 7 dLMHF cultures. MSCs seeded on TCPS were used as a reference. Calcein staining 2 h postseeding allowed analysis of both cell morphology and attachment. Observation of cell shape showed round cells on TCPS substrate, whereas cells displayed a stellar morphology on both control and LMHF matrices (Fig. 5a). No obvious morphological difference was observed at this stage between the two matrix conditions. The initial attachment of MSCs was also quantified 2 h postseeding. An increase in cell number was observed for cells seeded on both matrices compared to TCPS. Moreover, the number of adherent cells on LMHF matrix was significantly increased in comparison to control matrix. Focal contacts were assessed 6 h postseeding. Vinculin staining showed no focal contact for cells seeded on TCPS at this early time point, while focal adhesions were clearly visible on cells seeded on both matrices. Focal contact number was 50% higher in cells seeded on LMHF matrix compared to cells seeded on control matrix (Fig. 5b). After 16 h, morphological changes of C3H10T1/2 seeded on the various substrates were quantified using actin labeling. MSCs on both matrices exhibited more filopodia, lamellipodia, and cellular extensions compared to those seeded on TCPS. Cell morphology appeared similar on both matrices, but parameters measured by image analysis highlighted differences in cell shape between the matrices: LMHF matrices induced an increase in circularity and a decrease in perimeter and Feret diameter compared to control ECM, while no difference in cell area was observed.

Short-term behavior of C3H10T1/2 cells grown on matrix produced under LMHF stimulation. a Morphology and number of adherent C3H10T1/2 cells 2 h postseeding (calcein staining). Results are expressed as a ratio to control. P < 0.05: * vs. TCPS, # vs. matrix control. n = 3, 15 fields per dish, mean ± SEM. b Quantification of focal adhesion number (vinculin) per cell 6 h postseeding. n = 35 cells, mean ± SEM. c Actin staining 16 h postseeding. White arrows show cytoplasmic extensions. Quantitative analysis of the morphology of C3H10T1/2 cells cultured on TCPS (empty bars), control matrix (light gray bars), and strain matrix (dark gray bars). n = 200 cells, mean ± SEM, P < 0.05: * vs. TCPS, # vs. matrix control
MSCs Growth and Differentiation on Matrix Produced Under LMHF
Cell density of MSCs grown on TCPS, control matrix, and LMHF matrix was assessed by DNA quantification after 5 days in PM. At this time, the DNA content in both matrices (control and LMHF) were lower than on TCPS (Fig. 6a), showing a better proliferation on TCPS. The comparison of DNA content between matrices reveals no difference, showing that LMHF did not affect the proliferative quality of the matrix.

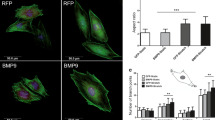
MSC differentiation on matrix produced under LMHF. a DNA quantification in C3H10T1/2 cells cultured on TCPS (empty bars), control matrix (light gray bars), and 7 dLMHF matrix (dark gray bars) for 2 days in preculture medium followed by 5 days in PM. n = 4, mean ± SEM. b PPARγ2 staining and quantification in C3H10T1/2 cells cultured on TCPS (empty bars), control matrix (light gray bars), and 7 dLMHF matrix (dark gray bars) for 2 days in preculture medium followed by 5 days in PM. Analysis of three dishes with 10 fields per dish. Mean ± SEM. c mRNA levels in C3H10T1/2 cells cultured on TCPS (empty bars), control matrix (light gray bars), and 7 dLMHF matrix (dark gray bars) for 2 days in preculture medium followed by 5 days in PM. Values were normalized to cyclophilin mRNA level and expressed as a ratio to controls (Ctrl). P < 0.05: * vs. TCPS, # vs. matrix control, n = 5, mean ± SEM
MSC differentiation was studied under the same conditions. The commitment toward the adipocyte lineage was assessed by analysis of PPARγ2 protein and mRNA levels. PPARγ2 protein was expressed equally on TCPS and control matrix. However, PPARγ2 protein expression was downregulated in cultures on LMHF matrix (Fig. 6b) compared to TCPS or control matrix. This was corroborated by a twofold decrease in PPARγ2 mRNA (Fig. 6c). The commitment of MSCs toward the osteoblastic lineage was revealed by increased levels of mRNA for osteoblast markers: Runx2 mRNA expression was higher on both matrices compared to TCPS, but the LMHF matrix did not improve Runx2 mRNA expression (Fig. 6c). Comparison between TCPS and control matrices showed that ALP and FN were not stimulated by the matrix and collagen was slightly increased. However, ALP, type I collagen, and FN were increased in MSCs grown on LMHF matrix compared to control ECM (Fig. 6c). Upregulation of these genes on LMHF matrix therefore indicates progression toward an osteoblastic phenotype.
Discussion
In this study, a mechanical loading system was developed to produce LMHF stimulations set in a range which has not been obtained by previously described devices. The mechanoresponsiveness of bone has been recognized for a long time, and it has been presumed that a threshold of 1,000 με would have to be exceeded to become anabolic [17], while strains below this level of deformation were considered insufficient to retain tissue morphology. Recent work also suggests an anabolic potential of vibratory mechanical signals (<5 με) [11]. Numerous techniques to induce mechanical deformation of osteoblasts in vitro have been described in the literature, but all devices generate high-magnitude deformation: strain magnitudes between 200 and 40,000 με [18, 19]. This study was conducted in a range of physiological microdeformations. With the device described here, osteoblastic responses were examined for deformations of about 20 με, which is much lower than the threshold considered osteogenic in the literature. This system has the advantage of being easy to set up as the buzzers and Petri dishes used are commercially available. Very precise measuring systems and modeling allow accurate determination of the microdeformations experienced by the cells. This system also presents certain limitations. LMHF stimulations were not completely homogeneous over the culture substrate. Mechanical loading systems used for cell cultures often displayed this drawback, and our system had the advantage to generate LMHF stimulations over a very narrow range. Moreover, this study was unable to consider in detail the physical differences between the effects of fluid shear stress and mechanical strain on bone cell deformation.
Bone cells respond to high-frequency strain (i.e., 400 Hz), in a range which is not usually described. With certain adaptations, this system could be used to generate lower frequencies, inducing deformations of similar magnitude, which would provide information about the threshold of responsiveness of osteoblasts in terms of frequency. However, the use of high frequencies for an in vitro study did not appear to be a problem as the primary objective was to produce an osteogenic matrix for MSCs and this frequency does not appear to be harmful to MC3T3 cells. This study demonstrates the responsiveness of osteoblasts to our LMHF signal, as reflected by NO release and cytoskeletal and focal adhesion modifications. Only one study [11] has reported in vitro osteoblast mechanosensing of an LMHF mechanical regimen. Although this mechanical regimen differed from that used in our study, as the magnitude of vibrations was less than 5 με and over a wide range of frequencies (5–100 Hz), Bacabac et al. [11] showed that NO release by MC3T3-E1 cells was significantly increased for the highest frequency. In our conditions, the amount of nitrite secreted after 1 dLMHF was below the limit of detection, but an increase of nitrite accumulated in the culture medium was assessed after a longer mechanical stimulation, demonstrating the threshold responsiveness of osteoblasts to LMHF. Mechanical strain is known to induce changes in cytoskeletal organization, and more specifically, actin bundles can be restructured by mechanical stretching [20]. MC3T3 cells displayed a well-organized actin fiber pattern on plastic support so that fiber reinforcement was not obvious after LMHF stimulation. However, the curling appearance of the actin cytoskeleton observed after LMHF stimulation clearly demonstrated that the polymerization/depolymerization process was affected. A similar actin fiber pattern has been observed in MC3T3 cells after exposure to ultrasound [21]. Vinculin, a plasma membrane-associated protein found in adhesion complexes [22], was selected as a marker of LMHF responsiveness based on previous investigations showing that focal contacts can be reorganized by mechanical stretching [23]. Three days of LMHF stimulation increased the size of vinculin-positive focal contacts, consistent with this previous study that reported a sustained increase in vinculin spot area in stretched osteoblasts. This increase resulted from clustering of small and probably less mature contacts. The number of spots was not affected at this time point, suggesting that the mechanically induced increase in vinculin spot number is early but transient, as noted [23]. Stress fibers and their associated focal adhesions therefore play a role in the LMHF signal-transduction mechanism.
LMHF loading was used to alter matrix synthesis. MC3T3-E1 cells were chosen for their ability to produce an abundant ECM and for their responsiveness to a large panel of mechanical stimulations [10, 24, 25]. Information can be provided by both quantitative and qualitative ECM modifications. In this study, LMHF stimulations initially enhanced OPN, BSP, and FN gene expression and then collagen mRNA. The increase in OPN gene expression was confirmed at the protein level. While the quantitative aspect is generally taken into account [26, 27], alteration of the matrix network by mechanical stimulation is still poorly documented. One study [28] reported thickening of FN fibers on osteoblastic cultures submitted to physiological mechanical stimulation. The image analysis used here allowed the detection of a characteristic pattern. LMHF stimulation led to larger lacunae in matrices, and this altered pattern was not an artifact due to decellularization as it was observed before and after removing the cells. We concluded that LMHF stimulation not only enhanced expression of major bone matrix proteins (collagen, OPN, BSP, FN) but also induced an obvious reorganization of the FN network.
To evaluate the potential functional role of LMHF stimulation of the ECM, we studied the behavior of MSCs seeded onto decellularized matrices. Murine C3H10T1/2 cells are good candidates to test matrix-induced osteogenic differentiation as their pluripotent differentiation capability is comparable to that of primary bone marrow-derived MSCs [29].
An explanation for the positive effect of the LMHF matrix on MSC osteogenesis required analysis of MSC initial adhesion and focal contact structures, which both influence cell behavior [30, 31]. Natural and biomimetic coatings are known to exert a proadhesive effect for a wide range of cells [32, 33], but this effect has not yet been documented for MSCs. The proadhesive quality of the in vitro generated bone-like ECM was demonstrated here: MSC initial adhesion (i.e., 2 h after seeding) was more efficient on control matrices compared to TCPS, and focal contacts were already present after 6 h but were not detected on TCPS. Thus, matrices retained their adhesive properties despite the decellularization process and, as expected, supported a better short-term adhesion of MSCs. Most importantly, LMHF stimulation provided additional adhesive properties to the ECM as reflected by enhanced MSC attachment at the very beginning of the culture (2 h) and by the larger number of focal contacts compared to control ECM. These improved adhesive properties of LMHF matrices were confirmed with another cell line (MC3T3-E1, data not shown).
Cell morphology was then analyzed as an indicator of the commitment toward osteoblastic/adipogenic lineage [34]. A round cell shape was associated with adipogenic differentiation [35], while a stellate MSC morphology, as observed on both matrices, indicated loss of the adipogenic phenotype.
The composition of the ECM could affect MSC recruitment: OPN and BSP have been implicated as key proteins for mediating cellular adhesion, notably at bone–implant interfaces [36]. The LMHF regimen enhanced BSP and OPN gene expression. Both proteins might play important roles in cell binding to the collagen matrix. However, Bernards et al. [37] showed that MC3T3 E1 cell binding on collagen-coated TCPS is enhanced on substrates with adsorbed OPN compared to BSP. The present study clearly demonstrated that LMHF stimulation increased OPN in the insoluble fraction, suggesting that it is incorporated into the ECM and could therefore improve MSC adhesion. Matricellular proteins including OPN modulate cell function but do not appear to contribute per se to the organization or physical properties of ECM structures [38]. However, the binding of OPN to structural ECM proteins such as collagen and FN [39, 40] will orient one protein with respect to another. This matrix-bound OPN would gain a preferential orientation/conformation for cell adhesion after LMHF stimulation.
The involvement of structural proteins, specifically FN, a glycoprotein promoting cellular attachment, was considered quantitatively and topologically in this study. During MSC osteogenesis processes in vivo, the ECM surrounding the cells is dynamically remodeled [41]. FN is observed early in the region of mesenchymal condensation [42]. One hypothesis for the matrix-induced cell commitment toward the osteoblastic lineage is that reorganization of the FN network in LMHF cultures facilitates the specific engagement of α5β1 integrin of MSCs. Previous studies [43, 44] have shown that modulation of FN conformation, including alterations in the cell binding domain to FN, directs cell proliferation and differentiation. Recently, Martino et al. [45] indicated that α5β1 integrin influences human MSC behavior: Blockade of α5 and β1 led to decreased ALP activity, demonstrating the capacity of α5β1 integrin-specific engagement to drive intracellular signaling events toward cell differentiation.
The molecular mechanisms which dictate the osteogenic differentiation of MSCs are largely unknown. Inducers of differentiation toward one lineage inhibit cell differentiation toward alternative lineages. For example, the canonical Wnt/beta-catenin pathway induces osteoblastogenesis and inhibits adipogenesis, whereas PPAR-γ is a prime inducer of adipogenesis and, as shown in recent studies, inhibits osteoblastogenesis [46, 47]. Indeed, we observed, 1 week after MSC plating, upregulation of type I collagen, ALP, and FN with concomitant downregulation of PPARγ2. Runx2 expression was not altered at this time, but this does not preclude an earlier and transient upregulation [48]. We suggest that, after 1 week, remodeling of the acellular matrix via de novo synthesis of ECM proteins had occurred and probably played an important role in orienting toward osteogenic rather than adipogenic differentiation.
The complexity of matrices raises a difficult challenge to precisely identify the added value of LMHF matrix in terms of composition and topography. Various strategies have been proposed to further our understanding of the role of environment in MSC differentiation. Simplified models using substrate coating with ECM components can be used to study the influence of the nature of substrate. For example, adhesion to ECM components such as laminin, collagen type I, or vitronectin can induce osteogenic differentiation of hMSCs [14, 49]. The influence of nano- or microtopography on cell commitment was studied on synthetic substrates with ultraprecise nanofeatures [50]. For example, nanotopography of various symmetries and with varying degrees of disorder affect MSC adhesion and osteoblastic differentiation: Random nanotopographies increase osteospecific differentiation compared to ordered nanotopographies [51]. Similarly, osteoblast differentiation is induced on poly(l-lactic acid) nanofibrous matrix vs. afibrillar substrate [52]. Nevertheless, these models fail to reproduce the complex structure necessary for the natural environment of stem cells. In the present study, the starting material was a natural matrix, with its complex composition and ultrastructure; and these two aspects of the matrix can be subtly modified by physical intervention, close to physiological conditions. This work showed that LMHF is an effective and useful strategy to modify the cell-produced ECM to modulate MSC behavior. ECM partially constitutes the stem cell niche. The use of 3D configured ECM will significantly enhance our understanding of how stem cells interact with and respond to the ECM niche. Studies currently under way to further characterize these natural ECMs and to understand their structure, composition, and function could provide an important contribution to the optimal synthesis of biomaterials designed to replace or regenerate tissues.
Conclusion
This in vitro study demonstrates the anabolic effects of LMHF stimulations on osteogenesis, especially on the ECM produced by osteoblasts. The mechanical stimulation described in this study can be used as a tool to improve ECM qualities such as MSC capture and to direct stem cell differentiation toward osteogenesis. Low-magnitude stimulation is fully adapted to the field of bone tissue engineering for the development of scaffolds such as macroporous ceramics or bioglass that display poor mechanical resistance. LMHF stimuli could therefore be an approach to functionalize breakable biomaterials by providing osteoinductive properties via the production of an osteogenic ECM interface.
References
Frost HM (2003) Bone’s mechanostat: a 2003 update. Anat Rec A Discov Mol Cell Evol Biol 275:1081–1101
Torcasio A, van Lenthe GH, Van Oosterwyck H (2008) The importance of loading frequency, rate and vibration for enhancing bone adaptation and implant osseointegration. Eur Cell Mater 16:56–68
Fritton SP, McLeod KJ, Rubin CT (2000) Quantifying the strain history of bone: spatial uniformity and self-similarity of low-magnitude strains. J Biomech 33:317–325
Rubin C, Turner AS, Bain S, Mallinckrodt C, McLeod K (2001) Anabolism, low mechanical signals strengthen long bones. Nature 412:603–604
Judex S, Boyd S, Qin YX, Turner S, Ye K, Muller R, Rubin C (2003) Adaptations of trabecular bone to low magnitude vibrations result in more uniform stress and strain under load. Ann Biomed Eng 31:12–20
Qin YX, Rubin CT et al (1998) Nonlinear dependence of loading intensity and cycle number in the maintenance of bone mass and morphology. J Orthop Res 16:482–489
Rubin C, Recker R, Cullen D, Ryaby J, McCabe J, McLeod K (2004) Prevention of postmenopausal bone loss by a low-magnitude, high-frequency mechanical stimuli: a clinical trial assessing compliance, efficacy, and safety. J Bone Miner Res 19:343–351
Verschueren SM, Roelants M, Delecluse C, Swinnen S, Vanderschueren D, Boonen S (2004) Effect of 6-month whole body vibration training on hip density, muscle strength, and postural control in postmenopausal women: a randomized controlled pilot study. J Bone Miner Res 19:352–359
Prisby RD, Lafage-Proust MH, Malaval L, Belli A, Vico L (2008) Effects of whole body vibration on the skeleton and other organ systems in man and animal models: what we know and what we need to know. Ageing Res Rev 7:319–329
Bacabac RG, Van Loon JJ, Smit TH, Klein-Nulend J (2009) Noise enhances the rapid nitric oxide production by bone cells in response to fluid shear stress. Technol Health Care 17:57–65
Bacabac RG, Smit TH, Van Loon JJ, Doulabi BZ, Helder M, Klein-Nulend J (2006) Bone cell responses to high-frequency vibration stress: does the nucleus oscillate within the cytoplasm? FASEB J 20:858–864
Salgado AJ, Coutinho OP, Reis RL (2004) Bone tissue engineering: state of the art and future trends. Macromol Biosci 4:743–765
Cukierman E, Pankov R, Stevens DR, Yamada KM (2001) Taking cell-matrix adhesions to the third dimension. Science 294:1708–1712
Salasznyk RM, Williams WA, Boskey A, Batorsky A, Plopper GE (2004) Adhesion to vitronectin and collagen I promotes osteogenic differentiation of human mesenchymal stem cells. J Biomed Biotechnol 2004:24–34
Mao Y, Schwarzbauer JE (2005) Stimulatory effects of a three-dimensional microenvironment on cell-mediated fibronectin fibrillogenesis. J Cell Sci 118:4427–4436
Usson Y, Guignandon A, Laroche N, Lafage-Proust MH, Vico L (1997) Quantitation of cell-matrix adhesion using confocal image analysis of focal contact associated proteins and interference reflection microscopy. Cytometry 28:298–304
Turner CH, Forwood MR, Rho JY, Yoshikawa T (1994) Mechanical loading thresholds for lamellar and woven bone formation. J Bone Miner Res 9:87–97
Brown TD (2000) Techniques for mechanical stimulation of cells in vitro: a review. J Biomech 33:3–14
Tanaka SM (1999) A new mechanical stimulator for cultured bone cells using piezoelectric actuator. J Biomech 32:427–430
Sato K, Adachi T, Matsuo M, Tomita Y (2005) Quantitative evaluation of threshold fiber strain that induces reorganization of cytoskeletal actin fiber structure in osteoblastic cells. J Biomech 38:1895–1901
Yang RS, Lin WL, Chen YZ, Tang CH, Huang TH, Lu BY, Fu WM (2005) Regulation by ultrasound treatment on the integrin expression and differentiation of osteoblasts. Bone 36:276–283
Mierke CT (2009) The role of vinculin in the regulation of the mechanical properties of cells. Cell Biochem Biophys 53:115–126
Guignandon A, Akhouayri O, Usson Y, Rattner A, Laroche N, Lafage-Proust MH, Alexandre C, Vico L (2003) Focal contact clustering in osteoblastic cells under mechanical stresses: microgravity and cyclic deformation. Cell Commun Adhes 10:69–83
Jackson RA, Kumarasuriyar A, Nurcombe V, Cool SM (2006) Long-term loading inhibits ERK1/2 phosphorylation and increases FGFR3 expression in MC3T3-E1 osteoblast cells. J Cell Physiol 209:894–904
Tang L, Lin Z, Li YM (2006) Effects of different magnitudes of mechanical strain on osteoblasts in vitro. Biochem Biophys Res Commun 344:122–128
Rath B, Nam J, Knobloch TJ, Lannutti JJ, Agarwal S (2008) Compressive forces induce osteogenic gene expression in calvarial osteoblasts. J Biomech 41:1095–1103
Ignatius A, Blessing H, Liedert A, Schmidt C, Neidlinger-Wilke C, Kaspar D, Friemert B, Claes L (2005) Tissue engineering of bone: effects of mechanical strain on osteoblastic cells in type I collagen matrices. Biomaterials 26:311–318
Di Palma F, Guignandon A, Chamson A, Lafage-Proust MH, Laroche N, Peyroche S, Vico L, Rattner A (2005) Modulation of the responses of human osteoblast-like cells to physiologic mechanical strains by biomaterial surfaces. Biomaterials 26:4249–4257
Zhao L, Li G, Chan KM, Wang Y, Tang PF (2009) Comparison of multipotent differentiation potentials of murine primary bone marrow stromal cells and mesenchymal stem cell line C3H10T1/2. Calcif Tissue Int 84:56–64
Buckbinder L, Crawford DT, Qi H, Ke HZ, Olson LM, Long KR, Bonnette PC, Baumann AP, Hambor JE, Grasser WA 3rd, Pan LC, Owen TA, Luzzio MJ, Hulford CA, Gebhard DF, Paralkar VM, Simmons HA, Kath JC, Roberts WG, Smock SL, Guzman-Perez A, Brown TA, Li M (2007) Proline-rich tyrosine kinase 2 regulates osteoprogenitor cells and bone formation, and offers an anabolic treatment approach for osteoporosis. Proc Natl Acad Sci USA 104:10619–10624
Kim JB, Leucht P, Luppen CA, Park YJ, Beggs HE, Damsky CH, Helms JA (2007) Reconciling the roles of FAK in osteoblast differentiation, osteoclast remodeling, and bone regeneration. Bone 41:39–51
Garcia AJ, Keselowsky BG (2002) Biomimetic surfaces for control of cell adhesion to facilitate bone formation. Crit Rev Eukaryot Gene Expr 12:151–162
Roessler S, Born R, Scharnweber D, Worch H, Sewing A, Dard M (2001) Biomimetic coatings functionalized with adhesion peptides for dental implants. J Mater Sci Mater Med 12:871–877
McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS (2004) Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell 6:483–495
Koutnikova H, Auwerx J (2001) Regulation of adipocyte differentiation. Ann Med 33:556–561
Puleo DA, Nanci A (1999) Understanding and controlling the bone–implant interface. Biomaterials 20:2311–2321
Bernards MT, Qin C, Ratner BD, Jiang S (2008) Adhesion of MC3T3-E1 cells to bone sialoprotein and bone osteopontin specifically bound to collagen I. J Biomed Mater Res A 86:779–787
Bornstein P, Sage EH (2002) Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol 14:608–616
Mukherjee BB, Nemir M, Beninati S, Cordella-Miele E, Singh K, Chackalaparampil I, Shanmugam V, DeVouge MW, Mukherjee AB (1995) Interaction of osteopontin with fibronectin and other extracellular matrix molecules. Ann NY Acad Sci 760:201–212
Chen Y, Bal BS, Gorski JP (1992) Calcium and collagen binding properties of osteopontin, bone sialoprotein, and bone acidic glycoprotein-75 from bone. J Biol Chem 267:24871–24878
Nakamura M, Sone S, Takahashi I, Mizoguchi I, Echigo S, Sasano Y (2005) Expression of versican and ADAMTS1, 4, and 5 during bone development in the rat mandible and hind limb. J Histochem Cytochem 53:1553–1562
Sasano Y, Li HC, Zhu JX, Imanaka-Yoshida K, Mizoguchi I, Kagayama M (2000) Immunohistochemical localization of type I collagen, fibronectin and tenascin C during embryonic osteogenesis in the dentary of mandibles and tibias in rats. Histochem J 32:591–598
Stephansson SN, Byers BA, Garcia AJ (2002) Enhanced expression of the osteoblastic phenotype on substrates that modulate fibronectin conformation and integrin receptor binding. Biomaterials 23:2527–2534
Keselowsky BG, Collard DM, Garcia AJ (2005) Integrin binding specificity regulates biomaterial surface chemistry effects on cell differentiation. Proc Natl Acad Sci USA 102:5953–5957
Martino MM, Mochizuki M, Rothenfluh DA, Rempel SA, Hubbell JA, Barker TH (2009) Controlling integrin specificity and stem cell differentiation in 2D and 3D environments through regulation of fibronectin domain stability. Biomaterials 30:1089–1097
Muruganandan S, Roman AA, Sinal CJ (2009) Adipocyte differentiation of bone marrow-derived mesenchymal stem cells: cross talk with the osteoblastogenic program. Cell Mol Life Sci 66:236–253
David V, Martin A, Lafage-Proust MH, Malaval L, Peyroche S, Jones DB, Vico L, Guignandon A (2007) Mechanical loading down-regulates peroxisome proliferator-activated receptor gamma in bone marrow stromal cells and favors osteoblastogenesis at the expense of adipogenesis. Endocrinology 148:2553–2562
Pregizer S, Baniwal SK, Yan X, Borok Z, Frenkel B (2008) Progressive recruitment of Runx2 to genomic targets despite decreasing expression during osteoblast differentiation. J Cell Biochem 105:965–970
Klees RF, Salasznyk RM, Kingsley K, Williams WA, Boskey A, Plopper GE (2005) Laminin-5 induces osteogenic gene expression in human mesenchymal stem cells through an ERK-dependent pathway. Mol Biol Cell 16:881–890
Dalby MJ, Gadegaard N, Curtis AS, Oreffo RO (2007) Nanotopographical control of human osteoprogenitor differentiation. Curr Stem Cell Res Ther 2:129–138
Dalby MJ, Gadegaard N, Tare R, Andar A, Riehle MO, Herzyk P, Wilkinson CD, Oreffo RO (2007) The control of human mesenchymal cell differentiation using nanoscale symmetry and disorder. Nat Mater 6:997–1003
Hu J, Liu X, Ma PX (2008) Induction of osteoblast differentiation phenotype on poly(l-lactic acid) nanofibrous matrix. Biomaterials 29:3815–3821
Acknowledgements
V. D. held a research fellowship from La Region Rhone Alpes.
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors have stated that they have no conflict of interest.
Rights and permissions
About this article
Cite this article
Dumas, V., Ducharne, B., Perrier, A. et al. Extracellular Matrix Produced by Osteoblasts Cultured Under Low-Magnitude, High-Frequency Stimulation is Favourable to Osteogenic Differentiation of Mesenchymal Stem Cells. Calcif Tissue Int 87, 351–364 (2010). https://doi.org/10.1007/s00223-010-9394-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-010-9394-8