
Abstract
Mesenchymal stem cells (MSCs) reside in many types of tissue and are able to differentiate into various functional cells including osteoblasts. Recently, adipose tissue–derived MSCs (AMSCs) have been shown to differentiate into many lineages, and they are considered a source for tissue regeneration. The purpose of this study was to compare the osteogenic differentiation capability of MSCs from bone marrow (BMSCs), MSCs from periosteum (PMSCs), and AMSCs using in vitro culture and in vivo implantation experiments. We harvested these MSCs from 7-week-old rats. The cells were seeded and cultured for 7 days in primary culture to assay a colony-forming unit. The frequency of the unit was the smallest in the BMSCs (P < 0.001). After primary culture, subculture was performed under osteogenic differentiation conditions for 1 and 2 weeks to detect mineralization as well as the bone-specific proteins of alkaline phosphatase and osteocalcin as osteogenic markers. BMSCs and PMSCs showed distinct osteogenic differentiation capability in comparison with other MSCs (P < 0.001). For the in vivo assay, composites of these cells and hydroxyapatite ceramics were subcutaneously implanted into syngeneic rats and harvested after 6 weeks. Micro-computed tomographic (CT) and histological analyses demonstrated that new bone formation was detected in the composites using BMSCs and PMSCs, although it was hard to detect in other composites. The CT analyses also demonstrated that the bone volume of BMSC composites was more than that of AMSC composites (P < 0.001). These results indicate that BMSCs and PMSCs could be ideal candidates for utilization in practical bone tissue regeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Mesenchymal stem cells (MSCs) are multipotent cells that differentiate into various functional cell types of mesodermal tissues [1–3]. It has been shown that many tissues contain MSCs. With rapid advancement in tissue engineering, a multitude of applications for cultured cells in the construction of regenerative tissues have been demonstrated [3–5]. Recently, adipose tissue has been considered a source for tissue engineering and regenerative medicine [6], and multipotent MSCs have been found in adipose tissue [7–10]. Adipose tissue–derived MSCs (AMSCs) have been shown to differentiate into many lineages, including chondrogenic [8, 11, 12], osteogenic [13–17], myogenic [9, 18], cardiomyogenic [19, 20], neurogenic [10, 21, 22], angiogenic [23–26], and hepatic [27] lineages. The chief advantage of adipose tissue includes the huge amount of fatty tissue available for cosmetic surgery, such as liposuction. It is expected to be a practical cell source for regenerative medicine. However, it is not well investigated whether AMSCs have a similar potential for osteogenesis as do MSCs from other types of tissue, especially bone marrow–derived MSCs (BMSCs).
The purpose of this study was to compare the in vitro as well as the in vivo bone-forming potential of BMSCs, AMSCs, and MSCs from the periosteum (PMSCs). Using 7-week-old rats, we determined the number of stem cells, mineralization capability, alkaline phosphatase (ALP) activity, and bone-specific osteocalcin production of these MSCs using in vitro culture assay and analyzed the new bone formation capability of the cells using in vivo implantation assay.
Materials and Methods
Materials
Cell culture media, antibiotics, and dexamethasone were purchased from Nacalai Tesque (Kyoto, Japan). Fetal bovine serum (FBS) was purchased from JRH Bioscience (Lenexa, KS). Phosphate-buffered saline (PBS) without Ca and Mg, trypsin-ethylenediaminetetraacetic acid (EDTA) solution (0.05% trypsin, 0.53 mM EDTA-4Na), and Hoechst 33258 were purchased from Invitrogen (Carlsbad, CA). β-Glycerophosphate was purchased from Calbiochem (San Diego, CA). l-Ascorbic acid 2-phosphate magnesium salt n-hydrate (ascorbic acid-2-phosphate) and collagenase were purchased from Wako Pure Chemical Industries (Osaka, Japan). Calcein was purchased from Dojindo Laboratories (Kumamoto, Japan). p-Nitrophenylphosphate (PNPP) substrate was purchased from Zymed Laboratories (San Francisco, CA). Tissue culture flasks and plates were purchased from BD Biosciences (San Jose, CA) or Asahi Techno Glass (Chiba, Japan). Other regents were purchased from Sigma-Aldrich (St. Louis, MO) or Nacalai Tesque.
Animals
We used nine Fischer 344 7-week-old male rats purchased from Japan SLC (Shizuoka, Japan). Three of them were used for in vitro experiments and six (three donors, three recipients) were used for in vivo implantation. All procedures used in the animal experiments were governed by the Guidelines for the Care and Use of Laboratory Animals from the National Institute of Advanced Industrial Science and Technology of Japan.
Isolation and Culture of Stem Cells
Three rats were killed for in vitro assays by placing them in a glass jar, where they breathed a mixture of 2% halothane (Fluothane; Takeda Pharmaceuticals, Osaka, Japan) in oxygen-enriched air for 10 minutes. Femora, tibiae, adipose tissue, and abdominal dermis were obtained from these rats. Bone marrow was obtained from both femora (total six femora). Both ends of the rat femora were cut away from the epiphysis. Bone marrow was flushed out using 10 mL of culture medium (minimal essential medium [MEM] with Earle’s salts and l-Gln) containing 15% FBS and 1% antibiotics (100 U/mL penicillin G, 100 μg/mL streptomycin sulfate, and 0.25 μg/mL amphotericin B) and then dispersed through an 18-gauge needle. The culture medium including the bone marrow was filtered by a 40 μm cell strainer (BD Bioscience) and centrifuged at 400 × g for 5 minutes. After centrifugation, the supernatant portion of the medium was discarded and the pellet was used as BMSCs. The periosteum was isolated from both diaphyseal areas of femora and tibiae (total six femora and six tibiae), and adipose tissue was obtained from an inguinal region (total three samples). Abdominal dermis was obtained from the abdomen (total three samples). The average weights of the periosteum from both femora and tibiae, inguinal adipose tissue, and abdominal dermis were 224 ± 24, 297 ± 11, and 104 ± 4 (mean weight [mg] ± standard deviation [SD], n = 3), respectively. The periosteum, adipose tissue, and abdominal dermis were each treated with 10 mL of PBS containing 3 mg/mL collagenase for 1 hour. They were then centrifuged at 400× g for 5 minutes after filtration by 40 μm cell strainers. After centrifugation, the supernatants of the periosteum and abdominal dermis were discarded and their pellets were used as PMSCs and fibroblasts, respectively.
After centrifugation, the supernatants of all tissues except for the adipose tissue were discarded. However, both the supernatant and pellet were used in the adipose tissue preparation. The supernatant portion from the adipose tissue was named “MSCs from the adipose supernatant portion” (ASUP). The pellet portion from the adipose tissue was named “MSCs from the adipose pellet portion” (APPT). This method of preparation was essentially the same as that used by Hauner et al. [28] with some modifications. Samples derived from BMSCs, PMSCs, APPT, ASUP, and fibroblasts were placed into culture dishes for primary culture for 7 days. All cultures were maintained in a humidified atmosphere of 95% air and 5% CO2 at 37°C; the culture medium was renewed three times per week. Previously, we reported that dermal fibroblasts did not exhibit mineralization and osteogenic ability under the same culture conditions used in this study [29]. Therefore, we used fibroblasts as a negative control for all further analyses. The cell surface antigens of these primary cultured cells were positive for mesenchymal markers (CD29, -90) and negative for hematopoietic markers (CD45, data not shown). This indicated that these cultured cells were of the mesenchymal type [30].
Colony-Forming Assays
Nucleated cells of freshly prepared BMSCs, PMSCs, APPT, ASUP, and fibroblasts were counted by use of a NucleoCounter (Chemometec, Allerød, Denmark). BMSCs were seeded at a rate of 1.0 × 104, 105, or 106 cells/cm2 on 20 cm2 dishes. PMSCs, APPT, ASUP, and fibroblasts were seeded at a rate of 1.0 × 102, 103, or 104cells/cm2 on 20 cm2 dishes. After 6 days of primary culture, three dishes from each cell concentration were washed twice with PBS, fixed with 10% paraformaldehyde, and stained with Giemsa solution (Nacalai Tesque) for 5 minutes. The colonies from each dish were counted under an optical microscope and CFU frequencies (colony-forming units per cell number) of various MSCs were calculated.
In Vitro Analysis
Osteogenic Differentiation of MSCs from Bone Marrow, Periosteum, and Adipose Tissue
After primary culture, various MSCs were trypsinized and subcultured in every two 24-well tissue culture plates at a cell density of 1.0 × 104/cm2. Under undifferentiated conditions, medium supplemented with 10 mM β-glycerophosphate was renewed three times per week. Under osteogenic differentiation conditions, the medium was further supplemented with 0.28 mM ascorbic acid-2-phosphate and 10 nM dexamethasone. In both cases, the cells were subcultured for 1 and 2 weeks [1].
Quantification of Mineralization (Fluorescence Intensity of Calcein)
To visualize the mineralized extracellular matrix of subcultured cells, we used the calcein uptake assay according to our previous report [29]. For the calcein assays, 1 μg/mL of calcein was added each time the culture medium was renewed. Prior to the assays, subcultured cell layers were washed twice with PBS. Calcein incorporated into the mineralization of the matrix was observed using a fluorescence microscope (model IX70; Olympus, Tokyo, Japan) and visualized and quantified using a Typhoon 8600 image analyzer (Molecular Dynamics, Sunnyvale, CA).
Measurements of ALP Activity and DNA Content
Measurements of DNA content and assays of ALP activity after the in vitro osteogenic differentiation were done according to the methods we previously reported [31]. The layers of subcultured cells after 1 and 2 weeks were washed twice with PBS and collected by scraping into a sample tube containing 500 μL of 10 mM Tris buffer (pH 7.4, 1 mM EDTA and 100 mM NaCl). The sample was sonicated and used for DNA quantification. The quantification of the DNA content was performed using Hoechst 33258. The sonicated suspension (20 μL) was mixed with 200 μL of 0.25 μg/mL Hoechst 33258 and incubated for 5 minutes at room temperature. Fluorescence was measured on a microplate reader (Wallac 1420 ARVOsx; Perkin-Elmer, Boston, MA). Standard DNA was prepared with salmon sperm DNA (Invitrogen). To measure ALP activity, the sonicated suspension remaining after DNA quantification was used. After centrifuging at 12,000 × g for 3 minutes at 4°C, 20 μL of supernatant was mixed with 100 μL PNPP substrate and incubated for 30 minutes at 37°C. The ALP activity represented by the amount of p-nitrophenol released was normalized to the DNA content. The remaining supernatant including the sonicated cell suspension was used for the osteocalcin assay by adding formic acid solution as described below.
Quantification of Osteocalcin
Measurements of osteocalcin content were done according to the methods we previously reported [29]. After the ALP/DNA measurement of the 2-week subcultured cells, 500 μL of 20% formic acid was added to the tube containing the remaining suspension to extract inorganic ions as well as organic matrix proteins and stored at 4°C for several days. It was then centrifuged at 15,000 × g for 10 minutes at 4°C. Gel filtration by 1.0 mL of 10% formic acid was applied to the supernatant to eliminate inorganic ions. Then, the gel-filtered samples were evaporated. The resulting concentrated samples were added to an enzyme immunoassay plate immobilized with anti-rat osteocalcin antibodies to measure the concentration of osteocalcin with an intact rat osteocalcin enzyme immunoassay kit (Biomedical Technologies, Stoughton, MA).
In Vivo Analysis
Subcutaneous Implantation of Rat MSCs from Bone Marrow, Periosteum, and Adipose Tissue/Hydroxyapatite Composites into a Syngeneic Rat
BMSCs, PMSCs, APPT, ASUP, and fibroblasts were harvested from donor rats (total three donors) by the same method as described above. Primary cultured MSCs were trypsinized and suspended with culture medium at a cell density of 1.0 × 107/mL. Each cell suspension was poured into a well of a 96-well tissue culture plate. We used a hydroxyapatite (HA) disk (5 mm in diameter and 2 mm thick, Cellyard; Pentax, Tokyo, Japan) as scaffold in vivo implantation. An HA disk was placed into each cell suspension including the MSCs and maintained in a humidified atmosphere of 5% CO2 at 37°C overnight for cell adhesion. An HA disk with only culture medium was used as a negative control. A syngeneic recipient rat was anesthetized by intraperitoneal injection of pentobarbital (Nembutal; Dainippon Pharmaceutical, Tokyo, Japan) at a final concentration of 3.5 mg/100 g of body weight. We implanted six composites (BMSCs/HA, PMSCs/HA, APPT/HA, ASUP/HA, fibroblasts/HA, and control HA) subcutaneously into the back of the recipient rat (total three recipients) at different sites, and the implants (total 18) were harvested after 6 weeks.
Micro-computed Tomographic Analysis
The capability of the composites for new bone formation was evaluated by micro-computed tomography (micro-CT, MCT-CB 100MF(Z); Hitachi Medical, Tokyo, Japan). After 6 weeks, HA implants were harvested and the round surface of the HA was placed on the surface of a jig and scanned at widths of every 10 μm at 60 kV and 110 μA. The analytical conditions were precision mode, ×7 magnification with an image intensifier field of 1.8 inches, and total 512 sagittal sections scanned. We evaluated the distribution of newly formed bone of the HA composites, and total volumes of the bone were measured using the software package TRI3D-BON (Ratoc System Engineering, Tokyo, Japan) according to the methods we previously reported [32].
Histological Analysis
After the micro-CT analysis, the composites were fixed with 10% paraformaldehyde, decalcified with K-CX solution (Falma, Tokyo, Japan) and embedded in paraffin. They were cut into sections 5 μm thick and stained with hematoxylin and eosin (Muto Pure Chemicals, Tokyo, Japan). The specimens were examined by light microscopy.
Statistical Assessment
Statistical analysis was done using StatMate III (ATMS, Tokyo, Japan). Quantification of CFU, calcein, osteocalcin, and bone volume was analyzed by one-way repeated analysis of variance (ANOVA), followed by Tukey’s honest significant difference (HSD) post hoc test. The quantification of ALP activity was analyzed using a two-way repeated ANOVA (cell source by week), followed by Tukey’s HSD post hoc test. Statistical significance was indicated by P < 0.05.
Results
Colony-Forming Assays
To examine the frequency of stem cells in each cell source, we observed the colony-forming capabilities of various MSCs and counted the number of colonies. The observation of colonies from each culture dish which were seeded at a rate of 1.0 × 104/cm2 is presented in Fig 1a, and the number of colonies per dish is shown in Fig 1b. It was not possible to count the colonies of BMSCs when seeded at more than 1.0 × 106 cells/cm2 and those of the others when seeded at more than 1.0 × 104 cells/cm2 because they proliferated to near confluence (Fig. 1b). Therefore, the frequency of CFUs of BMSCs was significantly lower compared with that of other MSCs (P < 0.001, Fig. 1c). It was determined that 1.0 × 106 cells/cm2 dish for BMSCs and 1.0 × 104 cells/cm2 dish for the other MSCs were the optimal initial cell densities for the subculture to be used in further analyses.

Colony-forming assays of 6-day primary cultured cells. Photographs of various cell sources seeded at a cell density of 1.0 × 104/cm2. Representative culture dishes stained with Giemsa solution are shown. Blue dots indicate colonies (a). Colony numbers at several cell densities were calculated using an optical microscope. Values are means ± SD (n = 3). We could not count the colonies due to confluence of the PMSCs, APPT, ASUP, and fibroblasts seeded at more than 1.0 × 104/cm2 (b). CFU per cell number of various cell sources. Data are expressed as a log scale. Values are means ± SD (n = 6). ✯ P < 0.001 vs. BMSCs (one-way ANOVA, Tukey test) (c)
In Vitro Analysis
Mineralized Matrix Formation Detected by Phase Contrast Microscopy
We observed mineralized deposition under phase contrast and fluorescence microscopy after 2-week subculture. As described in “Materials and Methods,” various MSCs were subcultured in the presence or absence of dexamethasone. As we previously reported [31, 33], dexamethasone induces mesenchymal cells to differentiate into osteoblasts and stimulates the development of mineralized matrices on a variety of materials. After 2-week subculture in the presence of dexamethasone (osteogenic differentiation condition), BMSCs and PMSCs showed mineral deposition around the cells to form nodular aggregates (left figures in Fig. 2).

In vitro mineralization after 2-week subculture of various MSCs under the osteogenic differentiation condition. Phase contrast microscopic images show extracellular bone mineral regions (brown areas). Scale bar = 200 μm. Fluorescence microscopic images show calcein uptake (green areas). Scale bar = 500 μm
Mineralized Matrix Formation Detected by Calcein Uptake
Previously, we reported a novel quantitative method for detecting mineralization by cultured cells utilizing a calcium-binding fluorescent dye, calcein. Calcein has been found to be specifically incorporated and deposited into extracellular bone matrix, as evidenced by costaining with alizarin red S [34]. To demonstrate the calcium deposition of various MSCs over time during the subsequent subculture period, we added calcein to the culture media. After 2-week subculture under osteogenic differentiation conditions, BMSCs and PMSCs showed strong calcein fluorescence in the matrix by fluorescence microscopy (right figures in Fig. 2) and the fluorescence intensities of the BMSCs and PMSCs were greater than those of APPT, ASUP, and fibroblasts (P < 0.001, Fig. 3a).

Quantification of mineralization and bone-specific protein levels. Calcein uptake (fluorescence intensity) of various MSCs subcultured under the osteogenic differentiation condition for 2 weeks. Values are means ± SD (n = 5). ✯ P < 0.001 vs. BMSCs, *P < 0.001 vs. PMSCs (one-way ANOVA, Tukey test) (a). ALP activity of various MSCs subcultured under the osteogenic differentiation condition for 1 and 2 weeks. Data are expressed as a log scale. Values are means ± SD (n = 5). ✯ P < 0.001 vs. BMSCs for 2 weeks, *P < 0.001 vs. PMSCs for 2 weeks (two-way ANOVA, Tukey test) (b). Osteocalcin content in various MSCs subcultured under the osteogenic differentiation condition for 2 weeks. Values are means ± SD (n = 5). ✯ P < 0.001 vs. BMSCs, *P < 0.001 vs. PMSCs (one-way ANOVA, Tukey test) (c)
Quantification of ALP Activity and Osteocalcin Content
ALP activity, which is recognized as an early osteoblastic marker, was measured. The DNA content, which reflects the cell numbers, was measured. After 1- and 2-week subculture under osteogenic differentiation conditions, ALP activities per DNA of the BMSCs for 2 weeks were significantly greater than those of the other cell types (P < 0.001, Fig. 3b). The activities of the PMSCs for 2 weeks were also greater than those of APPT, ASUP, and fibroblasts (P < 0.001, Fig. 3b).
Osteocalcin is a bone-specific protein and has been used as a late marker of osteogenic differentiation. We measured the osteocalcin content of the cell layers subcultured under the osteogenic differentiation condition for 2 weeks and found that the contents of BMSCs and PMSCs were significantly greater than those of APPT, ASUP, and fibroblasts (P < 0.001, Fig. 3c).
In Vivo Analysis
Micro-CT Analysis
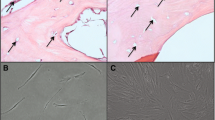
To observe and quantify the in vivo bone-forming capability of various primary cultured MSCs, we implanted MSC/HA composites and control HA. After implantation, they were examined by micro-CT analysis. We previously reported that BMSC/HA composites can show a high level of in vivo bone-forming ability [35–37]. According to the methods used in those reports, we prepared the composites and then implanted and harvested them after 6 weeks. The micro-CT images showed areas having high (white), medium (gray), and low (black) intensities. We defined the white, gray, and black areas as HA, newly formed bone (NB), and fibrovascular tissue with fat cells (F), respectively. There were more massive areas of NB (gray areas) in the BMSC/HA composite than in the other HA composites. Although some areas of NB could be found in the PMSC/HA composite, the areas were smaller than those of the BMSC/HA composite (Fig. 4). Furthermore, from 512 image sections of each composite, we calculated the total volume of newly formed bone. The volumes of BMSC/HA composites were significantly more than those of PMSC/HA composites (P < 0.001, Table 1), and only traces were detected in other composites (Table 1).

Micro-CT images of various MSC/HA composites and control HA after 6-week implantation. Inset: Higher magnifications of the areas indicated by the white square. Newly formed bone areas (gray, NB), hydroxyapatite (white, HA), fibrovascular tissue (black, F). Scale bar = 200 μm
Histological Analysis
After micro-CT analyses, MSC/HA composites and control HA were fixed and decalcified. The histology of implants demonstrated that the BMSC/HA and PMSC/HA composites showed newly formed bone together with active osteoblasts lining the pore areas of the composites. The BMSC/HA composite showed more bone areas than did the PMSC/HA composite. In contrast, no bone formation or osteoblasts were seen in the other composites and the control HA (Fig. 5).

Histological sections (hematoxylin and eosin stain) of various MSC/HA composites and control HA after 6-week implantation. Bone formation is indicated by arrow (red area). Black scale bar = 500 μm. Blue scale bar = 200 μm. Each figure at the right is a higher magnification of the rectangular area indicated by the black line in the corresponding figure at the left
Discussion
According to a previous report about AMSCs by Katz et al. [38], the pellet portion from adipose tissue after centrifugation consists of several different cell types and the supernatant portion includes both mature and immature adipocytes. Miyazaki et al. [39] obtained AMSCs that could differentiate into adipogenic, chondrogenic, and osteogenic lineages from not only the pellet portion but also the supernatant after centrifugation. Based on these reports, we obtained both APPT and ASUP and used them as cell (MSC) sources in this comparison study.
Sakaguchi et al. [40] demonstrated that the colony number of human BMSCs in primary culture was the lowest among the various MSCs. We also found that the CFU of the bone marrow was 100 times less than in the other types of cells in this study. The differences in the colony-forming cells might be caused by the variety of cell types residing in each tissue. Bone marrow consists of a huge amount of floating cells including hematopoietic cells and adherent cells on the bone surface. In contrast, the other cells used in the present study were derived from solid tissue.
For in vitro osteogenic comparison, we easily detected sufficient bone matrix and bone-specific protein (osteoclacin) in 2-week subculture of BMSCs and PMSCs under the osteogenic differentiation condition, although obvious matrix formation as well as osteocalcin were not detected in other subcultures. We also conducted real-time quantitative polymerase chain reaction (PCR) of ALP and osteocalcin mRNAs (data not shown). Both mRNA expression levels of the BMSCs under osteogenic conditions were greater than those of others. These in vitro results indicate that BMSCs and PMSCs contain many MSCs with osteogenic differentiation capability compared to AMSCs. This finding was supported by the in vivo implantation experiments. The in vivo analysis demonstrated that new bone formation was detected in BMSC/HA and PMSC/HA composites, although it was hard to detect in other composites.
Referring to previous reports that investigated the comparison of osteogenic ability between BMSCs and AMSCs, De Ugarte et al. [41] reported no significant difference in osteogenic ability between human BMSCs and AMSCs. In their report, quantification of ALP activity of 3-week subcultured BMSCs and AMSCs showed no significant difference. The ALP activity in their report was below 0.35 nmol/μg/minute, which was almost equivalent to that of 1-week subcultured ASUP in the present study (0.006 μmol/μg DNA/30 minutes). Our present study also detected 100 times more activity of 2-week subcultured BMSCs (1.08 μmol/μg DNA/30 minutes). In contrast, Gun-II et al. [42] reported that human AMSCs have inferior osteogenic capabilities compared to BMSCs. They quantified the percentage of ALP-positive cells under the osteogenic differentiation condition for both 2 and 3 weeks. The ALP-positive cells of the BMSCs were greater than those of the AMSCs, although the range of difference was less than that of the present study. Both Gun-II et al. and De Ugarte et al. used MSCs that had been passaged more than three times.
Concerning the osteogenic ability of periosteum, our study detected that PMSCs showed lower osteogenic ability compared to BMSCs. However, Yoshimura et al. [43] reported superior ability of PMSCs over BMSCs. They used MSCs passaged three times and subcultured for 2 weeks under the osteogenic differentiation condition. Sakaguchi et al. [40] also demonstrated the osteogenic comparison of various human MSCs passaged three times, and their result indicated that the osteogenic ability of BMSCs was similar to that of PMSCs. These previous reports of lower osteogenic ability of BMSCs used more passaged cells than the present study. Therefore, when comparing the cellular activity of different cell sources, we should regard the passage time. Sugiura et al. [44] also reported that the osteogenic ability of rat MSCs decreases with passaging. Their results showed that ALP activity of rat BMSCs at passage 1 was almost five times that of rat BMSCs at passage 3 under the osteogenic differentiation condition. Furthermore, these previous reports dealt only with in vitro culture experiments. In this study, we did in vivo implantation experiments, which confirmed the bone-forming ability of both BMSCs and PMSCs. As seen with in vitro culture experiments, the in vivo ability was not observed in other cells and the ability of BMSCs was more than that of PMSCs.
Regenerative medicine and tissue engineering have been rapidly developing for practical and global use. To achieve advances in regenerative medicine, the adaptability of various cell types and the investigation of the differentiation/proliferation potentials of adult stem cells from various cell sources are essential. We studied the potential of MSCs from bone marrow, periosteum, and adipose tissue. The potential on which we focused was osteogenesis, resulting in a demonstration of the clear potential of bone marrow and periosteum tissue. Therefore, adult MSCs from bone marrow and periosteum can be considered ideal candidates for regeneration of bone tissue.
References
Maniatopoulous C, Sodek J, Melcher AH (1988) Bone formation in vitro by stromal cells obtained from bone marrow of young adult rat. Cell Tissue Res 254:317–330
Le Douarin NH, Houssaint E, Jotereau FV, Belo M (1975) Origin of hemapoietic stem cells in embryonic bursa of Fabricius and bone marrow studied through interspecific chimeras. Proc Natl Acad Sci USA 72:2701–2705
Caplan AI, Bruder SP (2001) Mesenchymal stem cells: building blocks for molecular medicine in the 21st century. Trends Mol Med 7:259–264
Caplan AI, Ruben D, Haynesworth SE (1998) Cell-based tissue engineering therapies: the influence of whole body physiology. Adv Drug Deliv Rev 33:3–14
Ohgushi H, Caplan AI (1999) Stem cell technology and bioceramics: from cell to gene engineering. J Biomed Mater Res 48:913–927
Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, Mizuno H, Alfonso ZC, Fraser JK, Benhaim P, Hedrick MH (2002) Human adipose tissue is a source of multipotent stem cells. Mol Cell Biol 13:4279–4295
Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, Benhaim P, Lorenz HP, Hedrick MH (2001) Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng 7:211–228
Erickson GR, Gimble JM, Franklin DM, Rice HE, Awad H, Guilak F (2002) Chondrogenic potential of adipose tissue–derived stromal cells in vitro and in vivo. Biochem Biophys Res Commun 290:763–769
Mizuno H, Zuk PA, Zhu M, Lorenz HP, Benhaim P, Hedrick MH (2002) Myogenic differentiation of human processed lipoaspirate cells. Plast Reconstr Surg 109:199–209
Safford KM, Hiock KC, Safford SD, Halvorsen YD, Wilkinson WO, Gimble JM, Rice HE (2002) Neurogenic differentiation of murine and human adipose-derived stromal cells. Biochem Biophys Res Commun 294:371–379
Awad HA, Halvorsen YD, Gimble JM, Guilak F (2003) Effects of transforming growth factor beta 1 and dexamethasone on the growth and chondrogenic differentiation of adipose-derived stromal cells. Tissue Eng 9:1301–1312
Huang JI, Zuk PA, Jones NF, Zhu M, Lorenz HP, Hedrick MH, Benhaim P (2004) Chondrogenic potential of multipotential cells from human adipose tissue. Plast Reconstr Surg 113:585–594
Dragoo JL, Samimi B, Zhu M, Hame SL, Thomas BJ, Lieberman JR, Hedrick MH, Benhaim P (2003) Tissue-engineered cartilage and bone using stem cells from human infrapatellar fat pads. Adv J Bone Joint Surg Br 85:740–747
Cowan CM, Shi YY, Aalami OO, Chou YF, Mari C, Thomas R, Quatro N, Contag CH, Wu B, Longaker MT (2004) Adipose-derived adult stromal cells heal critical-size mouse calvarial defects. Nat Biotechnol 22:560–567
Halvorsen YC, Wilkison WO, Gimble JM (2004) Adipose-derived stromal cells—their utility and potential in bone formation. Int J Obes Relat Metab Disord 24 Suppl 4:S41–S44
Hicok KC, Laney TT, Zhou YS, Halvorsen YD, Hitt DC, Cooper LF, Gimble JM (2004) Human adipose-derived adult stem cells produce osteoid in vivo. Tissue Eng 10:371–380
Peterson B, Zhang J, Iglesias R, Kabo M, Hedrick M, Benhaim P, Lieberman JR (2005) Healing of critically sized femoral defects, using genetically modified mesenchymal stem cells from human adipose tissue. Tissue Eng 11:120–129
Rodriguez AM, Pisani D, Dechesne CA, Turc-Carel C, Kurzenne JY, Wdziekonski B, Villageosis A, Bagnis C, Breittmayer JP, Groux H, Aihaud G, Dani C (2005) Transplantation of a multipotent cell population from human adipose tissue induces dystrofin expression in the immunocompetent mdx mouse. J Exp Med 201:1397–1405
Planat-Bernard V, Menard C, Andre M, Puceat M, Perez A, Garcia-Veedugo JM, Penicaud L, Casteilla L (2004) Spontaneous cardiomyocyte differentiation from adipose tissue stroma cells. Circ Res 94:223–229
Strem BM, Zhu M, Alfonso Z, Daniels EJ, Schreiber R, Begyui R, Maclellan WR, Hendrick MH, Fraser JK (2005) Expression of cardiomyocytic markers on adipose tissue-derived cells in a murine model of acute myocardial injury. Cytotherapy 7:282–291
Ashjian PH, Elbarbary AS, Edmonds B, De Ugarte D, Zhu M, Zuk PA, Lorenz HP, Benhaim P, Hedrick MH (2003) In vitro differentiation of human processed lipoaspirate cells into early neural progenitors. Plast Reconstr Surg 111:1922–1931
Kang SK, Lee DH, Bae YC, Kim HK, Baik SY, Jung JS (2003) Improvement of neurological deficits by intracerebral transplantation of human adipose tissue–derived stromal cells after cerebral ischemia in rats. Exp Neurol 183:355–366
Miranville A, Heeschen C, Sengenes C, Curat CA, Busse R, Bouloumie A (2004) Improvement of postnatal neovascularization by human adipose tissue–derived stem cells. Circulation 110:349–355
Rehman J, Traktuev D, Li J, Merfeld-Clauss S, Temm-Grove CJ, Bovenkerk JE, Pell CL, Johnstone BH, Considine RV, March KL (2004) Secretion of angiogenic and antiapoptotic factors by human adipose stromal cells. Circulation 109:1292–1298
Planat-Benard V, Silvestre JS, Cousin B, Andre M, Nibbelink M, Tamarat R, Clergue M, Manneville C, Saillan-Barreau C, Duriez M, Tedgui A, Levy B, Penicaud L, Casteilla L (2004) Plasticity of human adipose lineage cells toward endothelial cells: physiological and therapeutic perspectives. Circulation 109:656–663
Cao Y, Shu Z, Liao L, Meng Y, Han Q, Zhao RC (2005) Human adipose tissue–derived stem cells differentiate into endothelial cells in vitro and improve potential neovascularization in vivo. Biochem Biophys Res Commun 332:370–379
Seo MJ, Suh SY, Bae YC, Jung JS (2005) Differentiation of human adipose stromal cells into hepatic lineage in vitro and in vivo. Biochem Biophys Res Commun 328:258–264
Hauner H, Entenmann G, Webitsch M, Gaillard D, Ailhaud G, Negrel R, Pfeiffer EF (1989) Promoting effect of glucocorticoids on the differentiation of human adipocyte precursor cells cultured in a chemically defined medium. J Clin Invest 84:1663–1670
Uchimura E, Machida H, Kotobuki N, Kihara T, Kitamura S, Ikeuchi M, Hirose M, Miyake J, Ohgushi H (2003) In-situ visualization and quantification of mineralization of cultured osteogenic cells. Calcif Tissue Int 73:575–583
Nagaya N, Fujii T, Iwase T, Ohgushi H, Itoh T, Uematsu M, Yamagishi M, Mori H, Kanagawa K, Kitamura S (2004) Intravenous administration of mesenchymal stem cells improves cardiac function in rats with acute myocardial infraction through angiogenesis and myogenesis. Am J Physiol Heart Circ Physiol 287:H2670–H2676
Ohgushi H, Dohi Y, Katsuda T, Tamai S, Tabata S, Suwa Y (1996) In vitro bone formation by rat marrow cell culture. J Biomed Mater Res 32:333–340
Nishikawa M, Myoui A, Ohgushi H, Ikeuchi M, Tamai N, Yoshikawa H (2004) Bone tissue engineering using novel interconnected porous hydroxyapatite ceramics combined with marrow mesenchymal cells: quantitative and three-dimensional image analysis. Cell Transplant 13:367–376
Ohgushi H, Dohi Y, Yoshikawa T, Tamai S, Tabata S, Okunaga K, Shibuya T (1996) Osteogenic differentiation of cultured marrow stromal stem cells on the surface of bioactive glass ceramics. J Biomed Mater Res 32:341–348
Hirose M, Kotobuki N, Machida H, Uchimura E, Ohgushi H (2003) Quantitative monitoring of in vitro mineralization process using fluorescent dyes. Key Eng Mater 240:715–718
Ohgushi H, Goldberg VM, Caplan AI (1989) Heterotopic osteogenesis in porous ceramics induced by marrow cells. J Orthop Res 7:568–578
Shimaoka H, Dohi Y, Ohgushi H, Ikeuchi M, Okamoto M, Kudo A, Kirita T, Yonemasu K (2004) Recombinant growth/differentiation factor-5 (GDF-5) stimulates osteogenic differentiation of marrow mesenchymal stem cells in porous hydroxyapatite ceramic. J Biomed Mater Res A 68:168–176
Noshi T, Yoshikawa T, Ikeuchi M, Dohi Y, Ohgushi H, Horiuchi K, Sugimura M, Ichijima K, Yonemasu K (2000) Enhancement of the in vivo osteogenic potential of marrow/hydroxyapatite composites by bovine bone morphogenetic protein. J Biomed Mater Res 52:621–630
Katz AJ, Llull R, Hedrick MH, Futrell JW (1999) Emerging approaches to the tissue engineering of fat. Tissue Eng 26:587–603
Miyazaki T, Kitagawa Y, Toriyama K, Kobori M, Torii S (2005) Isolation of two human fibroblastic cell populations with multiple but distinct potential of mesenchymal differentiation by ceiling culture of mature fat cells from subcutaneous adipose tissue. Differentiation 73:69–78
Sakaguchi Y, Sekiya I, Yagishita K, Muneta T (2005) Comparison of human stem cells derived from various mesenchymal tissues: superiority of synovium as a cell source. Arthritis Rheum 52:2521–2529
De Ugarte DA, Morizono K, Elabarbary A, Alfonso Z, Zuk PA, Zhu M, Dragoo JL, Ashjian P, Thomas B, Benhaim P, Chen I, Fraser J, Hedrick MH (2003) Comparison of multi-lineage cells from human adipose tissue and bone marrow. Cells Tissues Organs 174:101–109
Gun-II I, Shin YW, Lee KB (2005) Do adipose tissue–derived mesenchymal stem cells have the same osteogenic and chondrogenic potential as bone marrow–derived cells? Osteoarthritis Cartilage 13:845–853
Yoshimura H, Muneta T, Nimura A, Yokoyama A, Koga H, Sekiya I (2007) Comparison of rat mesenchymal stem cells derived from bone marrow, synovium, periosteum, adipose tissue, and muscle. Cell Tissue Res 327:449–462
Sugiura F, Kitoh H, Ishiguro N (2004) Osteogenic potential of rat mesenchymal stem cells after several passages. Biochem Biophys Res Commun 316:233–239
Acknowlegements
This work was supported by grants from the New Energy and Industrial Technology Development Organization, which was established by the Japanese government in 1980.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hayashi, O., Katsube, Y., Hirose, M. et al. Comparison of Osteogenic Ability of Rat Mesenchymal Stem Cells from Bone Marrow, Periosteum, and Adipose Tissue. Calcif Tissue Int 82, 238–247 (2008). https://doi.org/10.1007/s00223-008-9112-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-008-9112-y