
Abstract
Mature osteoclasts and their precursors are notoriously difficult to transfect using nonviral approaches, a limitation that represents a major technical obstacle in the study of osteoclast biology. Here, we describe a simple electroporation method using Amaxa® Nucleofector technology that results in efficient transfection of human blood-derived osteoclast precursors, which can be differentiated in subsequent culture to generate mature osteoclasts that retain expression of the transgene. Moreover, since these osteoclasts maintain the ability to resorb dentine, this technique could prove useful for assessing the role of specific genes/proteins in osteoclast function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Mature osteoclasts and their precursors are notoriously difficult to transfect using conventional lipid-based or electroporation approaches. This limitation represents a major technical obstacle in the study of osteoclast biology since the highly specialized nature of osteoclasts often does not permit extrapolation of data from other cell types. Although commercially available lipid-based transfection approaches have been successful in transfecting osteoclasts with siRNA [1, 2], these techniques are ineffective at delivery of dsDNA [3]. The only reliable method described that can efficiently transfect osteoclasts involves the use of virus-based delivery techniques such as adenovirus [4, 5], but this can be both time-consuming and technically demanding. Furthermore, there are concerns that adenovirus alone can activate signaling pathways (e.g., nuclear factor κB [NF-κB] [6]) that might affect osteoclasts. Electroporation techniques have been used to successfully transfect macrophage cell lines [7], and more recently the development of nucleofection (Amaxa® Biosystems, Cologne, Germany), an electroporation method that delivers the DNA directly into the nuclei, has enabled the successful transfection of not only macrophages [8] but also primary and terminally differentiated [9–11] cells since this technique does not rely on mitosis for entry of the DNA into the nucleus. As osteoclasts (and their direct precursors) are postmitotic cells derived from the same precursors as monocytes/macrophages, nucleofection approaches may also be suitable for transfection of osteoclasts.
Here, we describe an electroporation method using Amaxa® Nucleofector technology that allows efficient transfection of human blood-derived osteoclast precursors, which can be stimulated to differentiate in subsequent culture to generate transfected osteoclasts. Since the transfected osteoclasts maintain the ability to resorb dentine, this technique could prove useful for modulating osteoclast behavior and for assessing the role of specific proteins in osteoclast function, particularly at the single-cell level.
Materials and Methods
Isolation of Human Osteoclast Precursors
Venous blood was obtained from healthy volunteers using 9 mL ethylenediaminetetraacetic acid (EDTA)-coated Vacutainer® tubes (Becton Dickinson, Mountain View, CA). The collected blood was diluted 1:1 with phosphate-buffered saline (PBS) and layered over Lymphoprep™ (Axis-Shield, Kimbolton, UK) before centrifugation at 800 × g for 30 minutes with brake set to zero. Peripheral blood mononuclear cells (PBMCs) were removed from the plasma/lymphoprep interface using a 3 mL Pasteur pipette and diluted with PBS (at least twice the volume of the cell suspension) before centrifugation at 300 × g for 10 minutes. After repeating this wash step with PBS, the PBMCs were diluted to 1.5 to 2 × 106 cells/mL in α-minimal essential medium (MEM; +10% fetal calf serum [FCS], 100 U/mL penicillin, 0.1 mg/mL streptomycin, 2 mM L-glutamine) containing 20 ng/mL recombinant human macrophage colongy-stimulating factor (rhMCSF; R&D Systems, Minneapolis, MN) and seeded in 75 cm2 flasks (10 mL/flask). On day 3, the flasks were rinsed twice with PBS to remove nonadherent cells before replenishing with fresh medium containing rhMCSF.
RANKL Pretreatment and Cell Harvesting
Once the monocyte cultures reached 80–90% confluence (approximately 6 days), the medium was additionally supplemented with 100 ng/mL recombinant human receptor activator of NF-κB ligand (rhRANKL; Peprotech, London, UK) and the culture continued for a further 48 hours prior to transfection. Cells were then detached by incubation with 1 mg/mL trypsin for 15 minutes, transferred to a centrifuge tube, and fresh medium containing FCS was added to neutralize the trypsin. Since only approximately 50% of the adherent osteoclast precursors detached after this time, an additional 15-minute trypsin incubation was carried out, during which time the first batch of detached cells was kept on ice. To ensure removal of all cells, flasks were gently scraped after addition of fresh medium to the flask. Cell suspensions were pooled and centrifuged at 300 × g for 5 minutes.
Electroporation
Pelleted cells were resuspended in fresh medium and counted, then sufficient cells for all transfections (1 × 106 cells per transfection) were transferred to a fresh tube and centrifuged at 200 × g for 3 minutes. Cells were also retained from the cell suspension for use as untransfected controls (kept on ice while the other cells were transfected). After carefully removing the supernatant, the cell pellet was resuspended in 100 μL Mouse Macrophage Nucleofector Solution (Amaxa Biosystems) per transfection, then 100 μL was added to 2 μg of prealiquoted, endotoxin-free plasmid DNA (pmaxGFP [Amaxa Biosystems] or enhanced green fluorescent protein-conjugated Rab6 (EGFP-Rab6), a kind gift from Prof. Miguel Seabra, Imperial College London). The solution was added to Amaxa® electrode cuvettes and electroporated in an Amaxa® Nucleofector II using program Y-010. Immediately afterward, cells were diluted in 2.5 mL α-MEM (supplemented with 20% FCS, 100 U/mL penicillin, 0.1 mg/mL streptomycin, 2 mM L-glutamine) at room temperature (4 × 105 cells/mL), then cooled on ice for 5-10 minutes prior to supplementing with 20 ng/mL rhMCSF and 100 ng/mL rhRANKL. Cells were then seeded onto dentine slices in 96-well plates (4 × 104/well) and onto 9 mm glass coverslips in 48-well plates (1 × 105/well). Nontransfected control cells were seeded at half of this density. Medium was replaced after 48 hours with α-MEM (+10% FCS, 100 U/mL penicillin, 0.1 mg/mL streptomycin, 2 mM L-glutamine) containing 20 ng/mL rhMCSF and 100 ng/mL rhRANKL and every 3 days thereafter.
Visualization of Transfected Cells
Cells plated on glass coverslips or dentine slices were fixed at the indicated time points using 4% formaldehyde in PBS for 10 minutes, then permeabilized by incubation with 0.2% Triton X-100 in PBS for 15 minutes. Nuclei were counterstained by incubating with 0.1 μg/mL 4′,6-diamidino-2-phenylindole (DAPI) for 10 minutes, while in some cases the Golgi apparatus was stained by incubation with 2 μg/mL wheat germ agglutinin (WGA)-Alexa Fluor 633 conjugate (Invitrogen, La Jolla, CA) for 20 minutes. In cells cultured on dentine discs, F-actin was stained by incubation with 0.5 μg/mL tetramethylrhodamine isothiocyanate (TRITC)-phalloidin for 20 minutes, while the dentine surface was visualized by incubating for 20 minutes with a fluorescently conjugated bisphosphonate, ALN-AF633 (20 μM) [12], which has a high affinity for mineralized surfaces (our unpublished data). Following staining, coverslips and dentine discs were rinsed three times with PBS, then mounted on glass slides in VectaShield (Vector Labs, Burlingame, CA). Images were captured by light microscopy using a Zeiss (Oberkochen, Germany) Axiovert microscope equipped with an Optronics charge-coupled device camera and by confocal laser scanning microscopy using the Zeiss LSM 510 META system.
Assessment of Cell Viability and Transfection Efficiency
Nontransfected cells, sham-transfected cells (electroporated without DNA), and pmaxGFP-transfected cells were seeded into 96-well plates at 3 × 104/well in 100 μL (replicates of six). At 24 hours after transfection, 10 μL alamar blue (Biosource, Camarillo, CA) was added to each well and the culture continued for 3–4 hours before reading the fluorescence on a Biotek (Franklin Lakes, NJ) plate reader (emission 530 nm, excitation 590 nm). The cells were then fixed in 4% formaldehyde for 10 minutes and washed with PBS. Digital images (fluorescent and phase contrast merges) were taken of a random area within each well and transfected and untransfected cells manually counted to determine transfection efficiency. Data are expressed as the mean ± standard deviation (SD, n = 3).
Immunostaining for Rab6
Mature osteoclasts were isolated from neonatal rabbit bones as described previously [13] and seeded onto glass coverslips, cultured for 48 hours, then fixed in 4% formaldehyde. Cells were permeabilized by incubation with 0.2% Triton X-100 for 20 minutes, blocked with 10% FCS in PBS for 30 minutes, then incubated with 1:70 rabbit polyclonal Rab6 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) in PBS/5% FCS for 1 hour. Cells were washed five times in PBS, incubated with 1:200 Alexa Fluor 594-anti-rabbit immunoglobulin G for 1 hour, washed extensively again in PBS, then counterstained with WGA (as described above) before mounting in VectaShield and analysis by confocal microscopy.
Results
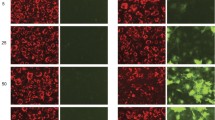
The Amaxa® nucleofection procedure resulted in successful transfection of human osteoclast precursors (which had been exposed to RANKL for 48 hours prior to transfection), with substantial numbers of mononuclear cells expressing high levels of GFP within 24 hours of transfection (Fig. 1A). Within 3 days of transfection, numerous GFP-expressing multinucleated osteoclasts had formed, and GFP expression persisted until 7 days posttransfection, at which stage most cells in the culture were multinucleated osteoclasts (Fig. 1A). The transfection procedure was also carried out on the M-CSF-dependent monocytes prior to exposure to RANKL, but little difference in transfection efficiency was found between these two approaches (data not shown). Since the cells require several days’ culture in RANKL to form mature osteoclasts, transfection of osteoclast precursors that have already begun to differentiate (by prior exposure to RANKL for 48 hours) is a better approach as this reduces the subsequent culture time required to generate osteoclasts.

Generation of osteoclasts from transfected precursors. (A) RANKL-pretreated cells were transfected with pmaxGFP, seeded onto glass coverslips, and cultured further with RANKL-supplemented medium. At 24 hours, 3 days, and 7 days posttransfection, cells were fixed and stained with DAPI (false colored red). Scale bars = 100 μm. (B) Cell viability 24 hours posttransfection was determined by alamar blue assay and expressed relative to nontransfected (NT) cells (mean ± SD, n = 3).
In order to assess the effect of nucleofection on cell viability, alamar blue assays were conducted on nontransfected, sham-transfected (no DNA), and pmaxGFP-transfected cells. A considerable loss of cell viability (∼45%) occurred in the transfected cells compared to the nontransfected cells, while electroporation alone (in the absence of dsDNA) resulted in a smaller but consistent loss of viability (∼20%, Fig 1B). However, the loss of viability resulting from transfection was easily compensated for, simply by seeding these cells at twice the density of the nontransfected control cells. Moreover, there was little difference in the ability of the transfected and nontransfected cell cultures to form osteoclasts (data not shown).
Although the transfection efficiency of osteoclast precursors at 24 hours was only 19.9% (±6.5, n = 3), this improved with further culture (>3 days) due to the fusion of transfected and nontransfected precursors to form GFP-expressing osteoclasts, such that typically 40% of the osteoclasts expressed GFP (Fig. 1A). However, up to 80% transfected osteoclasts were observed in some experiments, which presumably reflects the innate variability between cultures from different donors.
The fundamental characteristic that defines an osteoclast is the ability to resorb calcified bone surfaces. To assess whether transfected osteoclasts retained this functional property, osteoclast precursors transfected with pmaxGFP were seeded onto discs of dentine (a mineralized tissue very similar in composition to bone) in medium containing sRANKL and rhMCSF. By day 4 posttransfection, GFP-expressing multinucleated cells had formed, polarized (indicated by the presence of an F-actin ring), and begun to show evidence of resorption, demonstrated by the small resorption pits associated with transfected osteoclasts (Fig. 2A, arrowheads). By day 7 posttransfection, osteoclasts maintained expression of GFP (as they had when cultured on glass coverslips) and had resorbed much larger pits. Moreover, these cells continued to resorb and showed evidence of migration, demonstrated by the resorption front within crescent-shaped F-actin rings and the presence of resorption trails (Fig. 2B and C). This transfection approach is therefore ideal for analyzing the localization and function of individual proteins in resorbing human osteoclasts at the single-cell level.

GFP-transfected osteoclast precursors can differentiate into functional osteoclasts. Osteoclast precursors were transfected with pmaxGFP and seeded onto dentine slices. After 4 days (A) and 7 days (B and C), dentine slices were fixed, then F-actin and the dentine surface visualized by staining with TRITC-phalloidin (red) and ALN-AF633 (blue), respectively, prior to confocal microscopy. A and B are 1 μm xy optical sections at the dentine surface; C is a z-series showing zx and zy sections and an xy extended focus projection of an individual resorbing osteoclast. Images are from separate cultures. Arrowheads indicate sites of resorption. Scale bars = 50 μm.
To confirm that the nucleofection procedure is suitable for use with other expression constructs, e.g., those encoding GFP-fusion proteins, osteoclast precursors were transfected with EGFP-Rab6. Rab6 is a member of the Rab family of small guanosine triphosphatases that localizes to the Golgi and plays a role in intra-Golgi transport [14]. Figure 3A shows that EGFP-Rab6 is expressed in multinucleated osteoclasts 4 days posttransfection and is correctly localized to the perinuclear Golgi compartment since it colocalizes completely with the Golgi marker WGA (which binds to glycosylated proteins in the Golgi). This localization is identical to that of endogenous Rab6 in rabbit osteoclasts, detected by immunostaining (Fig. 3B). This confirms not only that transgene delivery and expression was successful but also that the protein product is correctly prenylated, a posttranslational modification that is essential for the specific subcellular localization and function of Rab proteins [15].

Expression and localization of EGFP-Rab6 in transfected osteoclasts. (A) Human osteoclast precursors were transfected with a plasmid encoding EGFP-Rab6 and cultured in medium containing RANKL and rhMCSF for 4 days. Cells were then fixed and stained with WGA to identify the Golgi apparatus. Images show a nontransfected cell adjacent to the EGFP-Rab6-expressing cell. (B) Rabbit osteoclasts were immunostained for Rab6 and counterstained with WGA. Colocalization of WGA and Rab6 can be seen in osteoclasts with either endogenous Rab6 (B) or EGFP-Rab6 (A). Scale bars = 20 μm.
Discussion
In summary, we have developed a novel electroporation method using Amaxa® nucleofection for expressing transgenes in resorbing human osteoclasts. This technique offers an attractive alternative to adenoviral transfection since it has the advantages of being very simple (requiring only plasmid DNA) and extremely quick. Although the transfection efficiency is not as high as reported with viral approaches, this method nevertheless could prove enormously useful, particularly for single-cell analysis, to allow the study of specific proteins in osteoclastic bone resorption, and hence for elucidating further aspects of osteoclast biology.
References
Selinger CI, Day CJ, Morrison NA (2005) Optimized transfection of diced siRNA into mature primary human osteoclasts: inhibition of cathepsin K mediated bone resorption by siRNA. J Cell Biochem 96:996–1002
Kim MS, Day CJ, Selinger CI, Magno CL, Stephens SRJ, Morrison NA (2006) MCP-1-induced human osteoclast-like cells are tartrate-resistant acid phosphatase, NFATc1, and calcitonin receptor-positive but require receptor activator of NFκB ligand for bone resorption. J Biol Chem 281:1274–1285
Laitala-Leinonen T (2005) Unsatisfactory gene transfer into bone-resorbing osteoclasts with liposomal transfection systems. J Negat Results Biomed 4:5
Tanaka S, Takahashi T, Takayanagi H, Miyazaki T, Oda H, Nakamura K, Hirai H, Kurokawa T (1998) Modulation of osteoclast function by adenovirus vector-induced epidermal growth factor receptor. J Bone Miner Res 13:1714–1720
Bruzzaniti A, Neff L, Sanjay A, Horne WC, De Camilli P, Baron R (2005) Dynamin forms a Src kinase–sensitive complex with Cbl and regulates podosomes and osteoclast activity. Mol Cell Biol 16:3301–3313
Morelli AE, Larregina AT, Ganster RW, Zahorchak AF, Plowey JM, Takayama T, Logar AJ, Robbins PD, Falo LD, Thomson AW (2000) Recombinant adenovirus induces maturation of dendritic cells via an NF-κB-dependent pathway. J Virol 74:9617–9628
Thompson CD, Frazier-Jessen MR, Rawat R, Nordan RP, Brown RT (1999) Evaluation of methods for transient transfection of a murine macrophage cell line, RAW 264.7. Biotechniques 27:824–826
Marriott HM, Bingle CD, Read RC, Braley KE, Kroemer G, Hellewell PG, Craig RW, Whyte MK, Dockrell DH (2005) Dynamic changes in Mcl-1 expression regulate macrophage viability or commitment to apoptosis during bacterial clearance. J Clin Invest 115:359–368
Iversen N, Birkenes B, Torsdalen K, Djurovic S (2005) Electroporation by nucleofector is the best nonviral transfection technique in human endothelial and smooth muscle cells. Genet Vaccines Ther 3:2
Djurovic S, Iversen N, Jeansson S, Hoover F, Christensen G (2004) Comparison of nonviral transfection and adeno-associated viral transduction on cardiomyocytes. Mol Biotechnol 28:21–31
Maasho K, Marusina A, Reynolds NM, Coligan JE, Borrego F (2004) Efficient gene transfer into the human natural killer cell line, NKL, using the Amaxa nucleofection system. J Immunol Methods 284:133–140
Thompson K, Rogers MJ, Coxon FP, Crockett JC (2006) Cytosolic entry of bisphosphonate drugs requires acidification of vesicles after fluid-phase endocytosis. Mol Pharmacol 69:1624–1632
Coxon FP, Helfrich MH, Van’t Hof R, Sebti S, Ralston SH, Hamilton A, Rogers MJ (2000) Protein geranylgeranylation is required for osteoclast formation, function, and survival: inhibition by bisphosphonates and GGTI-298. J Bone Miner Res 15:1467–1476
Martinez O, Schmidt A, Salamero J, Hoflack B, Roa M, Goud B (1994) The small GTP-binding protein rab6 functions in intra-Golgi transport. J Cell Biol 127:1575–1588
Novick P, Zerial M (1997) The diversity of Rab proteins in vesicle transport. Curr Opin Cell Biol 9:496–504
Acknowledgement
This work was supported by an Oliver Bird Rheumatism Programme studentship grant awarded to A. T.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Taylor, A., Rogers, M.J., Tosh, D. et al. A Novel Method for Efficient Generation of Transfected Human Osteoclasts. Calcif Tissue Int 80, 132–136 (2007). https://doi.org/10.1007/s00223-006-0245-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-006-0245-6