
Abstract
Osteocyte apoptosis caused by load-induced microdamage is followed by osteoclastic bone remodeling, and a causal link between apoptosis and repair has been suggested. The objectives of the present study were to use a chick model to examine the incidence of osteocyte apoptosis and the presence of osteoclasts during the first 96 hours following an osteotomy, prior to extensive callus mineralization. Osteotomies were performed on the right radii of 24 chicks at 23–24 days of age. The left radii served as controls. Radii were collected and processed at six time points following surgery (0, 12, 24, 48, 72, and 96 hours). Decalcified bone tissue sections were stained either for apoptosis using a modified TUNEL procedure or for tartrate-resistant acid phosphatase to identify osteoclasts in the intracortical and periosteal envelopes. The percentage of apoptotic osteocytes, as well as osteoclast counts (n/mm or n/mm2) were quantified in four regions (0–1, 1–2, 2–4, and 4–8 mm from the site of the osteotomy; regions 1–4, respectively) in the osteotomized radii and in the same measured areas in the control radii. Data for osteocyte apoptosis and osteoclasts in the control limb were subtracted from the osteotomized limb data to identify differences due to surgical influence. The incidence of osteocyte apoptosis was significantly higher at 12, 24, 48, and 72 hours versus 0 hours following osteotomy, and the response was highest in region 1; however, there was no interaction between time and region. Intracortical osteoclast counts (n/mm2) were elevated after 48 hours, and the response was similar in all regions. The data demonstrate that osteocyte apoptosis occurs within 24 hours in response to an osteotomy and temporally precedes an increase in osteoclast presence. Hence, osteocyte apoptosis may play a role in signaling during the bone healing process.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Fracture healing consists of complex cellular and intercellular responses, which control and mediate the repair process. Thousands of genes, both known and novel, are activated during the fracture healing process [1]. An increased understanding of the factors regulating bone repair at the cellular level will provide opportunities to influence the healing process. The general functions of most bone cells in bone repair are understood; however, the contribution of osteocytes to bone healing has not been extensively examined.
Osteocytes are the most plentiful cell in skeletal tissue [2], and until recently it was believed that they were relatively dormant. Osteocyte response to various mechanical stimuli has been demonstrated both in vivo and in vitro. Mechanical loads in vivo have increased the number of osteocytes expressing messenger RNA for osteopontin [3] and insulin-like growth factor I [4], as well as the number of osteocytes with prostaglandin endoperoxide H synthases 1 and 2 [5]. Cultured osteocytes increased release of prostaglandin E2 [6, 7] and nitric oxide [8, 9] when stimulated mechanically. Osteocytes also respond to hormonal influence. Estrogen withdrawal, via ovariectomy, resulted in an increased incidence of osteocyte apoptosis in rat cortical and trabecular bone but was prevented by estrogen replacement [10].
Cell apoptosis was described by Kerr et al. [11] as a type of regulated cell death. Osteocyte apoptosis has been observed in sites of bone turnover, such as in tissue of young mice, rats, and hamsters near areas where bone is being resorbed [12]. Regions of bone turnover in human infant bones contained apoptotic osteocytes [13, 14], whereas regions in adult bones without bone turnover contained very few [13]. Osteocyte apoptosis has also been observed following fatigue load-induced microdamage [15, 16] and prior to intracortical remodeling [16], and the authors hypothesized that the cells are directly involved in microdamage repair. It is thus possible that osteocyte apoptosis is directly involved in bone fracture repair.
A chick model for bone repair was utilized in the present study to explore the involvement of osteocytes in bone healing by examining the temporal events of osteocyte apoptosis and the appearance of osteoclasts during the first 96 hours following osteotomy. The short time frame of the study was based on a previous study using peripheral quantitative computed tomography (pQCT), where rapid formation of a mineralized callus following right radial midshaft osteotomy in a 24–day-old chick model was observed. Cortical bone mineral content and area of the developing callus increased approximately 100% at 5–10 days postsurgery, beginning approximately 4–5 days following osteotomy [17]. The objective of the present study was to determine cellular response (osteocyte apoptosis and osteoclast presence) prior to extensive callus mineralization; therefore, a 0–4 day time period was chosen with tissue sampling at 0, 12, 24, 48, 72, and 96 hours following osteotomy. The sequences of appearance of apoptotic osteocytes and osteoclasts were determined in four regions extending 0–8 mm away from the osteotomy site using histological sections. Based on the work of Verborgt et al. [15] and Noble et al. [16], it was hypothesized that the incidence of osteocyte apoptosis would be elevated within 24 hours of osteotomy and that this would be followed by an increase in the number of osteoclasts.
Materials and Methods
Animals
The study was conducted in two stages, approximately 2 months apart. For each stage, newly hatched male white Leghorn chicks (Hy-Line W36, n = 50 for the first stage and 30 for the second stage; Hy-Line International, Spencer, IA) were used. They were placed into a heated battery brooder with raised wire floors and provided feed (University of Wisconsin Chick Mash) and water ad libitum. Chicks were provided a constant lighting program (24 hours light:0 hours dark) for the first 3–4 days and then switched to a 12 hours light:12 hours dark program to entrain circadian rhythms. All animal procedures were approved by the University of Wisconsin-Madison College of Agriculture and Life Sciences Animal Care and Use Committee.
For each of the two stages, chicks were weighed at 22 days of age, and 12 chicks were randomly chosen for surgery from a group of birds with similar body weights. Surgeries at each stage occurred over 2 days (six birds/day). On each day, a group of six birds (23 or 24 days old) was fasted (2–4 hours) and weighed prior to surgery (mean ± standard deviation [SD] 198 ± 6 g, n = 24). Osteotomies were performed on the midshaft of the right radii of 24 chicks, six chicks per day, 2 days for each stage. The left radii were left untouched to serve as controls.
Butorphanol (2 mg/kg, 0.04 mL intramuscular [IM]) was administered prior to anesthesia for preemptive analgesia. Normal surgical procedures were used. A Dremel® tool (Robert Basch Tool Corporation, Racine, WI) with a cutting wheel (#20, 1 mm thick, 7 mm diameter) was used to cut most of the way through the radius (<1 second). The last segment was severed using scissors in order to avoid soft tissue damage. The osteotomized bone was aligned axially, and the skin was closed. Fixation was not used in order to mimic the previous study where it would have interfered with pQCT scans [17]. Hence, interfragmentary displacement caused an overlap of osteotomized ends. The ulna was left intact to provide an internal splint and thus minimize pain. Both wings were placed in figure-eight bandages postoperatively so that any effect of wing immobilization on apoptosis would also be accounted for in the control radii. Bandages were left on for approximately 48 hours. Hematomas, present during the initial phase of fracture healing, were observed but not measured or monitored. Birds were put into separate pens within the battery brooder following surgery to prevent collateral damage of the surgery site. Placement in the battery was in a manner that allowed visual contact in order to minimize isolation stress.
Butorphanol (2 mg/kg, 0.04 mL IM) was administered for analgesia at 4 and 8 hours following surgery. Birds were monitored for pain following the surgery every 3–4 hours and scored using a numerical rating scoring (NRS) method (Table 1) adapted from that used for pigeon studies (J. Paul-Murphy, unpublished data). The NRS method, along with subjective observations, was used to determine if additional pain medication was required (if NRS scores were >5). Additional pain medication was not required, based on NRS scores of 0 or 1. After two observation periods without medication had passed, birds were returned to a once-daily observation schedule. All birds received the same amount of butorphanol, except those killed at 0 hours.
Birds were killed at six time points following surgery using Beuthanasia® (Schering-Plough Animal Health Corp., Union, NJ) (0.1 ml intravenous [IV], containing 390 mg/mL pentobarbital and 50 mg/ml phenytoin): 0, 12, 24, 48, 72, and 96 hours. These time points allowed tissue collection prior to extensive callus mineralization [17]. Osteotomized radii were collected and the distal radial fragments kept for analysis. A 1–2 mm piece of the proximal fragment, including muscle tissue, was left attached to the distal fragment in order to reduce disruption of the osteotomy site. For each bird, the control radius was cut at the same measured location as the osteotomized radius in order to minimize site-specific variation between the osteotomized and control radii.
Tissue Processing
Radial fragments were processed according to recommendations by Hao et al. [18]. Bone tissue fragments were placed into cold Zamboni’s fixing solution [19] (phosphate-buffered paraformaldehyde and picric acid, pH 7.3) for 24–48 hours (4°C). The bone tissues were then placed individually into tissue cassettes, suspended in a 20% ethylenediaminetetraacetic acid (EDTA)-5% sucrose-0.1M Tris solution for decalcification. Samples were kept at 4°C and agitated during decalcification. EDTA solution was changed every 2–3 days. Decalcification took 6–7 days and was confirmed using radiography (26 kVp for 2 minutes; Hewlett-Packard Faxitron model 43855A, McMinnville, OR). Following decalcification, bone tissues were placed into phosphate-buffered saline (PBS) for at least 2 hours and then transferred to a 30% sucrose/PBS solution for storage (4°C) prior to cryosectioning and analysis.
Longitudinal sections (10 μm) from the control and osteotomized radii were obtained using a cryostat (Microm Vacutome HM500 Micron International GmbH, Walldorf, Germany). Two sections were placed on each slide (ProbeOnTM Plus; Fisher Scientific, Pittsburgh, PA). Slides were dried and stored until staining.
Following staining for apoptosis and tartrate-resistant acid phosphatase (TRAP) (described below), sections were examined using a Nikon (Tokyo, Japan) Eclipse E600 microscope connected to a Sony (Tokyo, Japan) color video camera (3CCD DSP, model DXC-390). Fields of view were saved, and cell counts and bone tissue areas were analyzed using Scion Image for Windows software (Scion, Frederick, MD; release Beta 4.0.2). All analyses were completed by one investigator (W.D.C.).
A previous osteotomy study estimated that the maximum callus length of the healing radius was 15 mm [17]. Therefore, approximately half of this length was selected for analysis in the distal radial fragments in this study. Data were collected from four regions extending away from the osteotomy site (or dissection cut in the control radii). The regions of measurement were set as: 0–1, 1–2, 2–4, and 4–8 mm from the osteotomy site (regions 1–4, respectively, Fig. 1).

Diagram of osteotomy and region sites where data were sampled in longitudinal bone tissue sections. Regions were measured and marked on the coverslips using a dissecting microscope. For TUNEL-stained sections, regions 1 and 2 were sampled in their entirety (as many fields of view were saved as necessary to include the whole region); in regions 3 and 4, six and four fields of view were randomly sampled, respectively. For TRAP-stained sections, all regions were sampled in their entirety. Published with permission [38].
Staining for Apoptosis
A modified terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) procedure was used to stain sections for apoptotic osteocytes. The TdT-FragEL™ DNA Fragmentation Detection Kit (Oncogene Research Products, Boston, MA) was used to stain two slides per radius for apoptosis. The kit used terminal deoxynucleotidyl transferase (TdT) to attach biotin-labeled and unlabeled nucleotides to the DNA fragments, which are present during cell apoptosis. A streptavidin-horseradish peroxidase conjugate was used in conjunction with diaminobenzidine to give the reaction an insoluble color precipitate. Cells positive for apoptosis stain brown. Staining procedures for negative controls eliminated the TdT enzyme during the labeling step. Positive control slides from the kit were also used.
The kit directions were followed with the following modifications. PBS was used instead of Tris-buffered saline for the rehydration and rinsing/soaking steps. The labeling mixture was used to dilute the TdT enzyme an additional 1:16 in order to reduce background staining, as recommended by the manufacturer. The incubation step with the blocking buffer was increased from 10 to 20 minutes and the incubation with diaminobenzidine reduced to 5 minutes to reduce the amount of background staining. Erlich’s hematoxylin was used as the counterstain rather than methyl green.
Data were obtained for TUNEL-positive osteocytes, total osteocytes, and cortical bone areas at ×400 magnification (empty lacunae were not counted). Data for each region were summed, and the percentage of TUNEL-positive osteocytes was calculated [(number of TUNEL-positive osteocytes/number of total osteocytes) * 100]. Data were collected from two sections per radius and then averaged to give a single data point for each of the regions within each radius.
Regions 1 and 2 were sampled in their entirety (fields of view saved covered the whole region), and regions 3 and 4 were subsampled. The number of fields of view to be subsampled from regions 3 and 4 was determined by the moving average method used by Vashishth et al. [20]. Data from regions 3 and 4 of six tissue sections (one osteotomized radius per time point) were randomized (% TUNEL-positive osteocytes, n/mm2 TUNEL-positive osteocytes, and n/mm2 total osteocytes). A running average was calculated (the mean kept incorporating the next data point into the expanded data set). The stabilization of these means indicated that six and four randomly chosen fields of view would adequately represent regions 3 and 4, respectively.
TRAP Staining
TRAP staining was used to identify osteoclasts in the bone tissue. Two slides per radius were stained for TRAP within a week of cryosectioning, using methods described by Muir et al. [21]. Briefly, slides were incubated for 2–3 hours (37°C) in a reaction mixture containing naphthol AS-BI phosphate as the substrate. Negative controls were stained using the same solution without the substrate. Slides were counterstained using alcian blue. TRAP-positive cells stained red.
Data were obtained for intracortical and periosteal osteoclast (TRAP-positive cells) counts using ×100 and ×200 magnification. Periosteal lengths (mm) and cortical bone areas (mm2) were measured using ×100 magnification. All regions were examined in their entirety for osteoclast counting. Calculations from the summed data for each region gave the number of intracortical osteoclasts per millimeter squared of cortical bone and the number of osteoclasts per millimeter of periosteal length. Data collected for marrow and endocortical TRAP-positive cells were inadequate for statistical analysis due to folding or missing tissue. Data from two sections per radius were averaged to give a single data point for each of the regions within each radius.
Coefficients of Variation
To test the precision of data collection, coefficients of variation (CVs) were calculated from six fields of view for TUNEL-stained slides and for area and length measurements on TRAP-stained slides. Two complete sections were used to calculate CVs for periosteal and intracortical osteoclast cell counts. CV calculations were based on four sampling times, 4–21 days apart. CVs for TUNEL-positive and total osteocyte cell counts were 11.1% and 2.5%, while periosteal and intracortical osteoclast counts were 8.1% and 11.2%, respectively. CVs for area measurements for TUNEL- and area and length measurements for TRAP-stained sections were 0.5%, 1.0%, and 0.8%, respectively.
Statistical Analyses
A total of 24 birds underwent surgery. The four surgery days were treated as blocks. Data for percent TUNEL-positive osteocytes and osteoclast counts (n/mm, n/mm2) were adjusted for the general effect of surgery by subtracting values for the control radii from those for osteotomized radii (adjusted data). Because the adjusted data did not follow normal distributions, these differences were ranked. The ranked data were then analyzed as a split-plot using Proc Mixed procedures of SAS Version 8.2 software (SAS Institute, Cary, NC, 1999–2001), with time and block in the whole plot and region and region*time in the subplot. Time refers to the number of hours following surgery before the tissue was collected. Region and time were treated as fixed effects. Block and bird (time*block) were treated as random variables. The mid-region distance from the osteotomy site was treated as a repeated measure to account for autocorrelation between regions.
Ranked data for the control radii were analyzed as a split-plot using the methods described above in order to discover if there were effects of surgery on the control limb. Additionally, ranked data for the 0-hour time group were analyzed alone to compare the osteotomized to control radii in the regions (two-way analysis of variance [ANOVA]). A relationship between percent TUNEL-positive osteocytes and intracortical osteoclasts (n/mm2) postsurgery was investigated for each region using the ranked adjusted data (data for time 0 not included). Percent TUNEL-positive osteocytes was used as a covariate to analyze intracortical osteoclasts using one-way ANOVA with time as the main effect. Testing the relationship using a covariate analysis allowed the blocking and time effects to remain in the model.
Data are presented as raw means from the original data set plus or minus standard errors, with least significant difference post hoc analyses where appropriate on the ranked data.
Results
No postoperative complications were observed. None of the chicks required additional pain medication beyond the standard three doses of butorphanol. NRS scores at postoperative observation periods were 0 in all cases except that two birds at 8 hours postsurgery had a score of 1. Subjective observations also failed to note any cases of bird discomfort. A minor amount of pecking at their own bandages was observed by some birds on both the control and the osteotomized wings. All chicks were active, consumed feed, and gained weight after surgery. Body weights at the time of death averaged 220 ± 13 (SD) g for the 24 birds (a gain of approximately 11% during the postoperative period).
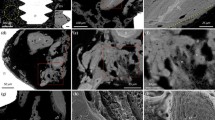
In Table 2, the differences between the osteotomized radii and the control radii are represented by the terms Tundif (apoptotic osteocytes, percent TUNEL-positive osteocytes in osteotomized radii minus control radii), Cortdif (n/mm2 intracortical osteoclasts in osteotomized radii minus control radii), and Peridif (n/mm periosteal osteoclasts in osteotomized radii minus control radii). The differences between osteotomized and control radii were analyzed to account for any effects of surgery and baseline cell activity. Examples of TUNEL and TRAP staining are illustrated in Figure 2. TUNEL-positive osteocytes stained brown versus the blue nuclei observed in nonapoptotic cells. TRAP-positive cells (stained red) are shown intracortically and along the periosteum.

Representative images of TUNEL- and TRAP-stained sections. Top: Image showing TUNEL staining of osteocytes (image saved at × 400 magnification, scale bar = 20 μm). TUNEL-positive osteocytes stain brown (arrow points to a positive osteocyte). Middle: Image showing TRAP staining of intracortical osteoclasts (image saved at × 100 magnification, scale bar = 100 μm). TRAP-positive cells stain red (arrows point to positive osteoclasts in middle and bottom images). Bottom: Image showing TRAP staining of periosteal osteoclasts (image saved at × 100 magnification, scale bar = 100 μm). Published with permission [38].
There were significant effects of time and region on Tundif but no interaction (Table 2). The incidence of apoptosis significantly increased between 0 and 12 hours, and it remained higher than the incidence at 0 hours until 96 hours after surgery. Tundif was higher in region 1 than in regions 2–4. The effect of time appeared to be largest in region 1, where Tundif increased from near 0% at surgery (0 hours) to almost 10% within 12 hours (Fig. 3). Region 2 appeared to have a similar, although smaller, response. When data from the control radii were tested alone, there was no effect of time on the incidence of osteocyte apoptosis (Table 3). There was an effect of region, with apoptosis in region 1 approaching 4.5% versus <1% in the other regions.

Region and time differences for percent Tundif (% TUNEL-positive osteocytes in osteotomized radii minus control radii). Time by region interaction was not significant. Means ± standard errors were calculated from raw data (n = 4, except for time = 3/region = 4, where n = 3). Published with permission [38].
Cortdif was strongly affected by time (Table 2). Cortdif was low until 72 hours, when it was more than threefold higher than at 48 hours. Cortdif nearly doubled between 72 and 96 hours postsurgery, but the two times were not significantly different. Similar effects were seen in all regions (Fig. 4). When data from the control radii were analyzed, there was a significant effect of region (Table 3), with region 4 showing a higher number per millimeter squared of intracortical osteoclasts than regions 1–3.

Region and time differences for Cortdif (n/mm2 intracortical osteoclasts in osteotomized radii minus control radii). Time by region interaction was not significant. Means ± standard errors were calculated from raw data (n = 4). Published with permission [38].
Data for Peridif showed a significant effect of region, and the primary effect appeared to occur in regions 3 and 4 (Table 2, Fig. 5). The negative numbers indicate that there was a higher number of osteoclasts in the control radii than the osteotomized radii. A stronger (P < 0.0001) region effect is shown in the control radii (Table 3).

Region and time differences for Peridif (n/mm periosteal osteoclasts in osteotomized radii minus control radii). Time by region interaction P = 0.0965. Means ± standard errors were calculated from raw data (n = 4). Published with permission [38].
To ensure that baseline values were the same for osteotomized and control radii, data for 0 hours were analyzed as a 2 × 4 factorial (radii by region). There were no significant differences between the osteotomized and control radii at time 0 for percent TUNEL-positive osteocytes, intracortical osteoclasts (n/mm2), or periosteal osteoclasts (n/mm) (data not shown).
The results of covariate analysis by region indicate that the data in this experiment did not support a relationship between Tundif and Cortdif (P = 0.2668 for region 1). However, Figure 6 shows that in region 1 Tundif (%) peaked at 24 hours postsurgery and then decreased while Cortdif (n/mm2) began to increase at 48 hours and was largest at 96 hours following surgery.

Changes in Tundif (% TUNEL-positive osteocytes in osteotomized radii minus control radii) and Cortdif (n/mm2 intracortical osteoclasts in osteotomized radii minus control radii) over time for region 1. Means ± standard errors were calculated from raw data (n = 4). Published with permission [38].
Discussion
Osteocyte death following fracture may occur via apoptosis. An early description of “shrunken and degenerating” osteocytes 24 hours following fractures in rats by Enneking [22] is indicative of the morphological definition of apoptosis described by Kerr et al. [11]. Since osteocyte apoptosis has been documented following load-induced microdamage, it was hypothesized in the present experiment that osteocytes would undergo apoptosis following osteotomy. The data supported this hypothesis. Osteocyte apoptosis, adjusted for the effect of surgery (Tundif), increased from approximately 0% (4.5% in the osteotomized radii minus 5.0% in the control radii, region 1) to almost 10% by 12 hours within 1 mm of the osteotomy site. This is smaller in magnitude than that demonstrated by Verborgt et al. [15] with load-induced microdamage in adult rats. Estimations from their results suggest that 13% of the cells were TUNEL-positive at baseline and approximately 40% were TUNEL-positive near microcracks by 1 day postloading, indicating a difference of 27% in apoptosis between pre- and postload-induced microdamage. Tundif in region 1 was 12%, 7%, and 9% at 24, 48, and 72 hours, respectively, and then approached baseline levels (2%) by 96 hours following osteotomy. This was in contrast to continuing high levels of osteocyte apoptosis for 10 days near microcracks in fatigued adult rat bone [15]. Noble et al. [16] also observed a significantly higher percentage of apoptotic osteocytes in damaged versus control bones in rats 7 days postinjury (26.3 versus 3.3%, respectively). Differences in the duration and magnitude of the apoptotic response in these studies may be due to different factors of the injury, such as the hematoma and the multitude of compounds released during the inflammatory phase of fracture healing. Osteotomy damage to osteocytes may have been less extensive than fatigue damage, thereby creating a different pattern of response. Differences may also be due to species or age, although Noble et al. [16] used relatively young rats (weights ∼100 g). Alternatively, in the present study, the increase in intracortical osteoclast presence observed at 72 and 96 hours may reflect bone resorption and therefore removal of some of the apoptotic osteocytes during the beginning of the remodeling process to reshape the bone. There was no change in total osteocyte density (n/mm2) due to time (data not reported).
The largest response of osteocyte apoptosis occurred in region 1, while the response over time in region 2 appeared to be similar but to a lesser degree. Although there was no time by region interaction, there was little change in Tundif over time in regions 3 and 4. This is similar to the results of Verborgt et al. [23], where the number of TUNEL-positive osteocytes (n/mm2) was highest within 1.5 mm of the load-induced microcracks and not different from nonloaded bones out to 3.75 mm from the microcracks.
In the present study, the response of osteoclasts varied within bone envelopes (intracortical, periosteal). The largest response to the osteotomy was observed in intracortical osteoclasts (n/mm2). Cortdif (n/mm2 intracortical osteoclasts in the osteotomized radii minus control radii) was relatively low until 72 hours after surgery, when it was over threefold higher than that at 48 hours. This included region 4, where osteocyte apoptosis appeared to be essentially unaffected. Bone resorption during the early phase of bone repair has been suggested [24, 25], although these authors refer only to the resorption of dead or “nonvital” tissue. The timing of osteoclast appearance is similar to that observed with load-induced tooth movement [3], where increases in cell number per square millimeter and resorption pits were observed within 48–72 hours. Increased intracortical remodeling has also been reported to follow load-induced microdamage (0 versus 8 days) in dogs [26].
Periosteal osteoclasts appeared to be suppressed but not eliminated during bone healing in regions 3 and 4. Cell counts (n/mm) in the osteotomized radii in these two regions were approximately 25–50% of the control radii (data not shown). Osteoclast suppression may be due to cell proliferation in the periosteum during posttrauma intramembranous bone formation. Although not assessed in the present study, a periosteal response (cell proliferation) has been observed as early as 16–24 hours after fracture [22, 27, 28], and intramembranous bone formation affects the surface several millimeters from the injury site [28, 29]. Although this explanation of the periosteal response (suppression) seems reasonable, the data must be interpreted with caution. A large number of experimental units in the Peridif data set (osteotomy minus control) contained no osteoclasts and may have led to an unrealistically low variance and, hence, the significant response. It should be noted that a large number of Cortdif data points also equalled 0. However, the Cortdif results can be accepted with confidence because the cell response for time is highly significant (P < 0.0001) and the changes in cell counts (n/mm2) were of a larger magnitude than for Peridif.
The second hypothesis of this experiment was that the presence of osteoclasts would increase following osteocyte apoptosis, as demonstrated with increased remodeling following load-induced microdamage by Noble et al. [16]. Although a relationship between the two was not established statistically (possibly due to modest sample size), the presence of intracortical osteoclasts began to increase 36–60 hours after the increase in osteocyte apoptosis in region 1. This time frame is shorter than that observed by Noble et al. [16], who did not find intracortical remodeling 7 days following a microdamage-inducing load. They did find evidence of remodeling at their next time point, 14 days postloading. As indicated previously, this difference in timing could be due to the different nature of the injuries or the species used. Age is not likely a factor since Noble et al. [16] used relatively young rats. It should also be noted that although there was no significant time by region interaction for percent Tundif, the osteocyte apoptotic response was minimal in region 3 and virtually none was observed in region 4, while intracortical osteoclast presence was significantly higher by 72 hours postsurgery in all regions. It is possible that there are multiple mechanisms attracting osteoclasts following a traumatic injury such as osteotomy.
There was a significantly higher incidence of osteocyte apoptosis in the control radii in region 1 compared to regions 2–4 (4.27% versus <1%, respectively). The incidence of apoptosis observed at the 0 time point in region 1 in the osteotomized radii was also higher than expected (4.5%). Reasons for this immediate apoptotic response are unclear and can only be hypothesized. It is conceivable that the higher incidence of apoptosis in region 1 of the control radii was caused when the bones were cut after dissection, even though the samples were immediately placed into fixative. Apoptosis can be induced within 30 minutes [30] to a few hours [30, 31]. In fact, osteocytes specifically have demonstrated an apoptotic response within 30 minutes of incubation (in vitro) with hydrogen peroxide [32]. If apoptosis did occur, then the response to the osteotomy (osteotomy minus control) was likely underestimated in the present experiment. It is also possible that the osteocyte DNA fragmentation at time 0 was not due to apoptosis but to cell disruption and thus necrosis during the osteotomy or cutting postdissection. This seems unlikely as TUNEL-positive cells were not located consistently near the cut/osteotomy edge, as might be expected with necrosis, but were found throughout region 1. Regardless, the data were treated conservatively by subtracting the percentage of TUNEL-positive osteocytes in the control radii from the osteotomized radii within each region for each bird.
DNA fragmentation assays are a valuable method for quantifying apoptosis in situ [33]. However, a limitation of the DNA fragmentation assay is that it can detect fragmentation due to causes other than apoptosis [34, 35], including tissue processing and analysis [34, 36]. If fragmentation occurred during laboratory procedures in the present experiment, it was accounted for by subtraction of the percentage of TUNEL-positive osteocytes in the control radii from osteotomized radii. Apoptosis was not confirmed with additional tests in the present experiment; however, several laboratories have gathered evidence to corroborate that in vivo staining of osteocytes for DNA fragmentation is representative of apoptosis [13, 15, 37]. Additionally, dilution of the TdT enzyme by an additional 1:16 should have increased the specificity for apoptosis. An excess of TdT in the assay can result in false-positive cells [36].
In summary, the incidence of osteocyte apoptosis increased rapidly (within 12 hours) following osteotomy and was followed temporally by the appearance of intracortical osteoclasts. The apoptotic response occurred primarily within 1 mm of the osteotomy. Whether or not osteocyte apoptosis caused recruitment of osteoclasts to the site of healing was not tested in this experiment. However, the temporal relationship between osteocyte apoptosis and intracortical osteoclast appearance is compatible with the theory that osteocyte apoptosis may play a role in the bone healing process.
References
Hadjiargyrou M, Lombardo F, Zhao S, Ahrens W, Joo J, Ahn H, Jurman M, White DW, Rubin CT (2002) Transcriptional profiling of bone regeneration. Insight into the molecular complexity of wound repair. J Biol Chem 277:30177–30182
Nijweide PJ, Burger EH, Nulend JK, Van der Plas A (1996) The osteocyte. In: Bilezikian JP, Raisz LG, Rodan GA (eds), Principles of bone biology. Academic Press, San Diego, pp 115–126
Terai K, Takano-Yamamoto T, Ohba Y, Hiura K, Sugimoto M, Sato M, Kawahata H, Inaguma N, Kitamura Y, Nomura S (1999) Role of osteopontin in bone remodeling caused by mechanical stress. J Bone Miner Res 14:839–849
Lean JM, Jagger CJ, Chambers TJ, Chow JWM (1995) Increased insulin-like growth factor I mRNA expression in rat osteocytes in response to mechanical stimulation. Am J Physiol 268:E318–E327
Forwood MR, Kelly WL, Worth NF (1998) Localisation of prostaglandin endoperoxide H synthase (PGHS)-1 and PGHS-2 in bone following mechanical loading in vivo. Anat Rec 252:580–586
Klein-Nulend J, van der Plas A, Semeins CM, Ajubi NE, Frangos JA, Nijweide PJ, Burger EH (1995) Sensitivity of osteocytes to biomechanical stress in vitro. FASEB J 9:441–445
Westbroek I, Ajubi NE, Alblas MJ, Semeins CM, Klein-Nulend J, Burger EH, Nijweide PJ (2000) Differential stimulation of prostaglandin G/H synthase-2 in osteocytes and other osteogenic cells by pulsating fluid flow. Biochem Biophys Res Commun 268:414–419
Pitsillides AA, Rawlinson SCF, Suswillo RFL, Bourrin S, Zaman G, Lanyon LE (1995) Mechanical strain-induced NO production by bone cells: a possible role in adaptive bone (re)modeling? FASEB J 9:1614–1622
Klein-Nulend J, Semeins CM, Ajubi NE, Nijweide PJ, Burger EH (1995) Pulsating fluid flow increases nitric oxide (NO) synthesis by osteocytes but not periosteal fibroblasts - correlation with prostaglandin upregulation. Biochem Biophys Res Commun 217:640–648
Tomkinson A, Gevers EF, Wit JM, Reeve J, Noble BS (1998) The role of estrogen in the control of rat osteocyte apoptosis. J Bone Miner Res 13:1243–1250
Kerr JFR, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26:239–257
Bronckers ALJJ, Goei W, Luo G, Karsenty G, D’Souza RN, Lyaruu DM, Burger EH (1996) DNA fragmentation during bone formation in neonatal rodents assessed by transferase-mediated end labeling. J Bone Miner Res 11:1281–1291
Noble BS, Stevens H, Loveridge N, Reeve J (1997) Identification of apoptotic changes in osteocytes in normal and pathological human bone. Bone 20:273–282
Stevens HY, Reeve J, Noble BS (2000) Bcl-2, tissue transglutaminase and p53 protein expression in the apoptotic cascade in ribs of premature infants. J Anat 196:181–191
Verborgt O, Gibson GJ, Schaffler MB (2000) Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J Bone Miner Res 15:60–67
Noble BS, Peet N, Stevens HY, Brabbs A, Mosley JR, Reilly GC, Reeve J, Skerry TM, Lanyon LE (2003) Mechanical loading: biphasic osteocyte survival and targeting of osteoclasts for bone destruction in rat cortical bone. Am J Physiol 284:C934–C943
Clark WD, Smith EL, Linn KA, Paul-Murphy JR, Cook ME (2005) Use of peripheral quantitative computed tomography to monitor bone healing after radial osteotomy in 3-week-old chickens (Gallus domesticus). J Avian Med Surg 19:198–207
Hao Z, Kalscheur VL, Muir P (2002) Decalcification of bone for histochemistry and immunohistochemistry procedures. J Histotech 25:33–37
Stefanini M, De Martino C, Zamboni L (1967) Fixation of ejaculated spermatozoa for electron microscopy. Nature 216:173–174
Vashishth D, Verborgt O, Divine G, Schaffler MB, Fyhrie DP (2000) Decline in osteocyte lacunar density in human cortical bone is associated with accumulation of microcracks with age. Bone 26:375–380
Muir P, Hayashi K, Manley PA, Colopy SA, Hao Z (2002) Evaluation of tartrate-resistant acid phosphatase and cathepsin K in ruptured cranial cruciate ligaments in dogs. Am J Vet Res 63:1279–1284
Enneking WF (1948) The repair of complete fractures of rat tibias. Anat Rec 101:515–537
Verborgt O, Tatton NA, Majeska RJ, Schaffler MB (2002) Spatial distribution of Bax and Bcl-2 in osteocytes after bone fatigue: complementary roles in bone remodeling regulation? J Bone Miner Res 17:907–914
Hollinger J, Wong MEK (1996) The integrated processes of hard tissue regeneration with special emphasis on fracture healing. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 82:594–606
Remedios A (1999) Bone and bone healing. Vet Clin North Am Small Anim Pract 29:1029–1044
Mori S, Burr DB (1993) Increased intracortical remodeling following fatigue damage. Bone 14:103–109
Brand RA, Rubin CT (1987) Fracture healing. In: Albright JA, Brand RA (eds), The scientific basis of orthopaedics, 2nd ed. Appleton & Lange, East Norwalk, CT, pp 325–345
Lee FY-I, Choi YW, Behrens FF, DeFouw DO, Einhorn TA (1998) Programmed removal of chondrocytes during endochondral fracture healing. J Orthop Res 16:144–150
Einhorn TA (1998) The cell and molecular biology of fracture healing. Clin Orthop Rel Res 355S:S7–S21
Evan GI, Brown L, Whyte M, Harrington E (1995) Apoptosis and the cell cycle. Curr Opin Cell Biol 7:825–834
Ashe PC, Berry MD (2003) Apoptotic signaling cascades. Prog Neuropsychopharmacol Biol Psychiatry 27:199–214
Kikuyama A, Fukuda K, Mori S, Okada M, Yamaguchi H, Hamanishi C (2002) Hydrogen peroxide induces apoptosis of osteocytes: involvement of calcium ion and caspase activity. Calcif Tissue Int 71:243–248
Li G, White G, Connolly C, Marsh D (2002) Cell proliferation and apoptosis during fracture healing. J Bone Miner Res 17:791–799
Willingham MC (1999) Cytochemical methods for the detection of apoptosis. J Histochem Cytochem 47:1101–1109
Zakeri Z, Lockshin RA (2002) Cell death during development. J Immunol Methods 265:3–20
Huppertz B, Frank H-G, Kaufmann P (1999) The apoptosis cascade - morphological and immunohistochemical methods for its visualization. Anat Embryol 200:1–18
Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC (1998) Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest 102:274–282
Clark WD (2003) Bone healing in chick radii following osteotomy: callus formation/resorption, osteocyte apoptosis and osteoclast-like cell response [PhD thesis]. University of Wisconsin-Madison, Madison
Acknowledgment
Appreciation is extended to the following for their assistance with this experiment: Hy-Line International for donation of the chicks; B. Yandell, P. Crump, and T. Tabone for statistical advice; J. Fialkowski, L. Dunlavy, J. Matheson, J. Yu, B. Cruzen, and C. Johnson for assistance prior to and during surgeries; L. Dunlavy, J. Matheson, and C. Johnson for assistance with bird monitoring following surgeries; Z. Hao and V. Kalscheur for laboratory assistance; M. Ludwig and B. Walter for assistance with feeding and housing chicks.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Clark, W.D., Smith, E.L., Linn, K.A. et al. Osteocyte Apoptosis and Osteoclast Presence in Chicken Radii 0–4 Days Following Osteotomy. Calcif Tissue Int 77, 327–336 (2005). https://doi.org/10.1007/s00223-005-0074-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-005-0074-z