
Abstract
Previous in vitro and in vivo studies demonstrated that osteopontin (OPN) is an inhibitor of the formation and growth of hydroxyapatite (HA) and other biominerals. The present study tests the hypotheses that the interaction of OPN with HA is determined by the extent of protein phosphorylation and that this interaction regulates the mineralization process. Bone OPN as previously reported inhibited HA formation and HA-seeded growth in a gelatin-gel system. A transglutaminase-linked OPN polymer had similar effects. Recombinant, nonphosphorylated OPN and chemically dephosphorylated OPN, had no effect on HA formation or growth in this system. In contrast, highly phosphorylated milk OPN (mOPN) promoted HA formation. The mOPN stabilized the conversion of amorphous calcium phosphate (a noncrystalline constituent of milk) to HA, whereas bone OPN had a lesser effect on this conversion. Mixtures of OPN and osteocalcin known to form a complex in vitro, unexpectedly promoted HA formation. To test the hypothesis that small alterations in protein conformation caused by phosphorylation account for the differences in the observed ability of OPN to interact with HA, the conformation of bone OPN and mOPN in the presence and absence of crystalline HA was determined by attenuated total reflection (ATR) infrared (IR) spectroscopy. Both proteins exhibited a predominantly random coil structure, which was unaffected by the addition of Ca2+. Binding to HA did not alter the secondary structure of bone OPN, but induced a small increase of β-sheet (few percent) in mOPN. These data taken together suggest that the phosphorylation of OPN is an important factor in regulating the OPN-mediated mineralization process.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Osteopontin (OPN), also known as secreted phosphoprotein-1 (SPP1), urinary stone phosphoprotein, uropontin, and early T-cell activator (ETA-1), is a highly conserved multifunctional phosphorylated glycoprotein expressed in many mineralized and soft tissues including bone, dentin, elastin, muscle, tumors, and in body fluids (milk, inner ear, and urine) [1–4]. It is also a major component of the calcium carbonate–containing egg shell [5]. The complementary deoxyribonucleic acid of OPN was first cloned and sequenced using rat osteosarcoma cells [6]. Concomitantly, OPN was isolated and characterized from rat bone by Prince et al. [7]. OPN contains an integrin receptor-binding arg-gly-asp (RGD) sequence [8], and its messenger ribonucleic acid (mRNA) is upregulated in response to 1, 25(OH)2 vitamin D3, parathyroid hormone, and elevated phosphate levels [9–12]. OPN contains a polyaspartic acid sequence and sites of Ser/Thr phosphorylation, of which half are highly conserved. These phosphorylated residues are postulated to mediate OPN binding to hydroxyapatite (HA) mineral crystals [4]. OPN also contains transglutaminase-reactive glutamine residues (Q34 and Q36) that allow it to be cross-linked by transglutaminase enzymes. This cross-linking results in the formation of OPN polymers, which also bind to HA [13–17].
The concept that OPN is involved in regulation of bone mineralization arose on the basis of its tissue distribution, its affinity for calcium, its immunolocalization in regions of mineralization [18], and the regulation of OPN gene expression by calcitrophic hormones [11, 12, 19]. Bone OPN has been shown from both cell culture [20] and gene ablation experiments to be important for both the differentiation and recruitment of osteoclasts [21–24], and for the inhibition of HA formation and growth [25]. OPN also inhibits in vitro growth of calcium oxalate [26] and calcium carbonate [27] crystals. Although the effects on bone remodeling have recently been linked to OPN-regulation of vascularization rather than to effects on osteoclastogenesis [28], the role of OPN in regulating HA deposition through its association with HA crystals [29] is well accepted. However the significance of OPN variants (with differing phosphorylation levels) in this process is unknown.
Although the predominant bone OPN has about 13 of its potential 29 phosphorylation sites per mole occupied [30], the phosphorylation is variable when different tissues are compared [31, 32], and when protein extracts from the same tissue are fractionated [32, 33]. The importance of this variable phosphorylation for regulation of biomineralization is suggested by the observation that the binding of some phosphorylated proteins to HA is altered by dephosphorylation and/or by modification of carboxylate groups [34], and by earlier reports that dephosphorylation of bone OPN abolishes its ability to inhibit HA formation [35, 36]. Similarly, dephosphorylation of eggshell OPN eliminates its inhibitory effect on calcium carbonate precipitation [29].
The concept that regulation of phosphorylation of extracellular matrix proteins in general, and OPN in particular, is important for control of biomineralization, is supported by the different effects of dephosphorylated collagen on HA formation and growth [37], by the effects of inhibiting kinase and phosphatase activities on mineralization in cell culture [38, 39], and by the different abilities of phosphorylated and nonphosphorylated OPN to activate macrophages [39, 40]. A feature of many of the anionic extracellular matrix proteins that regulate initial formation and growth of HA crystals and other biominerals [3, 41] is their ability at low concentrations to stabilize the first formed “critical” HA nuclei facilitating initial mineral deposition and, at higher concentrations, to bind to crystals limiting their growth and proliferation. It is our hypothesis that the extent of protein phosphorylation (or other posttranslational modifications) is a significant factor in determining OPN effects on the mineralization process. One possible effect of these modifications is a change of protein secondary structure, such as occurs in other systems [42]. The purpose of the present study was to investigate the importance of OPN phosphorylation by comparing the effects of OPN with different amounts of phosphorylation on HA formation, and to probe the mechanism of these effects by determination of the secondary structure of bone and milk OPN in the presence and absence of HA crystals.
Materials and Methods
Materials
OPN from rat bone and recombinant OPN were prepared and purified as detailed elsewhere [43]. The bone preparation had ~13 phosphorylated residues (44% of all possible phosphorylation sites); the serine residues that were phosphorylated were either totally phosphorylated or variably phosphorylated in different preparations [43]. OPN from bovine milk (mOPN) was prepared and characterized as described elsewhere and had 28/29 (96%) of its potential sites phosphorylated [44]. Dephosphorylated OPN was prepared from both the rat bone OPN preparation and from mOPN by incubation with alkaline phosphatase beads [35]. Recombinant OPN containing no phosphate was a gift from Dr. Magnus Hook [45]. Organic phosphate analysis of representative dephosphorylated and nontreated samples was performed using the method of Baykov et al. [46]. The OPN polymer was kindly provided by Dr Mari Kaartinen, and had been prepared as described elsewhere [16] from mOPN. Protein mixtures combining OPN and osteocalcin (OCN) were prepared on a weight per weight using bovine osteocalcin kindly provide both by Dr. Hauschka and Dr. Gundberg [47].
Mineralization Assays
Gelatin-Gel System
The effects of the various forms of OPN on HA formation and growth were monitored in the gelatin-gel diffusion system previously described [48, 49]. This double-diffusion system is designed to mimic cell-controlled mineralization, in that initial mineral deposition occurs at approximately physiologic calcium and phosphate concentrations (calcium x phosphate ~ 5.5 mM2), and after the precipitate is formed the flow of ions into the precipitant band is maintained at steady state levels owing to the continuous deposition of ions in HA [49]. In brief, in this system the protein or proteins in question are sandwiched in a 100-μm-thick band in a 6-cm-long (3-mL) polystyrene tube between two layers of 10% gelatin (Bloom Gelatin, Fisher Chemicals, Springfield, NJ, USA) at the site where HA forms in the absence of added protein. Calcium chloride and ammonium acid phosphate circulate into opposite ends of the gelatin-filled tubes from 4 L vessels containing 100-mM solutions in pH 7.4 Tris-hydroxy-methyl-amino methane (Tris) buffer. The Tris buffer contains 0.1% sodium azide to prevent bacterial growth. The solutions are circulated under a constant nitrogen flow at room temperature. Because previous studies had shown that at 25 μg/mL bone OPN effectively inhibited HA formation and growth [35], each protein was first analyzed at this concentration. If results were inconclusive, higher and/or lower concentrations were evaluated.
For study of de novo HA formation, gels were removed from the apparatus at 3.5 days. Mineral formation and growth in the de novo system was monitored at 5.0 days. To assess effects of protein on HA growth, preformed HA seed crystals [50] characterized by wide-angle X-ray diffraction and chemical analysis of calcium-to-phosphate ratios, were included with the protein at a final HA concentration of 0.5 mg/mL. At the indicated time-points, gels were removed from the polystyrene tubes, the position of the precipitant band was noted, and the gels were positioned on a slicer that produced 0.3-mL volumes such that the precipitant band was in the central slice. The precipitant band was either examined by x-ray diffraction to confirm the presence of HA, or all the slices were analyzed for calcium [51] and inorganic phosphate (Pi) [52] content. To correct for Ca or Pi binding by the proteins, the ion concentrations of the single-diffusion gels were subtracted from those of the double-diffusion gels. Results from individual experiments (containing 3 experimental, 3 control, and 12 single-diffusion gels), were combined and expressed as the ratio of experimental to control. Each individual experiment was repeated 3 to 5 times and results expressed as mean ± SD for each set of experiments.
ACP-HA Conversion
Amorphous calcium phosphate (ACP) was prepared from Tris-buffered solutions of 4 mM calcium chloride and 3 mM sodium acid phosphate at 25°C pH=7.4, as described elsewhere [50]. After ripening for 5 minutes, the ACP was filtered through a 600 mL medium-porosity fritted-glass filter by vacuum filtration, washed with 150 mL of Milli-Q water the pH of which was adjusted to 10 with ammonia, washed 2 with acetone, and air dried. The presence of ACP and the absence of any crystalline phases was confirmed by wide angle X-ray diffraction by using Cu K-alpha radiation on a Bruker AX-8 powder diffractometer (Bruker, Madison, WI, USA).
The conversion of ACP into HA was monitored by suspending 200 mg ACP in 15 mL 0.15 M Tris buffer at 25°C with and without 5 to 25 μg/mL mOPN or bone OPN, added from a 1 mg/mL stock (pH = 7.6). At this pH the total ACP to HA conversion requires about 6 hours to complete, and the induction time (time for conversion to start) is 50 minutes [53]. Aliquots were removed a 0, 0.5, 1, 2, 3, 4, 6, and 24 hours. The ACP–HA mixtures were separated by microcentrifugation in the cold for 1 minute (14,000 rpm), and the precipitate was dried in acetone and used for X-ray diffraction and infrared (IR) determination of percentage conversion. In some cases the ACP was precoated for 30 minutes with the protein prior to the start of the experiment. For these studies the protein was prepared in pH 7.8 Tris buffer to retard conversion of ACP to HA (induction time at this pH ~2 hours [53]), and the absence of any conversion was confirmed by X-ray diffraction. Percentage conversion was expressed based on the sharpening of the broad amorphous X-ray diffraction (XRD) band from 24 to 31 degrees to the distinct formation of a 002 and 310 band. This was calculated as the percentage change in XRD line-width of the c-axis 002 peak over that of the 310 peak. Percentage conversion was also calculated from the IR data as the ratio of the depth of the splitting of the phosphate v2 peak over the height from the base of the trough to a tangent to the split peaks. To compare data, four parameters were tabulated from plots of percentage conversion versus time. The point at which a tangent to the steepest part of the slope crossed the X-axis was defined as the induction time, ti. The time-point at which 50% of the conversion (based on control values) was achieved was defined as the half-life (t1/2). The percentage conversion at 24 hours, relative to the control data, was defined based on both XRD and IR data.
Statistics
Results from 3 to 5 independent experiments were combined, and mean and standard deviations (SDs) were compared by analysis of variance (GraphPad, Carlsbad, CA, USA). These were then contrasted with values from experiments with different concentrations of proteins by using the Bonferroni multiple comparisons test with Bonferroni P < 0.05 accepted as statistically significant.
Fourier Transform Infrared Spectroscopic Secondary Structure Determination
Deuterated rat bone OPN and bovine mOPN, Tris buffer, HA, and calcium chloride solutions were prepared by repeated (4x) lyophilization of highly purified proteins and other materials in pH 7.4 Tris-buffered D2O solutions. Protein secondary determination is routinely carried out in buffered D2O solutions and lyophilization is an accepted method for achieving the necessary H/D exchange [54]. Deuterated solutions were then mixed to give a final concentration of 20 mg/mL and infrared spectra were recorded for osteopontin alone, in the presence of deuterated calcium chloride solution (variable concentration), or 1 mg/mL deuterated HA. FTIR experiments were carried out on a Bruker Tensor spectrometer (Billerica, MA, USA) equipped with a broad-band MCT detector and a Bruker BioATRII accessory. Interferograms were collected at 2 cm−1 resolution (512 scans, 20°C), apodized with a Blackman-Harris function, and Fourier transformed with one level of zero-filling to yield spectra encoded at 1 cm−1 intervals. To reduce water interference in the spectral region associated with the protein amide I bands, all spectra were processed by subtracting the rotational-vibrational water vapor bands. Final protein spectra were obtained by subtracting matched buffer spectra (subtraction factor 0.98 ≤ x ≤ 1.02) to yield an essentially flat baseline in the spectral region of interest. In comparison to circular dichroism (CD) measurements, ATR–Fourier transform infrared (FTIR) experiments offer the unique advantage that the results are largely unperturbed by the strong scattering of HA. Furthermore, the use of IR spectroscopy is advantageous for the conformational analysis of largely unstructured proteins.
Results
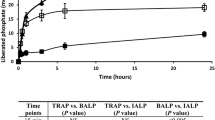
As previously reported for bovine bone OPN [35, 55], rat bone OPN was an effective inhibitor of both de novo HA formation (not shown) and HA-seeded growth (Fig. 1A). The transglutaminase cross-linked bovine mOPN polymer was also an effective inhibitor of seeded growth (Fig. 1B), and was more effective on a weight per weight basis than the bone OPN monomer, since the polymer was a significant inhibitor at 10 μg/mL whereas the bone OPN caused significant inhibition at 25 μg/mL. Neither the recombinant OPN, which had no phosphorylation (Table I), nor the chemically dephosphorylated OPN (not shown) affected de novo HA formation (Fig. 2) or seeded growth (not shown) at any concentration tested, in agreement with the earlier finding that dephosphorylated bone OPN had no effect on in vitro HA deposition [35, 55, 56]. In all the seeded growth studies with dephosphorylated OPNs the ratios of experimental to control values were not different from one (1.04 ± 0.06) for all conditions.

Effects of osteopontin on seeded apatite growth. A) Rat bone osteopontin inhibits hydroxyapatite (HA)–seeded growth in the gelatin gel system. B) Bovine osteopontin (OPN) polymer inhibits HA-seeded growth in the gelatin gel system. Relative yields of calcium and phosphate ions in the precipitant band at 5.0 days in the presence of increasing concentrations of protein and 0.5 mg/mL HA seed crystals. Values are compared to protein-free controls run at the same time, and represent mean ± standard deviation (SD) for 3 to 5 experiments, each containing three experimental and three control gels *P < 0.05 relative to OPN-free controls.

Effects of dephosphorylated osteopontin on apatite formation and growth. Chemically dephosphorylated rat osteopontin (OPN) had no effect on hydroxy apatite (HA) formation at 5 days. Relative yields of calcium and phosphate ions in the precipitant band in the presence of increasing concentrations of protein and 0.5 mg/mL. HA seed crystals. Values are compared to protein-free controls run at the same time, and represent mean ± standard deviation SD for 3 to 5 experiments, each containing three experimental and three control gels.
In contrast, the highly phosphorylated mOPN, promoted HA formation and growth in the gelatin-gel system (Fig. 3). However, at high concentrations (50–100 μg/mL) mOPN inhibited the accumulation of HA. To compare data with all forms of OPN, results for 25 μg/mL of the different OPNs with and without phosphorylation are summarized in Figure 4. A line is drawn through the control values, and significant differences are noted in the behavior of bone OPN and mOPN.

Effects of milk osteropontin (OPN) on de novo hydroxyapatite (HA) formation. Highly phosphorylated milk OPN (mOPN) promoted HA formation in the gelatin-gel system. Relative yields of calcium and phosphate ions in the precipitant band in the presence of increasing concentrations of protein. Values are compared to protein-free controls run at the same time, and represent mean ± standard deviation for 3 to 5 experiments, each containing three experimental and three control gels.

Effects of various forms of osteopontin
(OPN) on hydroxyapatite (HA) proliferation and growth. Ion accumulation in the presence of 25 μg/mL of protein. Relative yields of calcium and phoshate ions in the precipitant band at 5.0 days in the presence of increasing concentrations of protein and 0.5 mg/mL HA seed crystals. Values are compared to protein-free controls run at the same time, and represent mean ± standard deviation (SD) for 3 to 5 experiments, each containing three experimental and three control gels *P < 0.05 relative to OPN-free controls. deP-OPN, chemically dephosphorylated bone OPN; deP-mOPN, chemically dephosphorylated milk OPN.
Table II compares the effects of mOPN and bone OPN on the solution-mediated conversion of amorphous calcium phosphate to HA. This study was performed to test the hypothesis that the reason that a highly phosphorylated OPN is present in milk is to stabilize the ACP that is present [57]. The ACP granules in milk are located in micelles coated with casein, which would also prevent the conversion of ACP to HA [57]. At the lower concentrations tested, both mOPN and bone OPN had little effect on the conversion, but at higher concentrations, mOPN significantly retarded the conversion, whereas bone OPN did not. Furthermore the percentage conversion at 24 hours was always less for mOPN than that observed for bone OPN.
In vitro OPN associates with OCN [58], potentially masking certain domains in both proteins. The effects of synthetic mixtures of OPN and OCN at a total protein concentration of 25 μg/mL are compared in Figure 5. Both OCN alone and OPN alone inhibited HA formation and growth, but the mixtures promoted formation and growth of HA (Fig. 5A, B). Precoating the HA seeds with OCN before exposure to OPN increased the mineral yield, whereas precoating with OPN before exposure to OCN did not (Fig. 5C).

Effects of osteopontin–osteocalcin mixtures (total weight 25 mg/mL) on hydroxyapatite (HA) formation and growth A) Effects of mixtures of osteopontin (OPN) and osteocalcin (OCN) on de novo HA formation at 3.5 days.
Data are shown for Ca and Pi accumulation, mean ± standard deviation (SD) for 4 to 3 experiments. B) There was no effect of these mixtures on hydroxyapatite (HA)–seeded growth. Data for Ca accumulation only are presented, mean ± SD for n = 3 experiments.
Because the interaction between the phosphorylated OPNs and HA seemed to account for the observed effects of this relatively unstructured protein [59] on HA formation and growth, we used an FTIR technique to investigate this interaction. First we determined the effect of increasing calcium ion concentration on both milk and bone OPN secondary structures by using attenuated total reflection (ATR) FTIR spectroscopy (Fig. 6A). The structurally sensitive protein amide I band was found to be centered at ~1645 cm−1 and was largely unaffected by the addition of Ca2+ cations. The observed position and width of the amide I band is indicative of a predominantly unstructured (random coil) protein [60]. The spectral feature located at 1550 to 1600 cm−1 represents the OPN amide II band (protein elements that are not deuterium exchanged) as well as aspartic acid side-chain contributions ( va (COO-−) [61]. The decreasing intensity of this band feature might be attributable to a continued H/D exchange, which causes the amide II band to shift from 1550 to 1450, cm−1 (the latter band is obscured by the presence of the D2O scissoring band [61]). Although Ca2+ induced secondary structure changes would potentially accelerate H/D exchange, such a mechanism appears to be an unlikely contributing factor to the observed band intensity change because the analysis of the corresponding amide I band envelopes does not suggest alterations of the protein secondary structure.

Secondary structure determination: A) In presence and absence of ionic calcium: milk osteopontin (mOPN) amide I/amide II spectral region for increasing Ca2+ concentrations (0.15 mM Tris/D2O buffer). Spectra were obtained by subtraction of matched buffer spectra (subtraction factor 0.98 (≤ x ≤ 1.02). The spectra were, normalized to the peak height at 1645 cm−1. B. In presence and absence of hydroxyapatite (HA): amide I/amide II spectral region in the absence and presence of HA (2.5 mg/mL HA, 0.625 mM CaCl2, 16.25 mg/mL mOPN). Spectra were obtained by subtraction of matched buffer spectra (subtraction factor 0.98 ≤ x ≤ 1.02). The spectra were normalized to the peak height at 1645 cm−1.
In contrast to the mOPN interaction with Ca2+, mOPN interaction with HA induced a small but significant and reproducible shift of the mOPN amide I band envelope to lower wave numbers (Fig. 6B), which is just beyond the limit of secondary structure change detectable by current FTIR technology. The shift of the amide I band envelope is indicative of a slight increase (a few percent) in the differences
β-sheet content, and the protein remains largely unstructured. Although the interpretation of such small band shifts bears some risks, several lines of evidence support the notion of a band shift: The analysis of the osteopontin–Ca2+ interaction as well as experiments targeted at the investigation of bone osteopontin–hydroxyapatite interaction (not shown) revealed no changes in the amide I band envelope, which highlights the reproducibility of the experiments and the robustness of the data-processing protocol (particularly, with respect to the buffer subtraction).
Discussion
This study has shown that OPN phosphorylation regulates its interaction with HA and contributes to the OPN-mediated control of HA formation and growth. It also suggests that slight modifications in phosphorylated OPN conformation occur upon binding to HA and that these small conformational changes may be important for the regulation. As previously reported [35, 55, 56], the phosphorylated OPN isolated from bone was an effective mineralization inhibitor in the in vitro gelatin-gel system. The dephosphorylated forms had no significant effects, but the more highly phosphorylated form promoted HA formation and growth, as did mixtures of OPN and osteocalcin. These effects provide insight into the importance of OPN phosphorylation for the regulation of biomineralization.
The extracellular matrix proteins are differentially phosphorylated and dephosphorylated by casein kinases [31] and phosphatases, such as the bone-specific tartrate-resistant acid phosphatase (TRACP) [62]. In fact, bone OPN is a known substrate of TRACP, in that it is not modified by osteoblasts from TRACP-deficient mice [62]. Furthermore, the ability of bone sialoprotein (BSP)—a related RGD-containing phosphoglycoprotein with signaling properties—to induce osteogenesis is related to the relative extent of both OPN and BSP phosphorylation [31].
In contrast to bone OPN, which is only partially (38%) phosphorylated, the bovine mOPN is almost completely (96%) phosphorylated. Because the major phosphorylated proteins in milk are caseins, where all phosphate groups are attached to serines or threonines found in the sequence motif Ser/Thr-X-Glu/Ser(P), this suggests that the mammary gland enzyme (casein kinase) that is responsible for the phosphorylation has a high degree of specificity in the selection of phosphorylation sites. Although the regulation of the differential phosphorylation in mineralized tissues is still under investigation, results of the current study support our hypothesis that variations in phosphorylation regulate the mineralization process.
Earlier studies demonstrated that dephosphorylated bovine bone OPN bound with less affinity to HA than the bovine bone OPN itself [35], although there was some interaction through the polyaspartate residues [55, 63]. In the present study, what appeared to be most relevant for dictating the effects of OPN on the formation and growth of HA crystals in the gelatin-gel system (at any given concentration) was the extent of phosphorylation. Dephosphorylated OPN had no effect, bone OPN was an inhibitor, and highly phosphorylated mOPN promoted mineralization and inhibited the ACP to HA conversion more efficiently than did bone OPN. Recently, Ito et al. also found that mOPN was a HA nucleator when cross-linked to agarose beads, but an inhibitor when adsorbed on these beads [64]. Their data agree with our findings that lower concentrations of mOPN facilitate HA formation, and higher concentrations, presumably released from the beads when suspended in the mineralizing solution, could block any nucleation occurring on the beads.
It has been suggested [65] that the anionic OPN coats the HA (or other forming mineral crystals) and retards the further growth and proliferation of mineral. This is also seen when nonycrystalline calcium phosphates, such as ACP, are coated with bone and milk OPNs; therefore, the more highly phosphorylated mOPN stabilized ACP to a greater extent than bone OPN. Similarly OPN loses its ability to inhibit smooth muscle cell calcification when dephosphorylated [66]. The differences may be due not only to the extent of binding, but also to the small conformational differences between the proteins. Steitz et al. [65] also suggested that binding of OPN to HA alters the OPN conformation, facilitating recruitment and activation of macrophages that remove pathologic HA deposits.
In a recent study that monitored HA formation in a constant-composition titration system, Pampena found that all the OPN phosphorylated peptides synthesized to match the phosphorylation sites in OPN inhibited HA formation and growth; however, the most effective peptides were those that were the most anionic. The conformation studies suggest how this may occur. The more phosphorylated peptides or protein undergoes a slight conformational change when it interacts with HA, thereby increasing its affinity for sites on the crystal or crystal nucleus that would normally undergo growth. This, analogous to the binding of mOPN to agarose beads [64], may expose OPN domains that can nucleate HA. This is further suggested by the OPN and OCN mixtures that enhanced rather than blocked HA formation and growth. When HA crystals were precoated with OCN, and then mixed with bone OPN, there was a greater increase in mineral yield than when OPN was used first. This implies that the OPN sites that interact with OCN expose OPN domains that support HA-crystal nucleation and growth. Further structural studies will be required to determine the nature of those domains.
It is important to comment on the secondary structure analysis reported in this paper. Nuclear magnetic resonance (NMR) studies of the nonphosphorylated protein [59] had previously demonstrated that the protein had a flexible random coil structure and earlier CD evaluation indicated that OPN maintained its random coil conformation when calcium was added [67]. Such studies did not explain the failure of monoclonal antibodies that bound to OPN on plastic to bind to OPN immobilized on HA [67], findings interpreted as showing a conformational change when OPN bound to HA. Slight conformational changes were suggested by these investigators [65] when they looked at Ca-OPN interactions by FTIR secondary, structure determination. By using FTIR the present study revealed a slight but significant change in conformation when mOPN was bound to HA, whereas essentially no structural change was found for bone OPN. We did not examine unphosphorylated OPN because it neither bound to HA nor was reported to have any measurable conformation in the NMR studies [59].
Despite the slight increase in β-sheet content for mOPN in the HA-bound state, it is important to highlight that the protein remains largely unstructured. The resulting high flexibility of the protein structure, as previously suggested [59], might be a means to efficiently adapt to the HA surface upon adsorption and to ensure a tight binding to multiple sites both on HA, on cells, and within the matrix. It is noteworthy that only the highly phosphorylated OPN underwent a conformational change, suggesting that this change was brought about by phosphate interactions with a cationic surface on the HA crystals. This interaction most likely exposed the sites on the OPN structure that promoted HA formation. In the future it will be important to determine if other phosphorylated proteins that facilitate HA formation undergo a comparable change when they bind to HA.
In conclusion, based on these studies, we suggest the following mechanism for the OPN-regulated HA (and perhaps other) mineral formation processes. OPN is synthesized in response to calcitropic hormones. In soft tissues this includes response to elevated inorganic phosphate (and perhaps pyrophosphate), as OPN expression is decreased in mouse models with decreased extracellular pyrophosphate [68]. OPN is phosphorylated at specific sites under cell- and tissue-mediated controls. Small concentrations of highly phosphorylated OPN may act as nucleators, but for the most part, the concentration of partially phosphorylated OPN present is sufficient to coat HA nuclei and nascent crystals, thereby blocking their growth and proliferation. Removal of the phosphate groups releases the inhibitory effects. The ability of an anionic matrix protein to act both as an inhibitor and a nucleator, depending on state of phosphorylation and concentration, is in agreement with the data from other mineralizing systems [41].
References
D Denhardt M Noda (1998) ArticleTitleOsteopontin expression and function: role in bone remodeling J Cell Biochem 30-31 92–102 Occurrence Handle10.1002/(SICI)1097-4644(1998)72:30/31+<92::AID-JCB13>3.0.CO;2-A
CM Giachelli S Steitz (2000) ArticleTitleOsteopontin: a versatile regulator of inflammation and biomineralization Matrix Biol 19 615–622 Occurrence Handle10.1016/S0945-053X(00)00108-6 Occurrence Handle11102750
J Gokhale PG Robey AL Boskey (2001) The Biochemistry of bone R Marcus D Feldman J Kelsey (Eds) Osteoporosis, Second Edition vol 1 Academic Press San Diego 107–189
J Sodek B Ganss MD McKee (2000) ArticleTitleOsteopontin Crit Rev Oral Biol Med 11 279–303 Occurrence Handle11021631
MS Fernandez C Escobar I Lavelin M Pines JL Arias (2003) ArticleTitleLocalization of osteopontin in oviduct tissue and eggshell during different stages of the avian egg laying cycle J Struct Biol 143 171–180 Occurrence Handle10.1016/j.jsb.2003.08.007 Occurrence Handle14572472
A Oldberg A Franzen D Heinegard (1986) ArticleTitleCloning and sequence analysis of rat bone sialoprotein (osteopontin) cDNA reveals an Arg-Gly-Asp cell-binding sequence Proc Natl Acad Sci USA 83 8819–8823 Occurrence Handle3024151
CW Prince T Oosawa WT Butler M Tomana AS Bhown M Bhown RE Schrohenloher (1987) ArticleTitleIsolation, characterization, and biosynthesis of a phosphorylated glycoprotein from rat bone J Biol Chem 262 2900–2907 Occurrence Handle3469201
WT Butler (1995) ArticleTitleStructural and functional domains of osteopontin Ann N Y Acad Sci 760 6–11 Occurrence Handle7785926
PL Chang CW Prince (1994) ArticleTitle1 Alpha, 25-dihydroxyvitamin D3 stimulates synthesis and secretion of nonphosphorylated osteopontin (secreted phosphoprotein 1) in mouse JB6 epidermal cells Cancer Res 51 2144–2150
M Noda RL Vogel AM Craig J Prahl HF DeLuca DT Denhardt (1990) ArticleTitleIdentification of a DNA sequence responsible for binding of the 1,25-dihydroxyvitamin D3 receptor and 1,25-dihydroxyvitamin D3 enhancement of mouse secreted phosphoprotein 1 (SPP-1 or osteopontin) gene expression Proc Natl Acad Sci USA 87 9995–9999 Occurrence Handle2175918
GR Beck SuffixJr B Zerler E Moran (2000) ArticleTitlePhosphate is a specific signal for induction of osteopontin gene expression Proc Natl Acad Sci USA 97 8352–8357 Occurrence Handle10.1073/pnas.140021997 Occurrence Handle10890885
M Noda GA Rodan (1989) ArticleTitleTranscriptional regulation of osteopontin production in rat osteoblast-like cells by parathyroid hormone J Biol Chem 108 713–718
CW Prince D Dickie CL Krumdieck (1991) ArticleTitleOsteopontin, a substrate for transglutaminase and factor XIII activity Biochem Biophys Res Commun 177 1205–1210 Occurrence Handle10.1016/0006-291X(91)90669-X Occurrence Handle1676261
ES Sørensen LK Rasmussen L Møller PH Jensen P Hørup TE Petersen (1994) ArticleTitleLocalization of transglutaminase-reactive glutamine residues in bovine osteopontin J Biochem 304 13–16
MT Kaartinen A Pirhonen A Linnala-Kankkunen PH Mäenpää (1997) ArticleTitleTransglutaminase-catalyzed cross-linking of osteopontin is inhibited by osteocalcin J Biol Chem 272 22736–22741 Occurrence Handle10.1074/jbc.272.36.22736 Occurrence Handle9278432
MT Kaartinen A Pirhonen A Linnala-Kankkunen PH Mäenpää (1999) ArticleTitleTransglutaminase treated osteopontin exhibits increased collagen binding properties Biol Chem 274 1729–1735 Occurrence Handle10.1074/jbc.274.3.1729
MT Kaartinen S El-Maadawy NH Räsänen MD McKee (2002) ArticleTitleTransglutaminase and its substrates in bone J Bone Miner Res 12 2161–2173
S Ikeda A Nomura T Yamaguchi T Suda S Yoshiki (1992) ArticleTitleIn situ hybridization of bone matrix proteins in undecalcified adult rat bone sections J Histochem Cytochem 40 1079–1088 Occurrence Handle1619274
CW Prince WT Butler (1987) ArticleTitle1,25-Dihydroxyvitamin D3 regulates the biosynthesis of osteopontin, a bone-derived cell attachment protein, in clonal osteoblast-like osteosarcoma cells Collagen Rel Res 7 305–313
T Yamate H Mocharla Y Taguchi JU Igietseme SC Manoiagas E Abe (1997) ArticleTitleOsteopontin expression by osteoclast and osteoblast progenitors in the murine bone marrow: demonstration of its requirement for osteoclastogenesis and its increase after ovariectomy Endocrinology 138 3047–3055 Occurrence Handle10.1210/en.138.7.3047 Occurrence Handle9202251
M Ishijima SR Rittling T Yamashita K Tsuji H Kurosawa A Nifuji DT Denhardt M Noda (2001) ArticleTitleEnhancement of osteoclastic bone resorption and suppression of osteoblastic bone formation in response to reduced mechanical stress do not occur in the absence of osteopontin J Exp Med 193 399–404 Occurrence Handle10.1084/jem.193.3.399 Occurrence Handle11157060
H Ihara DT Denhardt K Furuya T Yamashita Y Muguruma K Tsuji KA Hruska K Higashio S Enomoto A Nifuji SR Rittling M Noda (2001) ArticleTitleParathyroid hormone-induced bone resorption does not occur in the absence of osteopontin J Biol Chem 276 13065–13071 Occurrence Handle10.1074/jbc.M010938200 Occurrence Handle11278791
SR Rittling HN Matsumoto MD McKee A Nanci XR An KE Novick AJ Kowalski M Noda DT Denhardt (1998) ArticleTitleMice lacking osteopontin show normal development and bone structure but display altered osteoclast formation in vitro J Bone Miner Res 13 1101–1111 Occurrence Handle9661074
H Yoshitake SR Rittling DT Denhardt M Noda (1999) ArticleTitleOsteopontin-deficient mice are resistant to ovariectomy-induced bone resorption Proc Natl Acad Sci USA 97 8156–8160 Occurrence Handle10.1073/pnas.96.14.8156
AL Boskey L Spevak E Paschalis SB Doty MD McKee (2002) ArticleTitleOsteopontin deficiency increases mineral content and mineral crystallinity in mouse bone Calcif Tissue Int 71 145–154 Occurrence Handle10.1007/s00223-001-1121-z Occurrence Handle12073157
SR Qiu A Wierzbicki CA Orme AM Cody JR Hoyer GH Nancollas S Zepeda JJ Yoreo ParticleDe (2004) ArticleTitleMolecular modulation of calcium oxalate crystallization by osteopontin and citrate Proc Natl Acad Sci USA 101 1811–1815 Occurrence Handle10.1073/pnas.0307900100 Occurrence Handle14766970
MT Hincke M St. Maurice (2000) Phosphorylation-dependent modulation of calcium carbonate precipitation by chicken eggshell matrix proteins M Goldberg A Boskey C Robinson (Eds) Chemistry and Biology of Mineralized Tissues, Am Acad Orthop Surg Rosemont IL 13–17
Y Asou SR Rittling H Yoshitake K Tsuji K Shinomiya A Nifuji DT Denhardt M Noda (2001) ArticleTitleOsteopontin facilitates angiogenesis, accumulation of osteoclasts, and resorption in ectopic bone Endocrinology 142 1325–1332 Occurrence Handle10.1210/en.142.3.1325 Occurrence Handle11181551
T Wada MD McKee S Steitz CM Giachelli (1999) ArticleTitleCalcification of vascular smooth muscle cell cultures: inhibition by osteopontin Circ Res 35 166–178
PJ Neame WT Butler (1996) ArticleTitlePosttranslational modification in rat bone osteopontin Connect Tissue Res 35 145–150 Occurrence Handle9084652
E Salih J Wang J Mah R Fluckiger (2002) ArticleTitleNatural variation in the extent of phosphorylation of bone phosphoproteins as a function of in vivo new bone formation induced by demineralized bone matrix in soft tissue and bony environments Biochem J 364 465–474 Occurrence Handle10.1042/BJ20011272 Occurrence Handle12023890
C Qin O Baba WT Butler (2004) ArticleTitlePost-translational modifications of sibling proteins and their roles in osteogenesis and dentinogenesis Crit Rev Oral Biol Med 15 126–136 Occurrence Handle15187031
JB Safran WT Butler MC Farach-Carson (1998) ArticleTitleModulation of osteopontin post-translational state by 1, 25-(OH)2-vitamin D3 Dependence on Ca2+ influx J Biol Chem 273 29935–29941 Occurrence Handle10.1074/jbc.273.45.29935 Occurrence Handle9792712
Y Fujusawa S Kuboki S Sasaki (1986) ArticleTitleChanges in interaction of bovine dentin phosphophory with calcium and hydroxyapatite by chemical modification Calcif Tissue Int 39 248–251 Occurrence Handle3024782
AL Boskey M Maresca W Ullrich SB Doty WT Butler CW Prince (1993) ArticleTitleOsteopontin-hydroxyapatite interactions in vitro: inhibition of hydroxyapatite formation and growth in a gelatin-gel Bone Miner 22 147–159 Occurrence Handle8251766
GK Hunter CL Kyle HA Goldberg (1994) ArticleTitleModulation of crystal formation by bone phosphoproteins: structural specificity of the osteopontin-mediated inhibition of hydroxyapatite formation Biochem J 300 723–728 Occurrence Handle8010953
A Endo MJ Glimcher (1989) ArticleTitleThe effect of complexing phosphoproteins to decalcified collagen on in vitro calcification Connect Tissue Res 21 179–190 Occurrence Handle2605942
AL Boskey SB Doty I Binderman (1994) ArticleTitleAdenosine 5-triphosphate promotes mineralization in differentiating chick limb-bud mesenchymal cell cultures Microsc Res Tech 28 492–504 Occurrence Handle10.1002/jemt.1070280605 Occurrence Handle7949395
GF Weber S Zawaideh S Hikita VA Kumar H Cantor S Ashkar (2002) ArticleTitlePhosphorylation-dependent interaction of osteopontin with its receptors regulates macrophage migration and activation J Leukoc Biol 72 752–764 Occurrence Handle12377945
S Razzouk JC Brunn C Qin CE Tye HA Goldberg WT Butler (2002) ArticleTitleOsteopontin posttranslational modifications, possibly phosphorylation, are required for in vitro bone resorption but not osteoclast adhesion Bone 30 40–47 Occurrence Handle10.1016/S8756-3282(01)00637-8 Occurrence Handle11792563
L Addadi S Weiner M Geva (2001) ArticleTitleOn how proteins, interact with crystals and their effect on crystal formation Z Kardiol 90 Suppl 3 92–98
S Winkler D Wilson DL Kaplan (2000) ArticleTitleControlling beta-sheet assembly in genetically engineereds silk by enzymatic phosphorylation/dephosphorylation Biochemistry 39 12739–12746 Occurrence Handle10.1021/bi001335w Occurrence Handle11027155
C Qin JC Brunn J Jones A George A Ramachandran JP Gorski WT Butler (2001) ArticleTitleA comparative study of sialic acid-rich proteins in rat bone and dentin Eur J Oral Sci 109 133–141 Occurrence Handle10.1034/j.1600-0722.2001.00001.x Occurrence Handle11347657
ES Sorensen P Hojrup TE Petersen (1995) ArticleTitlePosttranslational modifications of bovine osteopontin: identification of twenty-eight phosphorylation and three O-glycosylation sites Protein Sci4 2040–2049
K Jonsson D McDevitt MH McGavin JM Patti M Hook (1995) ArticleTitleStaphylococcus aureus expresses a major histocompatibility complex class II analog J Biol Chem 270 21457–21460 Occurrence Handle10.1074/jbc.270.37.21457 Occurrence Handle7545162
AA Baykov OA Evtushenko SM Avaeva (1988) ArticleTitleA malachite green procedure for orthophosphate determination and, its use in alkaline phosphatase-based enzyme immunoassay Anal Biochem 171 266–270 Occurrence Handle10.1016/0003-2697(88)90484-8 Occurrence Handle3044186
CM Gundberg PV Hauschka JB Lian PM Gallop (1984) ArticleTitleOsteocalcin: isolation, characterization, and detection Methods Enzymol 107 516–544 Occurrence Handle6094965
AL Boskey (1989) ArticleTitleHydroxyapatite formation in a dynamic gelatin gel system J Phys Chem 94 1628–1633 Occurrence Handle10.1021/j100341a086
L Silverman AL Boskey (2004) ArticleTitleDiffusion systems for evaluation of biomineralization Calcif Tissue Int 75 494–501 Occurrence Handle10.1007/s00223-004-0019-y Occurrence Handle15455184
NC Blumenthal F Betts AS Posner (1975) ArticleTitleEffect of carbonate and biological macromolecules on formation and properties of hydroxyapatite Calcif Tissue Res 18 81–90 Occurrence Handle1148899
JB Willis (1960) ArticleTitleDetermination of metals in blood serum by atomic absorption spectroscopy I. Calcium. Spectrocbim Acta 16 259–272
JK Heinonen RJ Lahti (1981) ArticleTitleA new and convenient colorimetric determination of inorganic orthophosphate and its application to the assay of inorganic pyrophosphates Anal Biochem 113 313–317 Occurrence Handle10.1016/0003-2697(81)90082-8 Occurrence Handle6116463
AL Boskey AS Posner (1973) ArticleTitleConversion of amorphous calcium phosphate to microcrystalline hydroxyapatite. A pH-dependent solution-mediated, solid-state conversion J Phys Chem 77 2313–2317 Occurrence Handle10.1021/j100638a011
M Diem (1993) Introduction to modern vibrational spectroscopy Wiley New York
AL Boskey (1995) ArticleTitleOsteopontin and related phosphorylated sialoproteins: effects on mineralization Ann NY Acad Sci 760 249–256 Occurrence Handle7785899
HA Goldberg KJ Warner MC Li GK Hunter (2001) ArticleTitleBinding of bone sialoprotein, osteopontin and synthetic polypeptides to hydroxyapatite Connect Tissue Res 42 25–37 Occurrence Handle11696986
TC McGann RD Kearney W Buchheim AS Posner F Betts NC Blumenthal (1983) ArticleTitleAmorphous calcium phosphate in casein micelles of bovine milk Calcif Tissue Int 35 821–823 Occurrence Handle6652558
NM Ritter MC Farach-Carson WT Butler (1992) ArticleTitleEvidence for the formation of a complex between osteopontin and osteocalcin J Bone Miner Res 7 877–885 Occurrence Handle1442202
LW Fisher DA Torchia B Fohr MF Young NS Fedarko (2001) ArticleTitleFlexible structures of SIBLING proteins, bone sialoprotein, and osteopontin Biochem Biophys Res Commun 280 460–465 Occurrence Handle10.1006/bbrc.2000.4146 Occurrence Handle11162539
JLR Arrondo FM Goni (1999) ArticleTitleStructure and dynamics of membrane proteins as studied by infrared spectroscopy Prog Biophys Mol Biol 72 367–405 Occurrence Handle10.1016/S0079-6107(99)00007-3 Occurrence Handle10605294
PI Haris D Chapman (1996) Fourier transform infrared spectroscopic studies of biomembrane systems HH Mantsch D Chapman (Eds) Infrared spectroscopy of biomolecules Wiley New York 239–279
AR Hayman TM Cox (2003) ArticleTitleTartrate-resistant acid phosphatase knockout mice J Bone Miner Res 18 1905–1907 Occurrence Handle14584904
DA Pampena KA Robertson O Litvinova G Lajoie HA Goldberg GK Hunter (2004) ArticleTitleInhibition of hydroxyapatite formation by osteopontin phosphopeptides Biochem J 378 1083–1087 Occurrence Handle10.1042/BJ20031150 Occurrence Handle14678013
S Ito T Saito K Amano (2004) ArticleTitleIn vitro apatite induction by osteopontin: interfacial energy for hydroxyapatite nucleation on osteopontin J Biomed Mater Res 69A 11–16 Occurrence Handle10.1002/jbm.a.20066
SA Steitz MY Speer MD McKee L Liaw M Almedia H Yang CM Giachelli (2002) ArticleTitleOsteopontin inhibits mineral deposition and promotes regression of ectopic calcification Am J Pathol 161 2035–2046 Occurrence Handle12466120
S Jono C Peinado CM Giachelli (2000) ArticleTitlePhosphorylation of osteopontin is required for inhibition of vascular smooth muscle cell calcification J Biol Chem 275 20197–20203 Occurrence Handle10.1074/jbc.M909174199 Occurrence Handle10766759
JP Gorski E Kremer J Ruiz-Perez GE Wise A Artigues (1995) ArticleTitleConformational analyses on soluble and surface bound osteopontin Ann N Y Acad Sci 760 12–23 Occurrence Handle7785891
K Johnson J Goding D Etten ParticleVan A Sali SI Hu D Farley H Krug L Hessle JL Millan R Terkeltaub (2003) ArticleTitleLinked deficiencies in extracellular PP (i) and osteopontin mediate pathologic calcification associated with defective PC-1 and ANK expression J Bone Miner Res 18 994–1004 Occurrence Handle12817751
Acknowledgements
The authors would like to thank the members of their research laboratories for their assistance with this study. Support was provided by NIH grants DE04141 (ALB); DE05092 (WTB & CQ), and NSF CHE-0215970 (AG). The support of Arla Foods, the Danish Dairy Research Foundation, and the Danish Research and Development Programme for Food Technology is acknowledged (ESS).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gericke, A., Qin, C., Spevak, L. et al. Importance of Phosphorylation for Osteopontin Regulation of Biomineralization. Calcif Tissue Int 77, 45–54 (2005). https://doi.org/10.1007/s00223-004-1288-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-004-1288-1