
Abstract
Growth plate chondrocytes are affected by 1,25(OH)2D3 and androgens, which may critically interact to regulate proliferation and differentiation during the male pubertal growth spurt. We investigated possible interactions of 1,25(OH)2D3 and the non-aromatizable androgen dihydrotestosterone (DHT) in primary chondrocyte cultures from young male rats. DHT and 1,25(OH)2D3 independently stimulated DNA synthesis and cell proliferation in a dose-dependent manner with maximally effective doses of [10-8 M] and [10-12 M], respectively. Both DHT and 1,25(OH)2D3 stimulated the expression and release of IGF-I, and the proliferative effects of each hormone were prevented by an IGF-I antibody. DHT and 1,25(OH)2D3 increased messenger RNAs (mRNAs) of their cognate receptors and of IGF-I receptor mRNA (IGF-I-R). 1,25(OH)2D3 also stimulated mRNA of the androgen receptor (AR), whereas DHT did not affect mRNA of the vitamin-D receptor (VDR). Coincubation with both steroid hormones did not stimulate receptor mRNAs more than either hormone alone. The proliferative effects of DHT and 1,25(OH)2D3 were completely inhibited by simultaneous incubation with both hormones, despite potentiation of IGF-I synthesis. In contrast, both hormones synergistically stimulated cell differentiation as judged by alkaline phosphatase activity, collagen X mRNA, and matrix calcification in long-term experiments. We conclude that DHT and 1,25(OH)2D3 interact with respect to chondrocyte proliferation and cell differentiation. The proliferative effects of both hormones are mediated by local IGF-I synthesis. Simultaneous coincubation with both hormones blunts the proliferative effect exerted by either hormone alone, in favor of a more marked stimulation of cell differentiation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The pubertal growth spurt involves regulated changes in endocrine systems such as the somatotropic and gonadotropic hormone axes, but also in the paracrine and autocrine milieu at the tissue level. Sex steroids influence epiphyseal growth both via modulation of pituitary growth hormone (GH) production [1] and by direct effects on chondrocytes [2] where androgen receptors (AR) have been identified [3]. Androgen-induced GH-independent pubertal growth has been described in children with GH deficiency [4] and in GH receptor-deficient Laron dwarfs [5]. It was hypothesized that androgens modulate cartilage growth via induction of local IGF-I synthesis [6].
1,25(OH)2D3 is another important regulator of enchondral growth [7, 8, 9, 10, 11]. Vitamin D deficiency per se is associated with impaired longitudinal growth [12]. Low doses of 1α,25(OH)2D3 stimulate proliferation in cultured growth plate chondrocytes [8, 9, 10, 11]. In contrast, high concentrations have an antiproliferative effect [9] and activate markers of the differentiation process [7, 13] in vitro. In vivo, high-dose 1,25(OH)2D3 inhibits GH-driven longitudinal growth both in the uremic rat model and in children with chronic renal failure [14, 15]. The mechanisms by which 1,25(OH)2D3 modulates proliferation and differentiation have not yet been identified.
Interactions between 1,25(OH)2D3 and androgens in the regulation of enchondral growth have been demonstrated in vitro [16, 17]. In cultured chondrocytes, pretreatment with 1,25(OH)2D3 or DHT synergistically enhanced each other’s proliferative capacity [17]. Simultaneous coincubation with both hormones has not yet been investigated.
In the present study we further investigated the mechanisms by which DHT and 1,25(OH)2D3 alone and in combination affect proliferation and differentiation of cultured growth plate chondrocytes. Moreover, we studied whether local IGF-I synthesis was regulated by the two hormones and whether their effects were dependent on IGF-I action. Finally, we investigated autologous and heterologous effects of the hormones on vitamin D, androgen and IGF-I receptor expression.
Materials and Methods
5α-dihydrotestosterone, low gelling temperature agarose, Triton X-100, tris-borate-EDTA-buffer, Actinomycin D and gel loading solution were obtained from Sigma Chemical Co (Munich, Germany). lα,25(OH)2D3 and 1ß,25(OH)2D3 were gifts from Drs. A. Calcanis and M. R. Uskokovic (Hoffmann-La Roche, Nutley, NJ and Grenzach, Germany). Fetal calf serum (FCS), PBS, Trypsin-EDTA and penicillin-streptomycin were purchased from Biochrom KG (Berlin, Germany). F-12 and DMEM medium, Na-Pyruvat and L-Glutamin were obtained from Bio Whittaker Europe (Verviers, Belgium), and charcoal FCS from ccpro (Neustadt, Germany). Clostridium collagenase, DNase I, trypan blue and ALP system for measurement of alkaline phosphatase (AP) were from Boehringer (Mannheim, Germany), L-Cystein was from Bachem (Heidelberg, Germany). [3H]-thymidine (25 Ci/mmol) was obtained from Amersham (Braunschweig, Germany). Standard-low-agarose-mr was from Bio-Rad (Richmond, VA, USA). CaCl2 and AgNO3 were obtained from Merck (Darmstadt, Germany). Polyclonal antibody for IGF-I (Ab-I) was supplied by W. Blum (Gießen, Germany). The IGF-I radioimmunoassay was from Mediagnost (Tuebingen, Germany). Rneasy mini kit and QIAshredder were from Quiagen (Hilden, Germany). MuLV reverse transcriptase, Rnase inhibitor, Ampli taq gold DNA polymerase, nucleotides and oligo d(T)16 were purchased from Perkin Elmer (Langen, Germany). Primers were from MWG-Biotech GMbH (Ebersberg, Germany) and 100 base-pair-ladder was obtained from Pharmacia Biotech (Freiburg, Germany).
Cell Cultures
Isolation of Chondrocytes
Epiphyseal chondrocytes from male 25–35-day-old Wistar rats (60–80 g) were isolated and cultured as described earlier [8, 10] using a modified technique of Benya et al. [18] and Lindahl et al. [19]. The procedure was in compliance with the Guiding Principles in the Care and Use of Animals, approved by the Council of the American Physiological Society.
Monolayer Cultures
First passage cells were cultured in 96-well plates (Nunc, Wiesbaden, Germany) for radiothymidine assays, 35-mm plastic dishes for determination of local IGF-I synthesis, 60-mm dishes for mRNA extraction, 24-well plates (Nunc, Roskilde, Denmark) for alkaline phosphatase (AP) measurements, and in a culture chamber mounted on a glass slide (Nunc) for light microscopy and von Kossa’s stain. At the start of each experiment 1st passage cells were subconfluent at a density of 40,000 cells/cm2. The medium used contained F-12/DMEM: 7/5, supplemented with 1 mM sodium pyruvate, 2 mM N-acetyl-L-alanyl-L-glutamine, 50 µg/ml penicillin-streptomycin, 1 mM L-cystein and 10% FCS at 37°C gassed with 95% air/5% CO2. The nominal calcium concentration, measured with an ion-selective electrode of a Fresenius Ionometer EF (Fresenius, Oberursel, Germany) was 1.2 mM. DHT and l,25(OH)2D3 were dissolved in ethanol (0.1% final concentration). All experiments in monolayer culture except von Kossa’s stain were performed under serum-free conditions.
Agarose-Stabilized Suspension Cultures
Cells were cultured in 60 mm dishes (Becton Dickinson) in agarose according to Benya and Schaffer [18] as described earlier [8, 10]. At the start of culture all cells were dissociated. Two milliliters of F12/DMEM medium containing 5% Ch-FCS, i.e., delipidated FCS, and hormones dissolved in ethanol or ethanol alone as solvent control were added on top and changed every other day.
Pellet Cultures
One ml media aliquots containing 1.5 × 105 first passage cells each were centrifuged in conical tubes (Becton Dickinson) forming a pellet, as described by Ballock et al. [20]. Chondrocytes were cultured in medium containing 25 µg/ml ascorbic acid and 2% Ch-FCS for 28 days. Medium was changed every other day and hormones were added as indicated. For RNA analysis 300 µl lysis buffer (RNeasy kit) was added at the end of culture and cells were homogenized in an ultrasound generator (Transsonic T700, Hans Schmidbauer KG, Singen, Germany). RNA was processed as described below. For von Kossa’s stain, pellets were cut into slices (5 µm thick) on a Jung Microtome at the end of culture and stained as described below.
Assays of Chondrocyte Growth and Proliferation
[3H]-Thymidine Assay
Incorporation of [3H]-thymidine was determined in serum-free cultures as described previously [8]. Before the start of the experiment cell cycles were synchronized in serum-free medium for 24 hours. Medium was changed to F-12/DMEM with 0.2% (w/v) BSA and hormones or solvent control. For the last 3 hours of a 48-hour incubation period, 2 µCi [3H]-thymidine was added to each well.
Colony Formation in Agarose-Stabilized suspension Culture
Suspension cultures were terminated at the times indicated. Colonies from parallel cultures were counted on a 2-mm grid of 100 squares using an inverted light microscope. A cell colony was defined as a cluster of cells as described previously [8, 19].
IGF-I RIA
IGF-I concentrations were measured in the supernatant of subconfluent, synchronized serum-free cultures (0.02% BSA) using a commercially available highly sensitive (0.02 ng/ml), specific IGF binding protein (IGFBP)-blocked RIA (abstract, 74th Annual Meeting of the American Endocrine Society 293, 1992).
Assays of Chondrocyte Differentiation
Assay of Alkaline Phosphatase (AP) Activity
Cells were cultured as described for the [3H]-thymidine assay. Cell cultures were exposed to DHT, 1α,25(OH)2D3 and their combination for a total duration of 24 hours. AP activity was analyzed as a function of release of para-nitrophenol from para-nitrophenol phosphate at pH 10.2.
Von Kossa’s Stain
First passage cells were cultured in monolayer on chamber slides (Nunc) or as pellets in conical tubes as described above. L-ascorbic acid (25 µg/ml) and ß-glycerophosphate (5 mM) were added starting on day 7. Cultures were incubated with DHT, 1,25(OH)2D3 and their combination for 5 weeks. Medium containing 6% Ch-FCS was changed every 48 hours. At the end of incubation, cultures were stained using von Kossa’s method, which visualizes focuses of calcified matrix as dark brown-black areas. Clusters of positive stained cells per visual field (magnification × 380) were assessed by an inverted light microscope.
Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
Subconfluent cells were kept in 3% FCS for 24 hours and thereafter were incubated in serum-free medium for 24 hours, and hormones were added to the medium for 8 hours. The RNeasy kit® was used for isolation of total RNA. RNA was quantified by measuring the absorbency at 260 nm.
Oligonucleotide primers were made for the following genes according to the rat-specific gene sequences published in Genbank: AR (M23264), CII (L48440) and CX (S79214), IGF-I and IGF-I-R [21] and VDR [22]. Total RNA (1 µg) was reversely transcribed with MuLV reverse transcriptase using oligo d(T)-primers. After cDNA synthesis, PCR amplification was performed using 4 µl cDNA template with specific primer pairs (IGF-I: F1: 5′-CAT TCG GAG GGC ACC ACA GA-3′ and R1: 5′-GCA GGT GTT CCG ATG TTT TG-3′; IGF-IR: F1: 5′-TCC ACA TCC TGC TCA TCT CC-3′ and R1: 5′-TCG CTT CCC ACA CAC ACT TG-3′; AR: F1: 5′-TGG CGA GAG ACA GCT TGT AC-3′ and R1; 5′-TCT TGC AAT AGG CTG CAC AG-3′; VDR: F1: 5′-GCC CAC CAC AAG ACC TAT-3′ and R1: 5′-CCT TTT GGA TGC TGT AAC TG-3′; CX: F1: 5′-TGC CTC TTG TCA GTG CTA AC-3′ and Rl: 5′-GCG TGC CGT TCT TAT ACA GG -3′; CII: F1: 5′-CTC CAG GTG TGA AGG GTG AG-3′ and R1: 5′-GAA CCT TGA GCA CCT TCA GG -3′). GAPDH cDNA was co-amplified as a control using specific primer pairs [23] (F1: 5′-GCT GGG GCT CAC CTG AAG GG-3′ and R1: 5′-GGA TGA CCT TGC CCA CAG CC-3′). Amplification was performed in a Perkin Elmer Gene Amp PCR System 9600 and consisted of 30 cycles (32 for VDR) of 30 s. denaturation at 95°C, 30 s. annealing at 58°C and 30 s. extension at 72°C (terminal extension 7 min at 72°C) after an initial denaturation step at 95°C for 10 min. The amplified products (IGF-I 303 bp, IGF-IR 490 bp, AR 490 bp, VDR 297 bp, CX 248 bp, CII 261 bp and GAPDH 343 bp) were visualized in a 1.5% agarose gel by staining with ethidium-bromide under ultraviolet transillumination. Optical densities were analyzed using the Molecular Analyst computed program (Bio-Rad, Vilbert Lourmat, France). Routine control reactions performed by omitting reverse transcriptase or cDNA template showed no reaction product. For each product of interest, amplification curves were obtained and the cycle number midway in the linear logarithmic phase of amplification was used in the experimental series. Moreover, linearity of the dose-PCR product relationship was confirmed by serial dilutions of total RNA (0.1–1 µg) for the IGF-1, IGF-1 receptor, androgen receptor, collagen X and collagen II products. For the vitamin D receptor, dose-product linearity had been shown previously [11]. Moreover, serial dilutions of standard DNA fragments (θ X-174 RF DNA-Hinc II Digest, 79-1057 bp) were co-amplified. The slope of the standard OD curve was identical with those of the IGF-1 and the IGF-1 receptor, and deviated slightly for the androgen receptor PCR product.
Statistics
Data are given as mean ± SEM. Data were checked for Gaussian distribution using the Kolmogorov-Smirnov test (Sigmastat, Jandel Cooperation, USA). Two-tailed unpaired Student’s t-tests were applied for comparison of two normally distributed groups; comparisons between more than two normally distributed groups were made by one-way ANOVA followed by pairwise multiple comparison (Student-Newman-Keuls method). For non-parametrically distributed data the Kruskal-Wallis test, followed by all-pairwise comparison (Dunn’s test) was used. P < 0.05 was considered statistically significant.
Results
Proliferation Assays
Incubation for 48 hours with increasing concentrations of DHT had a stimulatory effect on [3H]-thymidine incorporation by chondrocytes in serum-free monolayer culture, with a maximally effective dose of [10−8 M] (Fig. 1). Consistent with previous results of our group [7, 11], 1α,25(OH)2D3 had a dose-dependent biphasic effect on DNA synthesis. At 10−12 M, [3H]-thymidine incorporation was 175 ± 3.2%, at 10−10 M, 138 ± 3.7%, and at 10−8 M, 9l ± 3.1% of solvent control (each group P < 0.05 vs. control; [10−12 M] vs. [10−10 M]: P < 0.05; [10−12 M] vs. [10−8 M]: P < 0.05; n = 5 per group). Simultaneous coincubation of cultures at the maximally proliferative dose of 1α,25(OH)2D3 [10−12 M] with DHT prevented the stimulation of DNA synthesis induced by DHT alone at doses of [10−9 M] to [10−6 M] (Fig. 1). When coadministered at [10−10] or [10−8 M], 1α,25(OH)2D3 completely inhibited the proliferative action of DHT over the entire dose range studied (coincubation with 10−10 M 1α,25(OH)2D3: 103–116% of control; with 10−8 M 1α,25(OH)2D3: 91–105% of control; NS for each experiment).

Effect of combined administration of DHT and 1α,25(OH)2D3 on [3H]-thymidine incorporation. Subconfluent chondrocytes were incubated with DHT [10−10 M−10−5 M] alone (-●- solid line) or simultaneously together with 1α,25(OH)2D3 [10−12 M] (-▲- broken line) for 48 hours under serum-free conditions, and [3H]-thymidine incorporation was determined. Data are mean ± SEM, n = 5 per group, statistics by ANOVA, *P < 0.05 vs. solvent control; # P < 0.05 DHT vs. DHT + 1α,25(OH)2D3.
Similar results were obtained for colony formation in agarose-stabilized suspension culture. While DHT and 1α,25(OH)2D3 given alone stimulated colony formation dose-dependently with a maximum at 10−8 M and 10−12 M, respectively (data not shown), coincubation with maximally stimulatory concentrations of each hormone (DHT, [10−8 M]; 1α,25(OH)2D3, [10−12 M]) did not increase colony formation above control levels after 15 days of culture (Table 1). The effect of 1α,25(OH)2D3 was stereospecific, since the stereoisomer 1ß,25(OH)2D3 was inactive and a coincubation with DHT [10−8 M] and 1ß,25(OH)2D3 [10−12 M] did not attenuate DHT-driven colony formation (Table 1).
Local IGF-I Synthesis
We investigated whether the proliferative effects of DHT and 1α,25(OH)2D3 were mediated by stimulation of IGF-I synthesis. IGF-I mRNA levels were significantly increased after incubating serum-deprived chrondrocytes in monolayer with DHT [10−8 M] or 1α,25(OH)2D3 [10−12 M] for 8 hours. Coincubation of DHT [10−8 M] and 1α,25(OH)2D3 [10−12 M] increased IGF-I mRNA levels to a significantly higher degree than either hormone alone (Fig. 2). Earlier time-course studies had shown that IGF-I mRNA levels were maximally stimulated by both hormones after 6–8 hours of incubation and decreased again after longer periods of incubation (data not shown). Actinomycin D [2 µg/ml] prevented DHT [10−8 M]- and 1α,25(OH)2D3 [10−12 M] driven stimulation of IGF-I mRNA synthesis after an 8-hour incubation (control + actinomycin D, 100 ± 8.8%; DHT + actinomycin D, 111 ± 9.1%; 1α,25(OH)2D3 + actinomycin D, 115 ± 5.7%; DHT + 1α,25(OH)2D3 + actinomycin D, 121 ± 5.3%, mean ± SEM, no statistically significant difference between groups, n = 4).

Regulation of IGF-I mRNA by DHT and 1α,25(OH)2D3 (1,25 D). Chondrocytes were incubated for 8 hours under serum-free conditions with DHT [10−8M] and 1α,25(OH)2D3 [10−12 M] either separately or combined. IGF-I mRNA was analyzed by RT-PCR. The graph shows the ratio of IGF-I/GAPDH PCR products as percent of control. Data are mean ± SEM, n = 5, statistics by ANOVA, *P < 0.05 vs. solvent control, #P < 0.05 vs. DHT and 1α,25(OH)2D3 given alone.
Incubation with DHT [10−8 M] or 1α,25(OH)2D3 [10−12 M] for 20 hours increased IGF-I peptide levels in the supernatant seven- and nine-fold, respectively, compared to solvent control (Table 2). Coincubation with DHT [10−8 M] and 1α,25(OH)2D3 [10−12 M] raised IGF-I secretion 16-fold compared to solvent control. The effect of coincubation was significantly stronger than the effects of either hormone alone (Table 2). To assess whether the proliferative action of DHT and 1α,25(OH)2D3 was dependent on stimulation of IGF-I synthesis, [3H]-thymidine incorporation was determined in chondrocytes incubated with DHT [10−8 M] or 1α,25(OH)2D3 [10−12 M] in the presence or absence of a polyclonal IGF-I antibody (Ab-1). While the IGF-I antibody alone did not alter basal [3H]-thymidine incorporation compared to solvent control, it prevented DHT- and 1α,25(OH)2D3-driven DNA synthesis (Fig. 3).

IGF-I polyclonal antibody inhibits DHT- and 1α,25(OH)2D3-driven cell proliferation ([3H]-thymidine incorporation). The graph shows radiothymidine incorporation (counts per minute) after incubation with DHT [10−8 M] or 1α,25(OH)2D3 [10−12 M] and coincubation of each hormone with the polyclonal IGF-I antibody (Ab-1) [1 µg/ml] for 48 hours. Data are mean ± SEM, n = 4, statistics by ANOVA *P < 0.05 vs. solvent control, # P < 0.05 vs. DHT and 1α,25(OH)2D3 given alone.
Regulation of Androgen Receptor (AR), Vitamin D Receptor (VDR) and Type-1 IGF Receptor ( IGF-I-R) Expression
Chondrocytes in monolayer were incubated with DHT [10−8 M], 1α,25(OH)2D3 [10−12 M] and their combination for 8 hours under serum-free conditions. AR mRNA was upregulated by DHT, 1α,25(OH)2D3 and their combination (Fig. 4A). VDR mRNA was only slightly increased by DHT, but strongly stimulated by 1α,25(OH)2D3 and the combination of the two hormones (Fig. 4B). IGF-I-R mRNA was stimulated by DHT and significantly more by 1α,25(OH)2D3. Coincubation with both hormones also stimulated IGF-I-R mRNA; the effect was similar to that of DHT alone, but significantly less than that of 1α,25(OH)2D3 alone (Fig. 4C).

Regulation of the androgen receptor (AR)-, vitamin D receptor (VDR)- and type I IGF receptor (IGF-I-R)-mRNA by DHT, 1α,25(OH)2D3 (1,25) and their combination. The graph shows the expression of AR (A), VDR (B) and IGF-I-R (C), normalized for GAPDH expression, as percent of solvent control. Chondrocytes were incubated with DHT [10−8 M], 1α,25(OH)2D3 [10−12 M] and their combination for 8 hours under serum-free conditions. AR, VDR and IGF-I-R mRNA were analyzed by RT-PCR. Data are mean ± SEM, n = 3, statistics by ANOVA, *P < 0.05 vs. solvent control, § P < 0.05 vs. DHT, # P < 0.05 vs. 1α,25(OH)2D3 + DHT and DHT alone.
Regulation of AP Activity, Type II and X Collagen mRNAs and Matrix Calcification
Incubation of chondrocyte monolayers for 24 hours with DHT under serum-free conditions stimulated AP activity in a dose-dependent fashion with a maximally stimulatory effect at DHT [10−8 M] (Fig. 5). The maximally stimulatory effect of DHT was compatible with that of 1α,25(OH)2D3 [10−12 M]. Coincubation with the maximally stimulatory doses of both hormones increased AP activity significantly more than DHT or 1α,25(OH)2D3 alone (Fig. 5).

Induction of alkaline phosphatase activity by DHT, 1α,25(OH)2D3 and their combination. Chondrocytes cultured in serum-free medium were exposed to DHT [10−11–10−5 M] (-●- solid line) alone, or in combination with 1α,25(OH)2D3 [10−12 M] (-▲- broken line) for 24 hours. Blot illustrations are from one representative experiment. Data are mean ± SEM, n = 6 per group, statistics by ANOVA, *P < 0.05 vs. solvent control, # P < 0.05 vs. DHT alone.
Short-term experiments showed that type X collagen mRNA was already stimulated after 8 hours of incubation with DHT [10−8 M] or 1α,25(OH)2D3 [10−12 M], and even more by coincubation of the two hormones under serum-free conditions (data not shown). Similar results were obtained in long-term cultures. Type X collagen mRNA extracted from three-dimensional pellet cultures was clearly stimulated by DHT [10−8 M] or 1α,25(OH)2D3 [10−12 M] and even more by their combination after 28 days (Fig. 6A). Type II collagen mRNA was slightly stimulated by DHT [10−8 M] and 1α,25(OH)2D3 [10−12 M], and not affected by coincubation of DHT and 1α,25(OH)2D3 in short-term (8 hours; data not shown) as well as in long-term cultures (Fig. 6B).

Long-term regulation of type X collagen (CX)- and type II collagen (CII) mRNA by DHT, 1α,25(OH)2D3 and their combination in pellet culture. Chondrocytes were cultured in medium containing 2% Ch-FCS and incubated with DHT [10−8 M], 1α,25(OH)2D3 [10−12 M] and their combination for 28 days in three-dimensional pellet culture. Type X collagen (A) and type II collagen (B) mRNA were analyzed by RT-PCR. Data are mean ± SEM, n = 2 per group. Due to the small size of groups ANOVA could not be performed.
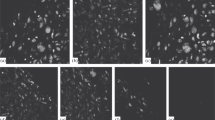
Formation of calcified matrix, visualized by von Kossa’s stain after 5 weeks of incubation in monolayer culture, was clearly induced by 1α,25(OH)2D3 [10−12 M], DHT [10−8 M], and their combination in an increasing order (Fig. 7). The amount of positive stained cell clusters per visual field was 1.6 ± 0.5 for control, 11.8 ± 0.6 for 1α,25(OH)2D3 [10−12 M], 17.8 ± 0.6 for DHT [10−8 M], and 23.0 ± 1.0 for DHT+1α,25(OH)2D3 (each P < 0.05 vs. control; P < 0.05, DHT vs. 1α,25(OH)2D3; P < 0.05, DHT+1α,25(OH)2D3 vs. each hormone alone, n = 5; mean ± SEM). Similar results were obtained in three-dimensional pellet cultures. After 21 days in culture the amount of calcified cell clusters per visual field was 21 ± 3.2 for control, 70 ± 3.0 for 1α,25(OH)2D3 [10−12 M], 101 ± 5.4 for DHT [10−8 M] and 149 ± 20 for DHT [10−8 M] + 1α,25(OH)2D3 [10−12 M] (each P < 0.05 vs. control, P < 0.05; DHT vs. 1α,25(OH)2D3; P < 0.05, DHT+1α,25(OH)2D3 vs. each hormone alone, n = 6; mean ± SEM).

Effect of DHT, 1α,25(OH)2D3 and their combination on calcification of intercellular matrix in monolayer culture. Chondrocytes were cultured in chamber slides for 6 weeks with medium containing 6% Ch-FCS. Cells were incubated with ethanol as solvent control (A), 1α,25(OH)2D3 [10−12 M] (B), DHT [10−8 M] (C) and their combination (D) for the last 5 weeks. Matrix calcification was visualized by von Kossa’s staining and appears dark brown-black (arrows). Calcification was clearly induced by 1α,25(OH)2D3, DHT, and by the combination of the two hormones in an increasing order. Magnification ×380 (left) and ×650 (right).
Discussion
We were able to demonstrate a proliferative effect of DHT on cultured growth plate chondrocytes of prepubertal male rats. Short-term cultures in serum-free medium and long-term cultures with low doses (5%) of charcoal-treated FCS showed a dose-dependent stimulatory effect on chondrocyte growth with a maximum at 10−8 M. Since DHT cannot be aromatized to estradiol, this is evidence for a direct, AR-mediated growth-promoting effect of male sex steroids. Our findings are consistent with reported effects of androgens on cultured human fetal chondrocytes [2], rat embryonic chondrocytes [16] and isolated mandibular cartilage [24]. Direct injection of testosterone increases the width of the epiphyseal growth plate in rats [25]. On the other hand, aromatizable testosterone did not induce proliferation in chondrocytes from rats older than the ones in our study [26], supporting the concept that androgen-mediated effects on growth plate cartilage are age-dependent [27, 28]. 5α-Reductase activity is highest during the time of gonadal maturation and decreases afterwards. In older animals, testosterone might therefore be preferentially aromatized to 17ß-estradiol, which hardly stimulates [24] or even inhibits [29] cartilage growth of male rats.
The dual action of 1α,25(OH)2D3 on epiphyseal cartilage with a maximal growth-stimulating concentration of 10−12 M, contrasting with an antiproliferative effect at higher doses, has previously been described by our group [9].
Both DHT and 1α,25(OH)2D3 are able to transactivate target genes by binding of specific nuclear receptor complexes to hormone response elements in the promotor region of regulated genes [30, 31]. We hypothesized that the gene encoding for IGF-I, a key regulator of physiological growth at the tissue level, may be a candidate for such regulatory mechanisms. Indeed, we found evidence that both DHT and 1α,25(OH)2D3 stimulate chondrocyte growth by induction of local IGF-I synthesis. Both hormones enhanced gene expression and peptide secretion of IGF-I. The stimulation of IGF-I mRNA levels by the two hormones was blocked by coincubation with actinomycin D, giving evidence for DHT and 1α,25(OH)2D3-induced de novo synthesis of IGF-I mRNA. In addition, their proliferative action was completely blocked by administration of a specific IGF-I antibody, indicating that DHT- and 1α,25(OH)2D3-driven proliferation are mediated by autocrine/paracrine IGF-I action.
Several in vitro studies have reported effects of androgens on IGF-I. In foreskin fibroblasts, testosterone stimulated IGF-I synthesis [32]. DHT was suggested to induce 5α-reductase activity via stimulation of local IGF-I synthesis in cultured fibroblasts [33]. In an osteoblast cell line, DHT increased IGF-1 gene expression [34]. According to Maor et al. [24] testosterone stimulates both growth and local IGF-I synthesis in mandibular condyle cartilage, but testosterone-mediated proliferation was not blocked by a monoclonal IGF-I antibody. This discrepancy with our findings may be due to methodological differences, such as antibody specificity, incomplete diffusion of the antibody in an isolated organ culture system, or model-specific differences in the interference of local IGF binding proteins with immunoblocking by IGF-I antibody. In addition, our experiments were performed under serum-free conditions, whereas Maor et al. used 2% FCS in their culture medium. In summary, our data strongly support the concept that DHT stimulates longitudinal bone growth by induction of local IGF-I synthesis.
Very few investigations have addressed a potential effect of 1,25(OH)2D3 on systemic or local IGF-I production. In osteoporotic women treated with 1,25(OH)2D3, IGF-I serum levels were increased [35]. In cultured osteoblasts, 1,25(OH)2D3 was found to stimulate IGF-I synthesis in some studies [36, 37], but suppressed it in others [38]. Our study is the first to investigate the influence of 1,25(OH)2D3 on IGF-I production in chondrocytes. It appears that 1,25(OH)2D3 at low, physiological concentrations has a facilitatory action on autocrine IGF-I synthesis and release by chondrocytes.
The molecular mechanisms by which androgens and vitamin D increase steady-state IGF-1 mRNA levels are as yet unknown. The P1 promoter, the predominant regulator of IGF-1 gene expression in extrahepatic tissues, contains binding sites for proteins of the C/EBP, AP-1, E2-F, and HNF transcription factor families as well as a cAMP binding protein (CREB) binding site, but lacks direct DNA binding sites for the vitamin D or the androgen receptor [39]. However, steroid hormone receptors can also affect transcriptional activities of target genes indirectly by affecting the expression and/or activity of other nuclear transcription factors. The vitamin D receptor complex interacts with AP-1 to transactivate target gene expression [40, 41]. The androgen receptor is a co-activator of the CREB binding protein (CBP) and modulates the action of other transcription factors by competing for CBP binding [42]. The activated glucocorticoid receptor complex suppresses IGF-1 gene expression via upregulating the repressive C/EBP-β and C/EBP-δ in osteoblasts [43]. Since the androgen receptor downregulates C/EBP-δ expression in androgen-dependent tissues [44], inverse regulation of this nuclear factor may constitute a mechanism for the contrasting effects of glucocorticoids and androgens on IGF-1 gene transcription.
We found that the intrinsic proliferative effects exerted by DHT and 1α,25-(OH)2D3 alone vanished completely when chondrocytes were coincubated with both hormones. 1α,25(OH)2D3 was recently shown to suppress the growth-promoting action of DHT in human ovarian cancer cells when administered at a pharmacological, intrinsically growth-inhibitory dose [45]. Here, we provide evidence that this effect also occurs in normal chondrocytes, and is present even at physiological doses of 1α,25(OH)2D3. Since 1α,25(OH)2D3 itself has an intrinsic growth-promoting effect at this concentration, the suppression of proliferation rates to the control level observed without either hormone upon coincubation demonstrates that DHT and 1α,25(OH)2D3 mutually inhibit their proliferative actions.
IGF-I synthesis was significantly higher during coincubation than it was under the influence of DHT or 1α,25(OH)2D3 alone. Interestingly, in vitro studies have shown that the dose-response curve of the proliferative effect of exogenous IGF-I on cartilage is not linear. With increasing concentrations of exogenous IGF-I, proliferation was decreasing or even inhibited [11, 46]. Exogenous and endogenous IGF-I action cannot be compared directly, but it is tempting to speculate that the lack of a proliferative effect of combined DHT and 1α,25(OH)2D3 administration was partly due to endogenous IGF-I production exceeding an optimal level.
We also investigated the possibility that the combination of the two hormones failed to stimulate proliferation because their corresponding receptors were downregulated. However, steady-state AR, VDR and IGF-I-R mRNA levels were not down- but upregulated by the combination of DHT and 1α,25(OH)2D3. Both DHT and 1α,25(OH)2D3 stimulated AR gene expression, in keeping with previous findings in osteoblasts [47, 48] and prostate carcinoma cells [49]. Moreover, we confirmed our previous finding of an autologous upregulation of VDR mRNA [11] by 1α,25(OH)2D3, whereas DHT had little effect on VDR gene expression.
1α,25(OH)2D3 and DHT also increased IGF-I-R gene expression. The combined administration of both hormones had a stimulatory effect similar to DHT alone, rendering downregulation of the IGF-I-R an unlikely mechanism of the observed inhibition of proliferation.
Consistent with previous observations in cultured chondrocytes or isolated cartilage [9, 15, 26, 27] DHT and 1α,25(OH)2D3 induced not only the proliferation, but also the differentiation of cultured chondrocytes. The differentiation process involves extracellular matrix synthesis, induction of ALP and matrix calcification [50, 51]. We investigated potential effects of DHT and 1α,25(OH)2D3 on the pattern of type II and X collagen gene expression, AP activity, and the induction of matrix calcification. Both DHT [10−8 M] and 1α,25(OH)2D3 [10−12 M] markedly stimulated collagen X mRNA levels in short-term (8 hours) and in long-term cultures (25 days), whereas collagen II mRNA levels were hardly affected by the two hormones. Coincubation with both hormones had an additive stimulatory effect on type X collagen mRNA levels, whereas type II collagen gene expression was not increased above control levels. DHT and 1α,25(OH)2D3 also induced AP activity and matrix calcification as visualized by von Kossa’s staining. Combined administration of both hormones had additive effects on these markers of chondrocyte differentiation.
Type II collagen is mainly expressed at earlier stages of chondrocyte maturation, with levels decreasing during hypertrophy and the onset of calcification [51, 52]. In contrast, type X collagen is localized in the hypertrophic region of the growth plate [52], and is apparently exclusively expressed in cartilage undergoing endochondral ossification [52]. AP activity increases with the beginning of differentiation and is maintained at the same level during mineralization [51]. Hence, our results suggest that DHT and 1α,25(OH)2D3 have synergistic effects on the differentiation of cultured chondrocytes, whereas they antagonize each other with respect to proliferation.
Our results were obtained from an animal cell culture model and cannot easily be compared with the in vivo situation. Interestingly however, low-dose testosterone treatment of boys with constitutional delay in growth and development effectively accelerated growth velocity without loss of height potential [53, 54]. In contrast, low-dose testosterone advanced skeletal maturation more than growth in boys with chronic renal failure and delayed puberty in those who received treatment with the vitamin D metabolites dihydrotachysterol or alfacalcidol, resulting in a loss of height potential [55]. The latter finding might indicate that the predominant cell differentiating effect of the combined application of DHT and 1α,25(OH)2D3 observed in our in vitro study might also be relevant in vivo.
In summary, our study demonstrates that DHT stimulates proliferation of cultured growth plate chondrocytes dose-dependently with a maximum at 10−8 M. DHT and 1α,25(OH)2D3 each induced proliferation via stimulation of local IGF-I synthesis. However, combined administration of the two hormones in their maximal stimulatory concentrations prevented the proliferative effect exerted by either hormone alone; this effect was not mediated via receptor or IGF-I downregulation. In contrast, differentiation was most effectively induced by coincubation with DHT and 1α,25(OH)2D3 and to a lesser extent by either hormone alone. We conclude that androgens and vitamin D interact to regulate growth and differentiation of enchondral cartilage.
References
N Mauras AD Rogol MW Haymond JD Veldhuis (1996) ArticleTitleSex steroids, growth hormone, insulin-like growth factor-1: neuroendocrine and metabolic regulation in puberty. Horm Res 45 74–80 Occurrence Handle1:CAS:528:DyaK28XpvFSjtg%3D%3D Occurrence Handle8742123
A Carrascosa L Audi MA Ferrandez A Ballabriga (1990) ArticleTitleBiological effects of androgens and identification of specific dihydrotestosterone-binding sites in cultured human fetal epiphyseal chondrocytes. J Clin Endocrinol Metab 70 134–140 Occurrence Handle1:CAS:528:DyaK3cXntVejsw%3D%3D Occurrence Handle2294127
H Ben-Hur HH Thole A Mashiah V Insler V Berman E Shezan D Elias A Zuckerman A Ornoy (1997) ArticleTitleEstrogen, progesterone and testosterone receptors in human fetal cartiligenous tissue: immunohistochemical studies. Calcif Tissue Int 60 520–526 Occurrence Handle10.1007/s002239900274 Occurrence Handle1:CAS:528:DyaK2sXjsFartLo%3D Occurrence Handle9164826
KM Attie NR Ramirez FA Conte SL Kaplan MM Grumbach (1990) ArticleTitleThe pubertal growth spurt in eight patients with true precocious puberty and growth hormone deficiency: evidence for a direct role of sex steroids. J Clin Endocrinol Metab 71 975–983 Occurrence Handle1:STN:280:By%2BA1czjtFA%3D Occurrence Handle2401720
Z Laron R Sarel A Pertzelan (1980) ArticleTitlePuberty in Laron type dwarfism. Eur J Pediatr 134 79–83 Occurrence Handle1:STN:280:Bi6D3czpslE%3D Occurrence Handle7408914
BS Keenan GE Richards SW Ponder JS Dallas M Nagamane ER Smith (1993) ArticleTitleAndrogen-stimulated pubertal growth: the effects of testosterone and dihydrotestosterone on growth hormone and insulin-like growth factor I in the treatment of short stature and delayed puberty. J Clin Endocrinol Metab 76 996–1001 Occurrence Handle1:STN:280:ByyB3s3hslA%3D Occurrence Handle8473416
Z Schwartz DL Schlader V Ramirez MB Kennedy BD Boyan (1989) ArticleTitleEffects of vitamin D metabolites on collagen production and cell proliferation of growth zone and resting zone cartilage cells in vitro. J Bone Miner Res 4 199–207 Occurrence Handle1:CAS:528:DyaL1MXktlOjurY%3D Occurrence Handle2786322
G Klaus J Merke H Eing U Huegel M Milde H Reichel E Ritz O Mehls (1991) ArticleTitle1,25(OH)2D3 receptor regulation and 1,25(OH)2D3 effects in primary cultures of growth cartilage cells of the rat. Calcif Tissue Int 49 340–348 Occurrence Handle1:CAS:528:DyaK38XnslSruw%3D%3D Occurrence Handle1664276
G Klaus B König U Huegel E Ritz O Mehls (1993) ArticleTitleIntermittent and continuous exposure to 1,25(OH)2D3 have different effects on growth plate chondrocytes in vitro. Kidney Int 44 708–715 Occurrence Handle1:CAS:528:DyaK2cXht1KjtL4%3D Occurrence Handle8258948
G Klaus BV Eichel T May U Huegel H Mayer E Ritz O Mehls (1994) ArticleTitleSynergistic effects of parathyroid hormone and 1,25-dihydroxyvitamin D3 on proliferation and vitamin D receptor expression of rat growth cartilage cells. Endocrinology 135 1307–1315 Occurrence Handle1:CAS:528:DyaK2cXmt1amuro%3D Occurrence Handle7523093
G Klaus L Weber J Rodriguez P Fernandez T Klein J Grulich-Henn U Huegel E Ritz O Mehls (1998) ArticleTitleInteraction of IGF-I and 1α,25(OH)2D3 on receptor expression and growth stimulation in rat growth plate chondrocytes. Kidney Int 53 1152–1161
SR Kreiter RP Schwartz HN Kirkman PA Charlton AS Calikoglu ML Davenport (2000) ArticleTitleNutritional rickets in African American breast-fed infants. J Pediatr 137 53–57 Occurrence Handle10.1067/mpd.2000.109009
Z Schwartz G Knight L Swain BD Boyan (1988) ArticleTitleLocalization of vitamin D3 responsive alkaline phosphatase in cultured chondrocytes. J Biol Chem 263 6023–6026 Occurrence Handle1:CAS:528:DyaL1cXitFKqtrw%3D Occurrence Handle3258864
CP Sanchez IB Salusky BD Kuizon P Abdella H Juppner WG Goodman (1998) ArticleTitleGrowth of long bones in renal failure: roles of hyperparathyroidism, growth hormone and calcitriol. Kidney Int 54 1879–1887 Occurrence Handle1:CAS:528:DyaK1cXotVagsr0%3D Occurrence Handle9853253
BD Kuizon WG Goodman H Juppner I Boechat P Nelson B Gales IB Salusky (1998) ArticleTitleDiminished linear growth during intermittent calcitriol therapy in children undergoing CCPD. Kidney Int 53 205–211 Occurrence Handle1:CAS:528:DyaK1cXnsFCiuw%3D%3D Occurrence Handle9453020
D Sömjen Y Weisman Z Mor A Harell AM Kaye (1991) ArticleTitleRegulation of proliferation of rat cartilage and bone by sex steroid hormones. J Steroid Biochem Mol Biol 40 717–723 Occurrence Handle10.1016/0960-0760(91)90296-H Occurrence Handle1958569
D Sömjen Y Weisman AM Kaye (1995) ArticleTitlePretreatment with 1,25(OH)2 vitamin D or 24,25(OH)2 vitamin D increases synergistically responsiveness to sex steroids in skeletal-derived cells. J Steroid Biochem Mol Biol 55 211–217 Occurrence Handle10.1016/0960-0760(95)00175-Y Occurrence Handle7495700
PD Benya JD Schaffer (1982) ArticleTitleDedifferentiated chondrocytes reexpress the differentiated collagen phenotype when cultured in agarose gels. Cell 30 215–224 Occurrence Handle1:CAS:528:DyaL38XlsVanur8%3D Occurrence Handle7127471
A Lindahl A Nilsson OGP Isaksson (1987) ArticleTitleEffects of growth hormone and insulin-like growth factor 1 on colony formation of rabbit epiphyseal chondrocytes at different stages of maturation. J Endocrinol 115 263–271 Occurrence Handle1:CAS:528:DyaL2sXmtFanurw%3D Occurrence Handle3437249
RT Ballock AH Reddi (1994) ArticleTitleThyroxine is the serum factor that regulates morphogenesis of columnar cartilage from isolated chondrocytes in chemically defined medium. J Cell Biol 126 IssueID5 1311–1318 Occurrence Handle1:CAS:528:DyaK2cXltVSgtrs%3D Occurrence Handle8063865
H Werner M Woloschak M Adam Y Shen-Orr CT Roberts Jr D LeRoith (1989) ArticleTitleDevelopmental regulation of the rat insulin-like growth factor I and receptor gene. Proc Natl Acad Sci USA 86 7451–7455 Occurrence Handle1:CAS:528:DyaL1MXmtFSjsrs%3D Occurrence Handle2477843
JK Burmester N Maeda HF Deluca (1988) ArticleTitleIsolation and expression of rat 1,25-dihydroxyvitamin D3 receptor DNA. Proc Natl Acad Sci USA 85 1005–1009 Occurrence Handle1:CAS:528:DyaL1cXhsVCmsbY%3D Occurrence Handle2829212
AED El Husseini JA Paterson RPC Shiu (1994) ArticleTitleBasic fibroblast growth factor (BFGF) and two of its receptors, FGFR 1 and FGFR 2: gene expression in the rat brain during postnatal development as determined by quantitative RT-PCR. Mol Cell Endocrinol 104 191–200 Occurrence Handle10.1016/0303-7207(94)90122-8 Occurrence Handle7527353
G Maor Y Segev M Phillip (1999) ArticleTitleTestosterone stimulates insulin-like growth factor-I and insulin-like growth factor-I-receptor gene expression in the mandibular condyle—a model for endochondral ossification. Endocrinology 140 1901–1910 Occurrence Handle1:CAS:528:DyaK1MXitFCqs7k%3D Occurrence Handle10098530
SG Ren S Malozowski P Sanchez DE Sweet DL Loriaux F Cassorla (1989) ArticleTitleDirect administration of testosterone increases rat tibial epiphyseal growth plate width. Acta Endocrinol 121 401–405 Occurrence Handle1:CAS:528:DyaL1MXmt1Gmsr4%3D Occurrence Handle2800918
Z Schwartz E Nasatzky A Ornoy BP Brooks WA Soskolne BD Boyan (1994) ArticleTitleGender-specific, maturation-dependent effects of testosterone on chondrocytes in culture. Endocrinology 134 1640–1647 Occurrence Handle1:CAS:528:DyaK2cXis1WlsLg%3D Occurrence Handle8137726
O Blanchard L Tsagris R Rappaport G Duval-Beaupere M Corvol (1991) ArticleTitleAge-dependent responsiveness of rabbit and human cartilage cells to sex steroids in vitro. J Steroid Biochem Mol Biol 40 711–716 Occurrence Handle10.1016/0960-0760(91)90295-G Occurrence Handle1:CAS:528:DyaK38XjsVOltg%3D%3D Occurrence Handle1958568
MT Corvol A Carrascosa L Tsagris O Blanchard R Rappaport (1987) ArticleTitleEvidence for a direct in vitro action of sex steroids on rabbit cartilage cells during skeletal growth: influence of age and sex. Endocrinology 120 1422–1429 Occurrence Handle1:CAS:528:DyaL2sXhs12gsb0%3D Occurrence Handle3830056
E Nasatzky Z Schwartz BD Boyan WA Soskolne A Ornoy (1993) ArticleTitleSex-dependent effects of 17-beta-estradiol on chondrocyte differentiation in culture. J Cell Physiol 154 359–367 Occurrence Handle1:CAS:528:DyaK3sXhsVSisrc%3D Occurrence Handle8425917
J Lindzey MV Kumar M Grossman C Young DJ Tindall (1994) ArticleTitleMolecular mechanism of androgen action. Vitam Horm 49 383–433 Occurrence Handle1:CAS:528:DyaK2MXjsVagsL0%3D Occurrence Handle7810074
MR Haussler CA Haussler PW Jurutka PD Thompson J-C Hsieh LS Remus SH Selznik GK Whitfield (1997) ArticleTitleThe vitamin D hormone and its nuclear receptor: molecular actions and disease states. J Endocrinol 154 57–73 Occurrence Handle9246938
WS Ashton BM Degnan A Daniel GL Francis (1995) ArticleTitleTestosterone increases insulin-like growth factor and insulin-like growth factor-binding protein. Ann Clin Lab Sci 25 381–388 Occurrence Handle1:CAS:528:DyaK2MXotlyhtLY%3D Occurrence Handle7486812
R Horton V Pasupuletti I Antonipillai (1993) ArticleTitleAndrogen induction of steroid 5α-reductase may be mediated via insulin-like growth factor-I. Endocrinology 133 447–451 Occurrence Handle1:CAS:528:DyaK3sXlvFKjsLs%3D Occurrence Handle8344190
F Gori LC Hofbauer CA Conover S Khosla (1999) ArticleTitleEffects of androgens on the insulin-like growth factor system in an androgen-responsive human osteoblastic cell line. Endocrinology 140 5579–5586 Occurrence Handle1:CAS:528:DyaK1MXns12htrs%3D Occurrence Handle10579321
I Zofkova RL Kancheva B Bendlova (1997) ArticleTitleEffect of 1,25(OH)2D3 on circulating insulin-like growth factor-I and ß2 microglobulin in patients with osteoporosis. Calcif Tissue Int 60 236–239 Occurrence Handle10.1007/s002239900221 Occurrence Handle1:CAS:528:DyaK2sXhsFKitL0%3D Occurrence Handle9069158
C Chenu A Valentin-Opran P Chavassieux S Saez PJ Meunier PD Delmas (1990) ArticleTitleInsulin-like growth factor I hormonal regulation by growth hormone and by 1,25(OH)2D3 and activity on human osteoblast-like cells in short-term cultures. Bone 11 81–86 Occurrence Handle1:CAS:528:DyaK3MXisVWhtL0%3D Occurrence Handle2357426
TL Chen JB Mallory RL Hintz (1991) ArticleTitleDexamethasone and 1,25(OH)2 vitamin D3 modulate the synthesis of insulin-like growth factor-I in osteoblast-like cells. Calcif Tissue Int 48 278–282 Occurrence Handle1:CAS:528:DyaK3MXlt1ynsLc%3D Occurrence Handle2059879
SH Scharla DD Strong S Mohan DJ Baylink TA Linkhart (1991) ArticleTitle1,25-dihydroxyvitamin D3 differentially regulates the production of insulin-like growth factor I (IGF-I) and IGF-binding protein-4 in mouse osteoblasts. Endocrinology 129 3139–3146 Occurrence Handle1:CAS:528:DyaK38Xhs1akuw%3D%3D Occurrence Handle1720089
PE Holthuizen PH Steenbergh JS Sussenbach (1999) Regulation of IGF gene expression. RG Rosenfeld CT Roberts (Eds) The IGF System. Humana Press Totowa NJ 37–61
F Aslam L McCabe B Frenkel AJ van Wijnen GS Stein JB Lian JL Stein (1999) ArticleTitleAP-1 and vitamin D receptor (VDR) signaling pathways converge at the rat osteocalcin VDR element: requirement for the internal activating protein-1 site for vitamin D-mediated trans-activation. Endocrinology 140 63–70 Occurrence Handle1:CAS:528:DyaK1MXhtFWgtw%3D%3D Occurrence Handle9886808
A Takeshita H Yasuda M Ishida K Ochiai (2002) ArticleTitlel,α25(OH)2D3 interferes with retinoic acid-induced inhibition of c-fos gene expression for AP-1 formation in osteoblastic cells. J Oral Sci 44 27–34 Occurrence Handle1:CAS:528:DC%2BD38XlvVSitL4%3D Occurrence Handle12058867
K Fronsdal N Engedal T Slagsvold F Saatcioglu (1998) ArticleTitleCREB binding protein is a coactivator for the androgen receptor and mediates cross-talk with AP-1. J Biol Chem 273 31853–31859 Occurrence Handle10.1074/jbc.273.48.31853 Occurrence Handle9822653
AM Delany D Durant E Canalis (2001) ArticleTitleGlucocorticoid suppression of IGF I transcription in osteoblasts. Mol Endocrinol 15 1781–1789 Occurrence Handle1:CAS:528:DC%2BD3MXnt12isbY%3D Occurrence Handle11579210
G Yang CW Gregory Q Shang DA O’Brien YL Zhang (2001) ArticleTitleDifferential expression of CCAAT/enhancer-binding protein-δ (C/EBPδ) in rat androgen-dependent tissues and human prostate cancer. J Androl 22 471–480 Occurrence Handle1:CAS:528:DC%2BD3MXjslKmt7c%3D Occurrence Handle11330648
MH Ahonen YH Zhuang R Aine T Ylikomi P Tuohimaa (2000) ArticleTitleAndrogen receptor and vitamin D receptor in human ovarian cancer: growth stimulation and inhibition by ligands. Int J Cancer 86 40–46 Occurrence Handle1:CAS:528:DC%2BD3cXitVegt78%3D Occurrence Handle10728592
G Maor Z Hochberg M Silbermann (1993) ArticleTitleInsulin-like growth factor I accelerates proliferation and differentiation of cartilage progenitor cells in cultures of neonatal mandibular condyles. Acta Endocrinol 128 56–64 Occurrence Handle1:CAS:528:DyaK3sXisVynsbg%3D Occurrence Handle8447195
M Takeuchi H Kakushi M Tohkin (1994) ArticleTitleAndrogens directly stimulate mineralization and increase androgen receptors in human osteoblast-like cells. Biochem Biophys Res Comm 204 905–911 Occurrence Handle10.1006/bbrc.1994.2545 Occurrence Handle1:CAS:528:DyaK2cXmslamsLw%3D Occurrence Handle7980559
C Kasperk A Helmboldt I Boercsoek S Heuthe O Cloos F Niethard R Ziegler (1997) ArticleTitleSkeletal site-dependent expression of the androgen receptor in human osteoblastic cell populations. Calcif Tissue Int 61 464–473 Occurrence Handle10.1007/s002239900369 Occurrence Handle1:CAS:528:DyaK2sXnvVWjurw%3D Occurrence Handle9383273
TY Hsieh CY Hg C Mallouh H Tazaki JM Wu (1996) ArticleTitleRegulation of growth, PSA/PAP and androgen expression by 1 alpha, 25-dihydroxyvitamin D3 in the androgen-dependent LNCaP cells. Biochem Biophys Res Comm 223 141–146 Occurrence Handle10.1006/bbrc.1996.0859 Occurrence Handle1:CAS:528:DyaK28XjsFGhtL8%3D Occurrence Handle8660360
N Balmain B von Eichel R Toury F Belquasmi M Hauchecorne G Klaus O Mehls E Ritz (1995) ArticleTitleCalbindin-D28K and D9K and 1,25(OH)2 vitamin D3 receptor immuno-localization and mineralization induction in long-term primary cultures of rat epiphyseal chondrocytes. Bone 17 37–45 Occurrence Handle10.1016/8756-3282(95)00132-W Occurrence Handle1:CAS:528:DyaK2MXnt1Omu7c%3D Occurrence Handle7577156
N Kergosien JM Sautier N Forest (1998) ArticleTitleGene and protein expression during differentiation and matrix mineralization in a chondrocyte cell culture system. Calcif Tissue Int 62 114–121 Occurrence Handle10.1007/s002239900404 Occurrence Handle1:CAS:528:DyaK1cXnslChsg%3D%3D Occurrence Handle9437044
RJ O’Keefe JE Puzas L Loveys DG Hicks RN Rosier (1994) ArticleTitleAnalysis of type II and type X collagen synthesis in cultured growth plate chondrocytes by in situ hybridization: rapid induction of type X collagen in culture. J Bone Miner Res 11 1713–1722
L Adan JC Souberbielle R Brauner (1994) ArticleTitleManagement of the short stature due to pubertal delay in boys. J Clin Endocrinol Metab 78 478–482 Occurrence Handle1:STN:280:ByuC2cbhsFI%3D Occurrence Handle7508951
RA Richman LR Kirsch (1988) ArticleTitleTestosterone treatment in adolescent boys with constitutional delay in growth and development. N Engl J Med 319 1563–1567 Occurrence Handle1:STN:280:BiaD2s%2FmtFY%3D Occurrence Handle3200264
MW Van Steenbergen JM Wit . Donckerwolcke (1991) ArticleTitleTestosterone esters advance skeletal maturation more than growth in short boys with chronic renal failure and delayed puberty. Pediatrics 150 676–680 Occurrence Handle1:STN:280:By2D3cfhtVY%3D
Acknowledgements
Part of this work was presented at the 33rd Annual Meeting of the European Society for Paediatric Nephrology in Prague, Septemer 2–5, 1999, Czech Republic and at the 32nd Annual Meeting of the American Society of Nephrology in Miami Beach, Florida, November 4–8, 1999, USA
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Krohn, K., Haffner, D., Hügel, U. et al. 1,25(OH)2D3 and Dihydrotestosterone Interact to Regulate Proliferation and Differentiation of Epiphyseal Chondrocytes . Calcif Tissue Int 73, 400–410 (2003). https://doi.org/10.1007/s00223-002-2160-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-002-2160-9