
Abstract
A 16-year-old male patient with type II autosomal dominant benign osteopetrosis (ADO) was genotyped and found to harbor a novel mutation in exon 25 of the gene encoding for the osteoclast-specific chloride channel, CLCN7, inherited from the father, who was asymptomatic. The patient had normal biochemical findings and acid-base balance, except for increased serum levels of creatine kinase, lactic dehydrogenase, and the bone formation markers bone alkaline phosphatase isoenzyme, osteocalcin and N-terminal type I collagen telopeptide/creatinine ratio. Unusual generalized osteosclerosis was observed together with a canonical increase in vertebral and pelvis bone mass. An affected first grade cousin presented with normal biochemical findings and a milder osteosclerotic pattern of the pelvis. At the cellular level, cultured osteoclasts from the patient showed increased motility, with lamellipodia, membrane ruffling and motile pattern of podosome distribution, all of which could have contributed to functional impairment of bone resorption. The present report documents a novel mutation of the CLCN7 gene causing osteopetrosis in a radiologically uncertain form of the diseases, with apparent incomplete penetrance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Autosomal dominant osteopetrosis (ADO) is a rare disease characterized by dense bones and a vast variety of heterogeneous symptoms, including hematological and neural defects with diverse severity [1]. It is generally seen in adults, however, in children from affected families it can be recognized in earlier stages. This disease has been classified in two subclasses, type I and type II, each characterized by specific patterns. The hallmark of type I ADO is a generalized cortical and cancellous bone volume increase, but normal fracture pattern and biochemical properties [2]. Mutations of the LRP5 LDL receptor were found to affect two families with this form of ADO [3]. Type II ADO, also known as Albers-Schönberg disease, is characterized primarily by vertebral endplate thickening (“sandwich” or “rugger-jersey” vertebral appearance) which includes increased cortical but normal cancellous bone volume, fragile bones with multiple fractures and delayed healing [2,4]. Type II ADO is the most common form, with an estimated prevalence of 5.5/100,000. Recently, it has been noted that CLCN7 gene mutations underlie type II ADO [5]. This gene encodes for the type 7 chloride channel located in the osteoclast-ruffled border membrane [6,7] where it compensates for active proton extrusion by chloride release. Its mutations generate dominant negative proteins as it appears in type II ADO [5]. They may also lead to protein loss of function responsible for an extremely rare form of autosomal, recessive, infantile, malignant osteopetrosis also characterized by primary retinal degeneration [8]. Approximately 50% of patients with autosomal recessive malignant osteopetrosis harbor mutations of the ATP6i gene, also denominated TCIRG1, encoding for the osteoclast-specific a3 subunit of the vacuolar H+-ATPase [9].
Due to the heterogeneity of symptoms, many forms of osteopetrosis need to be specifically classified. Neural failure is described in osteopetrosis, which includes mental retardation, as well as visual and hearing impairment due to nerve compression syndrome caused by narrowing of nerve foramina [4]. Some forms of the disease also include calcification of the brain and renal tubular acidosis, as it has been described in the so-called “marble brain syndrome” [1]. This form is due to mutations of the CAII gene encoding for carbonic anhydrase type II [10]. Tubular acidosis has recently been described in a case of malignant osteopetrosis caused by a double gene mutation, one in the ATP6i gene, and the other in the ATPV1B1 gene encoding for the kidney-specific B1 subunit of the vacuolar H+-ATPase [11].
Materials and Methods
Proband
This study was performed with the informed consent of the individuals analyzed. The proband was a 16-year-old male admitted to our care unit with lumbar spine and pelvis pain associated with functional limitation. The patient was evaluated radiologically, clinically and biochemically and was diagnosed as having an unclassifiable benign osteopetrosis.
Materials
Cell culture media, serum and reagents were from Gibco (Uxbridge, U.K.). Sterile glass ware was from Falcon Becton Dickinson (Meylan, France). The anti-αvβ3 antibody, LM609, and the anti-Fc receptor (CD16) antibody were from Chemicon International Inc. (Tamecula, CA). DNA extraction kit was from Stratagene (Amsterdam, The Netherlands). Reagents for PCR were from Qiagen (Genenco, Florence, Italy). Superscript II - reverse transcriptase was from Life-Technologies (Milan, Italy). Dye Terminator cycle sequencing ready reaction mix was from Perkin-Elmer Applied Biosystem (Foster City, CA, USA). Recombinant human Macrophage Colony Stimulating Factor (M-CSF) and recombinant human receptor activator of NF-kB ligand (RANKL) were from Peprotech EC Ltd. (London, UK). All other reagents were of the purest grade from Sigma Chemical Co. (St. Louis, MO).
Mutation Analysis
Peripheral blood samples were collected from patients, family members and informed healthy donors. DNA was extracted from EDTA-blood samples using a DNA extraction kit. The entire coding region of the type 7 chloride channel subunit was amplified using intronic primers, as described by Kornak et al. [8]. PCR reactions were carried out using the Qiagen master mix kit, including 1× PCR buffer, 1× Q-solution, 200 µM dNTP, 0.5 µM primer pair and 2.5 units/reaction Taq DNA polymerase. PCR products were purified with the Qiagen PCR purification kit (cat. #28104) following standard protocols recommended by the manufacturer. Purified PCR (10 ng) was used for 100 bp of DNA cycle sequencing which was performed using a Perkin-Elmer dye terminator cycle sequencing ready reaction mix, according to standard procedure. Reactions were applied to a Perkin-Elmer ABI 377 DNA sequencer. Sequences were aligned using the NCBI BLAST 2 program.
Osteoclast Preparation from Peripheral Blood Monocytes
Peripheral blood mononuclear cells were prepared from human blood diluted in Hanks balanced salt solution (1:1). Diluted blood was layered over Histopaque 1077 solution, centrifuged at 400× g for 30 min, then washed twice with Hanks solution and centrifuged at speed as above for 10 min. Cells were resuspended in DMEM containing 4 mM L-glutamine, 100 U/ml penicillin, 100 µg/ml streptomicin and 10% FCS. Three × 106 cells/cm2 were plated on glass coverslips or bone slices and incubated at 37° C in a humidified atmosphere with 5% CO2. After 1 hour, cell cultures were rinsed to remove nonadherent cells and maintained in medium as described above in the presence of 25 ng/ml M-CSF, 30 ng/ml RANK-L and 100 nM parathyroid hormone (PTH) for 21 days. Medium and factors were replaced every 3–4 days.
Tartrate-resistant Acid Phosphatase (TRAP) Activity
Cells were fixed in 3% paraformaldehyde in 0.1 M cacodylate buffer for 15 min, then extensively washed in the same buffer. TRAP activity was detected histochemically using the Sigma-Aldrich kit n. 386, according to the manufacturer’s instruction.
Fluorescence Microscopy
Cells plated on glass coverslips were fixed with 4% paraformaldehyde in PBS, at 4°C for 10 min, washed three times in PBS, then permeabilized with 0.05% Triton X-100 in PBS at 4°C for 10 min and incubated in 1% BSA, at room temperature for 15 min. Microfilaments were decorated by incubation with 5 µg/ml TRITC-conjugated phalloidin, at 37°C for 1 hour. Samples were viewed with conventional light or epifluorescence microscopy in a Zeiss Axioplan microscope (Jena, Germany).
Results
Proband
Most biochemical findings and the acid-base balance were within normal range. However, increased serum levels of creatine kinase, lactic dehydrogenase, bone alkaline phosphatase isoenzyme and osteocalcin, as well as increased N-terminal type I collagen telopeptide/creatinine ratio (Table 1) were noted. The patient also showed low levels for age of testosterone (2.84 ng/ml, normal values: 2.8–9.0 ng/ml), luteinizing hormone (0.9 mIU/ml, normal values: 1.5–9.2 mIU/ml) and follicle-stimulating hormone (0.88 mIU/ml, normal values: 1–14 mIU/ml).
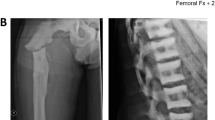
Radiographic analysis (Fig. 1) and bone mineral densitometry (not shown) demonstrated generalized osteosclerosis and increase of vertebral and pelvis bone mass, albeit with normal mineral content values. The patient suffered from recurrent bronchitis and otitis, and right ear deafness. A modest alteration of the acoustic nerve foramina was, however, more marked on the left side, which was not accompanied by size reduction. Therefore, this appearance was not thought to be pathological. Of note, cranial magnetic resonance imaging revealed a 5 cm diameter arachnoid cyst located at the level of the left temporal lobe with cortices compression. Electroencephalogram demonstrated cortical alteration, particularly in the left frontal-temporal area (not shown).

Radiographic analysis of the proband. (A) Humerus with sclerosis bands (arrow), (B) frontal and (C) lateral view of lumbar spine with “rugger-jersey” aspect (arrows), (D) hands’ phalange and (E) pelvis with “bone in bone” appearance (arrows).
Mutational Analysis
The patient was genotyped by direct DNA sequencing and found to harbor a novel mutation in the gene encoding for the osteoclast-specific chloride channel, CLCN7 [4] (Fig. 2). The mutation consisted of a heterozygous C/A transition in exon 25, codon 788, position 2401 of mRNA, predicted to lead to replacement of alanine (GCC) with aspartic acid (GAC). This genotype was not found in 50 control chromosomes.

Genetic analysis of CLCN7 in the available members of the kindred. (A) Pedigree structure of the kindred. Arrow indicates the proband Φ and indicate normal phenotype in members harboring the mutation. (B) Direct DNA sequencing of the proband (Left) and his unaffected mother (Right). Arrow indicates the site of heterozygous mutation (G/A transition, exon 25, codon 788).
Kindred Analysis
Family history was unremarkable. However, genotype of the family was performed and revealed the same heterozygous CLCN7 gene mutation in the father. Mother and sister were unaffected (Fig. 2). The father was asymptomatic, with clinical, biochemical (not shown) and radiographic (Fig. 3) analyses within normal ranges. Moreover, the mutation was also found in a first-grade cousin (Fig. 2) presenting with a mild diffuse osteosclerosis of the pelvis (Fig. 3), but otherwise normal patterns. He probably inherited the mutation from his asymptomatic mother, who was not available for DNA sequencing.

Radiographic analysis of relatives. (A) Pelvis and (B) lateral view of lumbar spine of the father. (C) Pelvis and (D) lateral view of lumbar spine of the first grade cousin. Note diffuse sclerosis in (C) and normal pattern in (A), (B) and (D).
Cell Study
At the cellular level, we noted that in our patient neutrophil counts and function, which could account for recurrent infections, were unremarkable. In contrast, relative to control subjects, cultured osteoclasts, generated from the peripheral blood monocytic fraction, showed a more motile phenotype compared to control osteoclasts, as demonstrated by the appearance of lamellipodia, membrane ruffling and motile pattern of podosome distribution (Fig. 4).

Micrographs of osteoclasts generated from peripheral blood monocytes. Peripheral blood mononuclear cells were separated and osteoclasts differentiated as described in Methods. (A) and (B): tartrate-resistant acid phosphatase histochemical detection. Original magnification 200×. (C) and (D): Rodhamine-conjugated phalloidin decoration of microfilaments. Original magnification 400×. (A) and (C): osteoclasts from a healthy donor; (B) and (D): osteoclasts from patient. Note the motile phenotype in patient’s versus control’s osteoclasts. Arrowheads: osteoclasts; small arrow: lamellipodium; large arrow: podosomes.
Discussion
Symptoms in osteopetrosis are quite heterogeneous even within the same class of the disease. ADO accounts for the majority of cases, with a generally benign outcome ranging from very mild, non-symptomatic, to severe forms characterized by multiple pathological fractures. In our patient, clinical classification of the disease was unclear. The generalized osteosclerosis and normal fracture pattern would have suggested type I ADO, whereas altered biochemical properties, recurrent infections and increased vertebral and pelvis bone mass would identify this form as type II ADO. Type II ADO was confirmed by direct DNA sequencing of the CLCN7 gene, which was found to be mutated in all patients with this form of osteopetrosis investigated so far [5].
The mutation of our patient is novel. Nine other mutations are presently known, 7 of which lead to type II ADO in 12 unrelated families (dominant negative effect), and 2 to autosomal recessive malignant osteopetrosis also characterized by primary retina degeneration (loss of function effect) [5,6]. Our mutation is located in exon 25, which encodes the C-terminal domain of the CLCN7 protein. Only one mutation was recognized to date in exon 25. It was observed in two type II ADO unrelated families, consisted in an AG deletion in position 2423 of the mRNA, and was predicted to lead to frame-shift resulting in a protein that would differ from wild-type CLCN7 only in the last 10 amino acids [5]. Our mutation is the second known to occur in exon 25, at position 2401 of mRNA, and is predicted to cause replacement of alanine with aspartic acid. This change does not seem to represent a gene polymorphism since it was not found in alleles from normal subjects, and appears to lead to a mild form of type II ADO, with some features also shared by the type I subtype, especially diffuse osteosclerosis and absence of pathological fractures. Penetrance in this family is thought to be incomplete as the carrier father showed an apparently normal phenotype, and the affected cousin showed a milder pattern of osteosclerosis and normal biochemical findings.
Interestingly, our proband showed increased markers of bone formation (i.e., bone alkaline phosphatase isoenzyme, osteocalcin and N-terminal type I collagen telopeptide/creatinine ratio). Whether or not enhancement of osteoblast activity occurs in osteopetrosis is controversial. In animal models of osteopetrosis, increased bone formation has been described [12], and in patients harboring mutations in the ATP6i gene encoding for the a3 subunit of the osteoclast V-ATPase [16] we observed active osteoblasts in bone biopsies and increased serum alkaline phosphatase activity [16]. The latter marker is often enhanced in several forms of osteopetrosis [1], consequently, it is feasible that bone formation can be affected in these individuals. Hence, we recommend measuring bone formation markers in osteopetrotic patients and analyzing osteoblast activity by bone histomorphometry and functional and molecular assays in vitro, to address whether or not this represents a hallmark of the disease or sporadically characterizes some individuals.
Osteopetrosis is often associated with neural failure, mostly due to nerve compression syndromes [1]. In our patient, we did observe right ear deafness, and evidence of compression of the auditory nerve was therefore investigated. However, changes in the auditory nerve foramina appeared very modest, with no narrowing of foramen size. We believe that this is not likely to account for the acoustic impairment. Therefore, alternative causes were considered and cranial magnetic resonance imaging revealed the presence of a broad arachnoid cyst located in the left temporal lobe, which caused compression of the neural cortices. The primary acoustic area is located in the temporal area, therefore this finding could indeed explain the severe controlateral acoustic loss observed in our patient, which appears to be independent of the osteopetrosis pattern.
At the cellular level, our results suggested that recurrent infections were independent of altered neutrophil counts or function as both parameters were normal. In contrast, osteoclasts appeared more motile relative to cells from normal subjects. Motility is a requirement for osteoclasts to approach the resorbing site and then move aside at the end of the resorption phase. At the time of resorption, mature osteoclasts arrest and acquire a unique morphological conformation featuring membrane polarization and the appearance of a peripheral adhesion area characterized by intense actin ring formed by countless podosomes [13,14,15]. In osteoclasts plated in culture dishes, a disk-like shape and peripheral ring-like distribution of podosomes are noted. Osteoclasts from our patient were instead elongated and presented extensive podosome-containing lamellipodia. We believe that this change could perturb their resorption ability, albeit with a mild end result, as suggested by the benign course of the disease in our proband. This is at variance with the morphological features observed in osteoclasts derived from osteopetrotic patients harboring mutations of the ATP6i gene, where no morphological changes were observed in bone biopsies and in cultured osteoclasts [16]. From our study it is not clear whether such a motile phenotype is a direct consequence of the mutation in the CLCN7 gene or whether it indirectly depends on the reduced bone resorption ability of the osteoclasts. Our findings in ATP6i-dependent osteopetrosis would rule out the latter possibility given that in these patients bone resorption is severely compromised. However, no data are yet available to conclude that the CLCN7 gene contributed to the morphological organization of the resorbing osteoclasts.
In conclusion, we presented a family affected by type II ADO showing a novel CLCN7 gene mutation and unusual clinical manifestations. We believe that this genotype-phenotype correlation may contribute to a better understanding of the clinical features of this class of genetically and phenotypically heterogeneous diseases.
References
MP Whyte (2002) Osteopetrosis. PM Royce B Steinmann (Eds) Connective tissue and its heritable disorders: medical, genetic, and molecular aspects, 2nd ed. Wiley-Liss New York 753–770
J Bollerslev PE Anderson Jr (1988) ArticleTitleRadiological, biochemical and hereditary evidence of two types of autosomal dominant osteopetrosis. Bone 91 7–13
L Van Wesenbeeck E Cleiren J Gram et al. (2003) ArticleTitleSix novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet 72 763–771 Occurrence Handle10.1086/368277 Occurrence Handle1:CAS:528:DC%2BD3sXitFajs7Y%3D Occurrence Handle12579474
O Bénichou JD Laredo MC de Vernejoul (2000) ArticleTitleType II autosomal dominant osteopetrosis (Albers-Schönberg disease): clinical and radiological manifestations in 42 patients. Bone 26 87–93 Occurrence Handle1:STN:280:DC%2BD3c%2FotlCmsw%3D%3D Occurrence Handle10617161
E Cleiren O Benichou E Van Hul et al. (2001) ArticleTitleAlbers-Schonberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the CLCN7 chloride channel gene. Hum Mol Genet 10 2861–2867 Occurrence Handle1:CAS:528:DC%2BD38XjtVegtg%3D%3D Occurrence Handle11741829
A-V Rousselle D Heymann (2002) ArticleTitleOsteoclastic acidification pathways during bone resorption. Bone 30 533–540 Occurrence Handle10.1016/S8756-3282(02)00672-5 Occurrence Handle1:CAS:528:DC%2BD38XisV2ksL8%3D Occurrence Handle11934642
G Stenbeck (2002) ArticleTitleFormation and function of the ruffled border in osteoclasts. Cell Dev Biol 13 285–292 Occurrence Handle10.1016/S1084952102000587 Occurrence Handle1:CAS:528:DC%2BD38XntVanu7w%3D
U Kornak D Kasper MR Bosl E Kaiser M Schweizer A Schulz W Friedrich G Delling TJ Jentsch (2001) ArticleTitleLoss of the CIC-7 chloride channel leads to osteopetrosis in mice and man. Cell 104 205–215 Occurrence Handle1:CAS:528:DC%2BD3MXis1Kmu7s%3D Occurrence Handle11207362
A Frattini PJ Orchard C Sobacchi et al. (2000) ArticleTitleDefects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nature Genet 25 343–346 Occurrence Handle10.1038/77131 Occurrence Handle1:CAS:528:DC%2BD3cXkvFKktLo%3D Occurrence Handle10888887
WS Sly PY Hu (1995) The carbonic anhydrase II deficiency syndrome: osteopetrosis with renal tubular acidosis and cerebral calcification. CR Scriver AL Beaudet WS Sly D Valle (Eds) The metabolic and molecular bases of inherited diseases, 7th ed, McGraw-Hill New York 4113–4124
KJ Borthwick N Kandemir R Topaloglu et al. (2003) ArticleTitleA phenocopy of CAII deficiency: a novel genetic explanation of inherited infantile osteopetrosis with distal tubular acidosis. J Med Genet 40 115–121 Occurrence Handle10.1136/jmg.40.2.115 Occurrence Handle1:CAS:528:DC%2BD3sXhslOgsLo%3D Occurrence Handle12566520
M Marzia NA Sims S Voit et al. (2000) ArticleTitleDecreased c-Src expression enhances osteoblast differentiation and bone formation. J Cell Biol 151 311–320 Occurrence Handle10.1083/jcb.151.2.311 Occurrence Handle1:CAS:528:DC%2BD3cXnsVaqsL0%3D Occurrence Handle11038178
M Zaidi HC Blair BS Moonga E Abe CL-H Huang (2003) ArticleTitleOsteoclastogenesis, bone resorption, and osteoclast-based therapeutics. J Bone Miner Res 18 599–609
JE Aubin (1992) ArticleTitleOsteoclast adhesion and resorption: the role of podosomes. J Bone Miner Res 7 365–368
A Teti PC Marchisio AZ Zallone (1991) ArticleTitleClear zone in osteoclast function: role of podosomes in regulation of bone-resorbing activity. Am J Physiol . C1–C7
A Taranta S Migliaccio I Recchia et al. (2003) ArticleTitleGenotype-phenotype relationship in human ATP6i-dependent autosomal recessive osteopetrosis. Am J Pathol 162 57–68 Occurrence Handle1:CAS:528:DC%2BD3sXlsFyruw%3D%3D Occurrence Handle12507890
Acknowledgements
We thank Prof. Paolo Menè for helpful discussions. The Telethon grant #E.0831 and the “Fondo per gli Investimenti per la Ricerca di Base” (FIRB) grant #RBAU01X3NH to A.T. are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Letizia, C., Taranta, A., Migliaccio, S. et al. Type II Benign Osteopetrosis (Albers-Schönberg Disease) Caused by a Novel Mutation in CLCN7 Presenting with Unusual Clinical Manifestations . Calcif Tissue Int 74, 42–46 (2004). https://doi.org/10.1007/s00223-002-1087-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-002-1087-5