
Abstract
The brains of Alzheimer’s disease patients show an increased number of senile plaques compared with normal patients. The major component of the plaques is the β-amyloid peptide, a cleavage product of the amyloid precursor protein (APP). Although the processing of APP has been well-described, the physiological functions of APP and its cleavage products remain unclear. This article reviews the multifunctional roles of an APP orthologue, the C. elegans APL-1. Understanding the function of APL-1 may provide insights into the functions and signaling pathways of human APP. In addition, the physiological effects of introducing human β-amyloid peptide into C. elegans are also reviewed. The C. elegans system provides a powerful genetic model to identify genes regulating the molecular mechanisms underlying intracellular β-amyloid peptide accumulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder that is characterized clinically by age-related cognitive dysfunction and pathologically by the accumulation of dense plaques and neurofibrillary tangles in the brains of AD patients (Kidd 1964; Krigman et al. 1965; Luse and Smith 1964; Terry et al. 1964). The major component of the plaques is the β-amyloid peptide (Aβ; (Glenner and Wong 1984; Masters et al. 1985), which is a cleavage product of the amyloid precursor protein (APP; Kang et al. 1987). Autosomal-dominant mutations in APP have been correlated with 10–15% of early-onset AD cases (Chartier-Harlin et al. 1991; Goate et al. 1991; Murrell et al. 1991; Bird 2010). The majority (30–70%) of the mutations correlated with early-onset AD, however, are in the presenilin genes, PS1 and PS2, which encode components of the γ-secretase complex that cleaves APP to produce Aβ (Kimberly et al. 2003; Takasugi et al. 2003; Bird 2010). These results suggest that Aβ plays a central role in the disease. The contribution of other APP cleavage products to the pathology of AD is unclear.
Function of the APP family
To ensure that therapeutic strategies do not interfere with essential functions of APP, the functions of APP have been addressed using mouse knockout (KO) models. Mice that carry a null allele of APP or that produce low levels of a truncated form of APP show several phenotypes, including a reduced body weight, reactive gliosis, agenesis of the corpus callosum, defects in spatial learning and exploratory behavior, and/or decreased locomotor activity and forelimb grip strength (Muller et al. 1994; Zheng et al. 1995). Overexpression of APP in mice also results in several phenotypes, which vary in severity, presumably because of varying levels of APP expression. These phenotypes include lethality, neophobia, impaired spatial alteration, reactive gliosis, and an increase in the number of synaptophysin and GAP-43 immunoreactive presynaptic terminals (Mucke et al. 1994; Hsiao et al. 1995).
Analyzing the in vivo functions of APP in mammals, however, is complicated by the presence of two APP-related genes, APLP1 and APLP2 (Wasco et al. 1992, 1993a, b; Sprecher et al. 1993; Slunt et al. 1994). The APP-related proteins are similar to APP, but do not contain Aβ. APP and APP-related proteins share a cysteine-rich region (E1) and a region rich in acidic residues (E2) in their extracellular domains; the highest sequence similarity among these proteins, however, is within the small intracellular domain (AICD) (Kang et al. 1987; Yamada et al. 1987; Wasco et al. 1992, 1993b; Sprecher et al. 1993; Slunt et al. 1994; Paliga et al. 1997; Lenkkeri et al. 1998).
The functions of the APP-related genes have also been analyzed through KO mouse models. APLP1 KO mice have a reduced body weight (Heber et al. 2000), while APLP2 KOs show no obvious defects (von Koch et al. 1997). Interestingly, double KOs of APLP2 and APP or APLP2 and APLP1 result in postnatal lethality (von Koch et al. 1997; Heber et al. 2000). Moreover, triple KO mice in which all three APP family members are inactivated show postnatal lethality and type II lissencephaly; cortical areas are disorganized and show irregular protrusions (Herms et al. 2004). Collectively, these results suggest that APP family members have essential and overlapping functions during development, including proper brain development, and these functions do not require Aβ. The molecular pathways in which APP participates during development, however, remain unknown.
Several genes implicated in Alzheimer’s disease (AD), such as APP and the presenilins, have orthologues in the nematode Caenorhabditis elegans (Daigle and Li 1993; Levitan et al. 1996; Li and Greenwald 1997; Westlund et al. 1999; Goutte et al. 2000; Francis et al. 2002); reviewed in (Ewald and Li 2010). Roughly 38% of the C. elegans genes have human orthologues (Shaye and Greenwald 2011); furthermore, an astonishing 40–50% of the identified human disease genes have C. elegans orthologues that show on average 50 and 70% amino acid identity and similarity, respectively (Ahringer 1997; Wheelan et al. 1999; Culetto and Sattelle 2000). Hence, C. elegans presents a powerful complementary genetic system to understand the function, processing, and regulation of APP and related proteins. In this review, we focus on the function of the C. elegans orthologue APL-1. In addition, we review using the power of C. elegans genetics to identify genes underlying the intracellular accumulation of the human β-amyloid peptide introduced by transgene expression.
Caenorhabditis elegans as a model system to examine neurodegenerative disorders
C. elegans is a multicellular eukaryotic organism with a short reproductive cycle of 3 days (Brenner 1974). From the fertilization of the egg to the adult hermaphrodite, each of the 959 somatic cells is characterized and delineated (Sulston and Horvitz 1977; Sulston et al. 1983). About one-third of all its somatic cells are neurons (302 neurons), which are connected by 5,000 chemical synapses, 2,000 neuromuscular junctions, and 600 electrical synapses (White et al. 1986). All of these synaptic connections have been analyzed by serial section electron micrograph reconstructions (White et al. 1986), establishing the first organism for which the entire neural wiring circuitry is known. Neurons use classical neurotransmitters, such as acetylcholine (Hosono et al. 1987), dopamine (Sulston et al. 1975), GABA (McIntire et al. 1993), glutamate (Lee et al. 1999), and serotonin (Horvitz et al. 1982), as well as numerous neuropeptides (Li and Kim 2008). Since C. elegans is transparent, neuronal structures and morphologies are easily traceable by expressing green fluorescent protein (GFP) in a neuron of choice, and the cellular localization of a protein can be visualized by tagging the protein with GFP (Chalfie et al. 1994). The function of individual neurons can be elucidated either by genetic alterations or by laser ablation of single cells (Chalfie et al. 1985; Bargmann and Horvitz 1991). Recently, optogenetic techniques have been adapted to C. elegans, such that the activity of neurons can be measured by monitoring calcium levels (Kerr et al. 2000) and neurons can be activated by blue light stimulation of channelrhodopsin (Nagel et al. 2005). Despite C. elegans adults being only about 1 mm in size and its neuronal cell bodies only one to two μm (White et al. 1986), electrophysiological methods to patch-clamp neurons have been successfully described (Goodman et al. 1998), although it is unclear whether C. elegans produces action potentials (Lockery and Goodman 2009). Lastly, C. elegans is easy to manipulate genetically (Brenner 1974) and generating transgenic animals is relatively fast and straightforward (Mello et al. 1991; Mello and Fire 1995). In summary, C. elegans is an easy, tractable and well-established system to investigate developmental and neuron-specific functions.
C. elegans APL-1 has an essential function
Like Drosophila (Rosen et al. 1989), C. elegans contains only one APP family member, APL-1 (Daigle and Li 1993). APL-1 shows high amino acid similarity within the extracellular domain (46% for E1 and 49% for E2) and the intracellular domain (71%) to human APP; however, similar to Drosophila APPL and mammalian APLP1 and APLP2, C. elegans APL-1 does not contain an Aβ sequence (Daigle and Li 1993). Hence, the Aβ sequence may be a vertebrate-specific development. Loss of apl-1 results in a completely penetrant early larval lethality (Hornsten et al. 2007; Wiese et al. 2010), showing that like the mammalian APP gene family, apl-1 also has an essential function. By contrast, Drosophila mutants in which appl was inactivated were viable and had subtle behavioral deficits (Luo et al. 1992). Hence, C. elegans can be a useful model organism to reveal the essential function of APP in development.
APL-1 expressed in neurons is essential for survival in C. elegans
Like mammalian APP family members, but unlike Drosophila, apl-1 is expressed in multiple cell types, including a subset of neurons, muscles, seam cells, hypodermal cells, and supporting cells (Hornsten et al. 2007; Niwa et al. 2008). By examining expression of APL-1 tagged at the C-terminus with GFP (APL-1::GFP), APL-1 appears to be continuously expressed starting from the 30-cell egg-stage through adulthood (Hornsten et al. 2007; Wiese et al. 2010). This broad expression pattern suggests that apl-1 is involved in multiple functions and may explain the pleiotropic phenotypes of apl-1 KOs. For instance, after hatching, C. elegans develops through four larval stages before arising as an adult; each larval stage transition is punctuated by a molt, whereupon the animals synthesize a new cuticle and undergo a stereotypic pattern of movements to emerge from the old cuticle, a process referred to as ecdysis (Singh and Sulston 1978). apl-1 KOs are unable to fully shed their old cuticle, leading to the lethality seen during the first to second larval stage transition (Hornsten et al. 2007; Wiese et al. 2010). However, by pinching a hole in the old cuticle with a needle, second larval stage apl-1 KO mutants can emerge and develop to the next larval stage, whereupon once again, the mutant is unable to molt properly and dies (C. Ewald and A. Silva, unpublished obs.). These data indicate that apl-1 activity is essential during the molting ecdysis process not just during the first to second larval stage transition, but also during later larval transitions. In addition, the apl-1 KO mutants have several other defects, including the formation of large vacuolar-like structures in hypodermal cells (Fig. 1b), the separation of organs from underlying basement membrane (Fig. 1d) and occasionally, the lack of posterior morphogenesis (Hornsten 2001; Hornsten et al. 2007). Germline transformation by introduction of full-length APL-1 or APL-1::GFP driven by its endogenous promoter completely rescued the apl-1 phenotypes (Hornsten et al. 2007; Wiese et al. 2010).

Loss of apl-1 leads to lethality, detachment of cells, and formation of vacuolar-like structures. apl-1 complete loss-of-function (KO) animals show a fully penetrant lethality due to a molting defect. KO animals also show other defects. a–d Show head regions of C. elegans at the first larval stage (L1; anterior left, ventral side down). apl-1(yn10) KO mutants (b) show vacuolar-like structures (arrow) in the hypodermis, which were not observed in wild-type animals (a). A heterozygous apl-1(yn10) animal (c) appears superficially wild type. By contrast, cells appeared detached (arrowheads) in an apl-1(yn32) KO mutant. Scale bar 10 μm
To pinpoint the cells in which apl-1 expression is critical for viability, APL-1 transgenes were constructed such that apl-1 expression could be driven in specific tissues. Driving apl-1 expression in hypodermal cells or muscle tissue did not rescue the lethality of apl-1 KOs (Hornsten et al. 2007). This result was somewhat surprising given that the cuticle is produced by hypodermal cells (Singh and Sulston 1978). By contrast, driving apl-1 expression in neurons rescued the lethality of apl-1 KO mutants, suggesting that APL-1 expression in neurons triggers the stereotypic pattern of movements to emerge from the old cuticle (Hornsten et al. 2007).
One noteworthy change in apl-1 expression is during the last molt, when animals transition from the fourth larval stage into adulthood. This developmental transition is regulated by the let-7 microRNA (miRNA), which regulates heterochronic transcription factors and the nuclear hormone receptor NHR-25 (Hayes et al. 2006; Hada et al. 2010). During this transition, seam cells, which act as stem cells to produce new hypodermal cells during molting, join the hypodermal syncytium (Sulston and White 1980). During the fourth larval stage, seam cells begin to express apl-1 (Niwa et al. 2008), and this apl-1 seam cell expression is regulated by the binding of NHR-25 to a 200-base pair (bp) enhancer element 6 kbp upstream of the apl-1 ATG (Niwa et al. 2008; Niwa and Hada 2010; Hada et al. 2010). Loss of let-7 miRNA results in extra hypodermal cell production, resulting in vulva bursting and lethality (Reinhart et al. 2000). RNAi of apl-1, which knocks down apl-1 mRNA levels by 60%, rescues the lethal phenotype of let-7 mutants (Niwa et al. 2008), suggesting that let-7 miRNA downregulates apl-1 to induce terminal differentiation of seam cells to join the hypodermal syncytium. Furthermore, double mutants of let-7 miRNA family members mir-48(n4097) and mir-84(n4037) are unable to shed their old cuticle during the fourth larval stage to adult transition; this phenotype is also suppressed by apl-1 knockdown (Niwa et al. 2008). These data suggest that several let-7 miRNA family members directly or indirectly regulate apl-1 expression during the larval to adult transition. apl-1 expression is necessary in neurons for the molting process to proceed properly, but its expression must be downregulated in seam cells to allow seam cell terminal differentiation. In mammals, the role of APP in differentiation is more equivocal. For instance, sAPP, as a co-factor with epidermal growth factor, promotes cell proliferation of ventricular zone cells, but does not affect differentiation (Caille et al. 2004). By contrast, APP immunoreactivity was detected in embryonic mouse neural tube (Salbaum and Ruddle 1994), suggesting a role during neural differentiation. Moreover, downregulation of APP decreased neurite differentiation in vitro (Allinquant et al. 1995). In advanced stage melanomas, which express high levels of APP, APP knockdown by RNAi causes the terminal differentiation of metastatic melanoma cells (Botelho et al. 2010), again suggesting that APP expression is downregulated to allow cell differentiation. However, in certain instances, APP induces cell differentiation. Transfection of human APP into human embryonic stem cells induced rapid differentiation of stem cells into neural cells (Freude et al. 2011). Hence, mammalian APP can induce or inhibit cell differentiation depending on cellular context. Whether sAPL-1 also functions as a regulator for neuronal differentiation in C. elegans remains to be determined.
Processing of APP and APL-1
In mammals, APP can be processed through two pathways. In the first pathway, α-secretase cleaves APP to release a large extracellular fragment sAPPα. The small remaining C-terminal transmembrane fragment APP-CTFα (also known as C83) is subsequently cleaved by the γ-secretase complex to release the p3 fragment extracellularly and the AICD fragment intracellularly (Haass et al. 1993; reviewed in Nunan and Small 2000). Because the initial α-secretase cleavage is within the β-amyloid peptide sequence, this pathway does not generate the β-amyloid peptide. In the second pathway a β-secretase cleaves APP N-terminal to the α-secretase site to release the extracellular fragment sAPPβ. Subsequently, the remaining C-terminal transmembrane fragment APP-CTFβ (also known as C99) is cleaved by γ-secretase to release the β-amyloid peptide extracellularly and AICD intracellularly (Haass et al. 1992; reviewed in Nunan and Small 2000).
The functions of the C. elegans orthologues have provided significant insights into the role of the mammalian counterparts. Mutations in the presenilin genes were initially identified in familial Alzheimer disease patients (Levy-Lahad et al. 1995; Rogaev et al. 1995; Sherrington et al. 1995); however, the functional role of the presenilins was informed by the C. elegans SEL-12 presenilin homologue, which was identified as a suppressor of Notch signaling (Levitan and Greenwald 1995). The human presenilins not only show amino acid homology to the C. elegans SEL-12, but also functional conservation, since introduction of human presenilin can rescue sel-12 deficiencies (Levitan et al. 1996). Moreover, PEN-2, a component of the γ-secretase complex, was first genetically identified in C. elegans in screens to enhance sel-12 presenilin mutant phenotypes (Francis et al. 2002). The mammalian orthologue was similarly found in the γ-secretase complex (Chen et al. 2002). Lastly, the function of nicastrin/APH-1 was determined in C. elegans specifically to provide insights into its function in mammals (Yu et al. 2000).
APL-1 is similarly cleaved by secretases. However, like mammalian APLP1 and APLP2, C. elegans APL-1 lacks sequence homology in the Aβ region (Daigle and Li 1993). Because only one sAPL-1 fragment was detected on Western blots (Hornsten et al. 2007), APL-1 is presumably processed by only one of the pathways. Bioinformatic searches of the sequenced C. elegans genome revealed no sequence homology to a β-secretase (C. Link, unpublished obs.). Furthermore, protein extracts from transgenic animals carrying human APP showed no detectable sAPPβ nor human Aβ peptide, suggesting that APL-1 is not cleaved by a β-secretase (Link 2006). These results suggest that APL-1 is first cleaved by an α-secretase to release sAPL-1 and subsequently cleaved by the γ-secretase complex (Hornsten et al. 2007).
The extracellular domain of APL-1 is essential for viability in C. elegans
In genetic screens to identify viable apl-1 alleles, the yn5 allele, which contains a deletion of the transmembrane domain, intracellular domain and large parts of the 3′UTR, was identified (Hornsten et al. 2007). apl-1(yn5) animals are fully viable, demonstrating that the extracellular domain is necessary and sufficient for viability. Furthermore, introducing a fragment containing only the extracellular domain of APL-1 corresponding to the yn5 mutation (APL-1EXT) was sufficient to rescue the lethality of apl-1 KOs. APL-1EXT is slightly larger than sAPL-1 because it encompasses the entire extracellular domain and does not appear to undergo further processing (Hornsten et al. 2007). Pan-neuronal expression of APL-1 EXT was also sufficient to rescue the apl-1 KO lethality (Hornsten et al. 2007), suggesting that neuronal release of sAPL-1 is sufficient for the molting ecdysis program to be activated. By contrast, introducing a fragment containing the APL-1 intracellular domain was not sufficient to rescue (Hornsten et al. 2007; Wiese et al. 2010). In mice, knockin of a fragment containing the extracellular domain of APP (sAPPα) was able to rescue the neuronal and behavioral deficits of APP −/− knockout mice (Ring et al. 2007). However, the lethality associated with APP −/− ;APLP2 −/− double knockout mice was not rescued by knockin of the extracellular domain corresponding to sAPPβ (Li et al. 2010a) or by knockin of APP carrying the Swedish, Arctic, and London mutations, a humanized Aβ, and a mutated intracellular domain [APP(Swe-Artic-London)/hAβ/mutC] (Li et al. 2010b). Although the secreted sAPP levels from primary hippocampal neuronal culture of these two knockin mouse strains were similar to wild-type secreted sAPP levels, both knockin APP levels were (20–40%) less than endogenous APP levels in vivo (Li et al. 2010a, b), which could have a physiological effect on viability. Moreover, the sAPPβ knockin had a C-terminal FLAG tag that could affect functionality; similarly, the APP(Swe-Artic-London)/hAβ/mutC knockin had numerous mutations that could also affect the extracellular domain functionality. By contrast to the pan-neuronal APL-1 expression rescue of apl-1 KOs, a conditional double KO of APP-APLP2 in neurons was not lethal (Li et al. 2010a). These data would argue that mammalian AICD is necessary for viability, suggesting a functional difference between the orthologous proteins. Knockin of the intracellular domain of APP, untagged sAPPβ, or sAPPα would resolve whether these APP domains rescue the APP −/− ;APLP2 −/− lethality.
To determine whether APL-1 is homologous to APP family members, the human and mouse orthologues were tested for rescue of the apl-1 KO lethality. Introducing human APP, murine APP, human APLP1, human APLP2, or human APP, APLP1, and APLP2 together did not rescue the apl-1 KO lethality (Hornsten 2001; Hornsten et al. 2007; Wiese et al. 2010); furthermore, swapping the extracellular domain of C. elegans APL-1 with human APP up to the putative α-secretase cleavage site also did not rescue the lethality (Hornsten 2001), suggesting that while APL-1 and APP share sequence similarities, slight changes in the structure affect protein interactions.
The extracellular domain of APL-1 is required for proper synaptic function at the neuromuscular junction
The subset of neurons in which apl-1 expression is detected includes neurons within the ventral nerve cord (Hornsten et al. 2007), which contains a large number of cholinergic excitatory motor neurons (Duerr et al. 2008). Upon stimulation, cholinergic neurons release acetylcholine, which is degraded by acetylcholinesterase in the synaptic cleft. Excessive acetylcholine in the synaptic cleft leads to constitutive activation of the acetylcholine receptor, resulting in constitutive muscle contraction that causes paralysis in C. elegans (Brenner 1974; Johnson et al. 1981). Similarly, pharmacological administration of aldicarb, an inhibitor of acetylcholinesterase, leads to C. elegans paralysis (Rand and Russell 1985; Nguyen et al. 1995). RNAi knockdown of apl-1 resulted in faster paralysis of aldicarb-treated animals compared with control animals (Wiese et al. 2010). This hypersensitivity to aldicarb of RNAi apl-1 worms was rescued by introducing full-length APL-1 or the extracellular domain of APL-1 (Wiese et al. 2010). These results suggest that lower levels of APL-1, and in particular the extracellular domain of APL-1, result in excessive acetylcholine levels, decreased acetylcholinesterase function, or enhanced acetylcholine receptor function. Whether the apl-1 knockdown hypersensitivity is caused by morphological defects is unknown. In APP−/−;APLP2−/− double KO mice, morphological defects of the neuromuscular junction displayed before the postnatal lethality were not rescued by knockin of sAPPβ (Li et al. 2010a), suggesting that sAPP might not function in the development of the neuromuscular junction in mice. However, sAPP in mammals might still function at the neuromuscular junction later in development or in adulthood, similar to sAPL-1 in C. elegans.
The extracellular domain of APL-1 binds heparin
In mammals, the extracellular fragment sAPPβ, but not sAPPα, is further cleaved in vitro by an unknown secretase to yield an E1 domain fragment and an E2 domain fragment (Nikolaev et al. 2009). In C. elegans, the presence of either E1 or E2 rescued the apl-1 KO lethality (Hornsten et al. 2007), suggesting a redundant function of the two domains. Both domains have putative binding sites to heparin, which is abundantly found in the extracellular matrix between cells (Kramer 2005). Deleting the putative heparin-binding site of E1 together with the entire APL-1 E2 domain did not rescue the apl-1 KO lethality (C. Ewald, unpublished obs.), suggesting an essential role for heparin binding. The APL-l E2 domain consists of six alpha helices (αA-αF; (Hoopes et al. 2010). Although only 34% of the amino acid sequence of APL-1 E2 is similar to human APP E2, the crystal structure of the APL-1 E2 domain almost perfectly overlays that of human APP E2, except that there is a 26 degree greater angle between the αC and αD helices of human APP E2 compared with C. elegans APL-1 (Hoopes et al. 2010). In C. elegans, the heparin binding to E2 was stronger at pH 5 than at neutral pH (Hoopes et al. 2010), suggesting that extracellular changes in pH regulate heparin binding and presumably function. Human APP E2 was present as dimers in X-ray crystal structures, and heparin-binding sites were close to the dimer interface (Wang and Ha 2004), whereas C. elegans APL-1 E2 in solution was found to be monomeric (Hoopes et al. 2010). However, C. elegans protein extracts contained APL-1 dimers by SDS-PAGE analysis, suggesting that APL-1 can dimerize (Hornsten et al. 2007). One possible model is that when APL-1 is at the cell surface, it homodimerizes upon heparin binding to aid in cell adhesion.
LRPs are putative receptors for APL-1
If sAPL-1 is released to initiate the molting ecdysis program or to ensure proper cell adhesion, then sAPL-1 is likely to bind a receptor to activate a downstream signaling pathway. By analogy to mammalian APP, which binds LRP (Kounnas et al. 1995) and LRAD3 (Ranganathan et al. 2011), possible sAPL-1 receptors are the low-density lipoprotein receptor-related proteins LRP-1 and LRP-2. The C. elegans orthologue LRP-1 regulates cholesterol metabolism and molting (Yochem et al. 1999). Hence, sAPL-1 could bind LRP-1 or LRP-2 to activate a downstream pathway. A second possible receptor would be a TNF-like receptor similar to DR6, a death domain containing receptor that binds and can be activated by mammalian sAPP (Nikolaev et al. 2009; Kuester et al. 2011). However, although a DR6-like orthologue has not been identified in C. elegans, a DR6 adaptor protein encoded by trf-1 TRAF has been found in C. elegans (Wormbase), suggesting that a functional equivalent to the DR6 receptor is present in C. elegans.
Intracellular domain of APL-1 has possible Goα and FEH-1-binding sites
The intracellular domain of APL-1 shares 71% sequence similarity to human AICD (Daigle and Li 1993). This similarity includes a potential Goα-binding site (MRGFIEVDVYTPEERHVAG), suggesting that APP and related proteins can act as receptors. C. elegans that overexpress APL-1 have locomotory defects and sluggish movements (Hornsten et al. 2007). These locomotory defects were suppressed by constitutive activation of Goα (G. O’Connor, unpublished obs.), suggesting a potential receptor function of APL-1. Such an APL-1 receptor function is independent of the role of APL-1 in activating the molting ecdysis program, as constructs that delete the Goα-binding site or the whole intracellular domain of APL-1 rescued the apl-1 KO lethality (Hornsten et al. 2007). In mammals, the intracellular domain of APP, particularly the YENPTY motif, binds the adaptor protein Fe65 (Borg et al. 1996) to traffic the complex to the cell surface (Sabo et al. 2001). The APL-1 intracellular domain also contains a YENPTY motif and binds to FEH-1, the C. elegans orthologue of Fe65 (Zambrano et al. 2002). Loss of feh-1 resulted in embryonic lethality or first larval stage arrest; RNAi knockdown of feh-1 or apl-1 led to hyperactive pharyngeal pumping compared with control worms, suggesting that feh-1 and apl-1 regulate pharyngeal pumping through a common pathway (Zambrano et al. 2002).
Overexpression of APL-1 causes lethality that is rescued by decreased sel-12 presenilin activity
Thus far, loss of APP function has not been correlated with AD. By contrast, duplication of the APP gene locus (Cabrejo et al. 2006; Rovelet-Lecrux et al. 2006; Sleegers et al. 2006) or Down syndrome, a trisomy of chromosome 21 on which APP is located (Glenner and Wong 1984), has been correlated with early-onset AD (Mann and Esiri 1989; Schupf et al. 1998; Korbel et al. 2009). These findings suggest that higher levels of APP may contribute to developing AD. Overexpression of APL-1 in C. elegans led to an incompletely penetrant early larval lethality, detachment of organs, and locomotive defects (Hornsten et al. 2007). This apl-1 overexpression lethality was not suppressed by mutations in ced-3, which encodes a caspase required for apoptosis (Yuan et al. 1993), or crt-1, which encodes calreticulin, which is required for necrotic cell death (Xu et al. 2001) in C. elegans (Hornsten et al. 2007); furthermore, APL-1::GFP generally did not co-localize with BEC-1, a component of the phosphatidylinositol class III kinase complex that regulates autophagy (Fig. 2b; (Meléndez et al. 2003). These data suggest that the apl-1 overexpression lethality is not due to ectopic activation of a cell death pathway. By contrast, reducing sel-12 presenilin activity, thereby decreasing γ-secretase activity, partially suppressed the apl-1 overexpression lethality (Hornsten et al. 2007). In a sel-12 reduction-of-function mutant background, APL-1 was still cleaved (Hornsten et al. 2007), and the relative ratio of protein levels of sAPL-1 to full-length APL-1 was similar compared with animals with functional SEL-12. However, overall APL-1 protein levels were lower in sel-12 presenilin mutants, suggesting that either APL-1 is being degraded more quickly or sel-12 presenilin regulates APL-1 levels (Hornsten et al. 2007).

APL-1 is expressed in multiple cell types and is not degraded via autophagy. a In wild-type animals, APL-1::GFP is expressed in neurons (SAA/SAB and RIG/RIF can be seen with faint expression) and processes in the nerve ring (nr). APL-1::GFP showed punctate expression in axonal processes (chevron) and head muscles in the anterior region of wild-type worms. b APL-1::GFP (green) shows little co-localization with RFP::BEC-1 (red). Overlapping areas are shown in white of the confocal z-stack. Scale bars 10 μm
Missense mutations in apl-1 destabilizes APL-1 and results in lethality
When screening for new alleles of apl-1, a missense mutation, yn32, which corresponds to a glutamic acid to lysine substitution at residue 371 (E371K), was identified (Hornsten et al. 2007). Germline transformation with an APL-1(E371K)::GFP transgene was unable to rescue the apl-1 KO lethality; although low levels of GFP fluorescence were present, protein extracts showed no detectable APL-1 protein by Western blot analysis, suggesting that yn32 is a null allele (Hoopes et al. 2010). APL-1(E371K) thermally unfolded faster than wild-type protein in stability assays in vitro, but compensatory mutations that restored wild-type properties in vitro did not rescue the apl-1 KO lethality in vivo (Hoopes et al. 2010). The E371 residue is in the αD helix where it forms a salt bridge with lysine residue 439 and a hydrogen bond with tryptophan residue 435, both of which are located in the αF helix; hence, the E371K mutation disrupts the αD to αF helix interaction and destabilizes the protein (Hoopes et al. 2010).
Trafficking of APL-1
APL-1 tagged at the C-terminus with GFP (APL-1::GFP) was seen in neuronal processes (Fig. 2), indicating that APL-1::GFP is transported to the axon terminal (Hornsten et al. 2007). Anterograde trafficking of APL-1::GFP to the axon terminal is dependent on the kinesin motor UNC-104 (Wiese et al. 2010; Arimoto et al. 2011), whereas retrograde transport of APL-1::GFP back to the cell body is dependent on DL1-1 dynein, which is transported to the axonal terminals by UNC-116 kinesin 1 (see model in Fig. 3; Arimoto et al. 2011). Reduction in unc-108/rab2 GTPase activity, which functions in vesicular trafficking and is specifically localized to the Golgi (Sumakovic et al. 2009), resulted in retention of APL-1::GFP fluorescence in the cell body where APL-1::GFP was co-localized with UNC-108::RFP (Wiese et al. 2010). By contrast, loss of rab-5 GTPase activity, which mediates transport of vesicles from the plasma membrane to early endosomes, caused decreased APL-1::GFP levels, presumably because APL-1::GFP at the cell surface is degraded and subsequently internalized (Wiese et al. 2010). In mammals, vesicles containing APP are trafficked to the nerve terminals by fast anterograde transport with kinesin motors (Koo et al. 1990; Kaether et al. 2000; Kamal et al. 2000). APP at the cell surface is internalized through clathrin-mediated endocytosis (Nordstedt et al. 1993). The vesicles are shuttled to endosomes with the help of Rab5 GTPase (Ikin et al. 1996), where APP is cleaved by β- and γ-secretase (Vassar et al. 1999), and the vesicle shuttled back to the cell surface for exocytosis (Koo et al. 1996). Hence, APL-1 transport in C. elegans neurons may be similar to APP transport in mammalian neurons. Because C. elegans is transparent, APL-1 transport can be directly monitored in vivo (Arimoto et al. 2011). Therefore, C. elegans might be a new useful approach to study APL-1/APP transport in vivo.

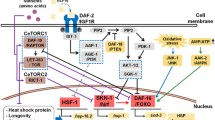
Model for Aβ and APL-1 trafficking and function. a Aβ in the endoplasmic reticulum (ER) is bound by the chaperone HSP-3, an orthologue of HSP70, to mediate Aβ folding or degradation. Soluble Aβ in the cytoplasm can lead to oxidative stress and presumably cellular damage or can form intracellular deposits. HSP-16.2, an orthologue of αΒ-crystallin, binds Aβ in the cytoplasm to target it for degradation. b By passing through the ER and Golgi, APL-1 is N-glycosylated before budding off in vesicles from the trans-Golgi-network (TGN) with the help of UNC-108/GTPase. The UNC-104/UNC-116 kinesin motors transport the APL-1 vesicles along microtubules in the anterograde direction, while the DL1-1 dynein motor transports the vesicle retrogradely; UNC-16 functions as a cargo adaptor for anterograde transport. On the cell membrane, APL-1 could potentially bind heparin, copper (Cu++), and zinc (Zn++) and be phosphorylated. APL-1 affects multiple functions: (1) locomotion via intracellular Goα; (2) pharyngeal pumping rate via binding of FEH-1 to the YENPTY site on APL-1; and (3) developmental progression, survival and molting by α/γ- secretase cleavage and release of the extracellular domain of APL-1 (sAPL-1). The remaining intracellular fragment and full-length APL-1 are endocytosed and transported via DLI-1/dynein along microtubules to the early endosome containing RAB-5 GTPase for degradation
Expression of human APP in C. elegans
To examine whether human APP can be expressed and processed in C. elegans, transgenic animals carrying a human APP transgene were generated (Link 2006). Whole animal lysates were probed with different antibodies against APP and Aβ; fragments corresponding to α- and γ- secretase cleavages, but not β-secretase cleavage of human APP, were detected (Link 2006). By contrast, human APP in Drosophila is cleaved by a β-secretase-like enzyme (Carmine-Simmen et al. 2009). These results indicate that C. elegans can produce human APP, but human APP is only processed by an α/γ-secretase pathway and, consequently, no Aβ is produced from APP introduced into C. elegans.
Expression of human β-amyloid peptide in C. elegans neurons causes learning defects
In familial AD cases, mutations of the APP gene have been linked to increased production of Aβ1–42, which is highly fibrillogenic and abundantly found in brain plaques of AD patients (Haass and Selkoe 1993). In addition, a number of transgenic mice lines that overexpress Aβ neuronally or triple transgenic mice (APP; Presenilin; Tau) formed intracellular Aβ deposits, which have been correlated with several defects, including neurodegeneration and a severe decrease in lifespan (LaFerla et al. 1995, 1997; Chui et al. 1999; Li et al. 1999; Gouras et al. 2000; Kuo et al. 2001; Wirths et al. 2001). To investigate the in vivo effects of Aβ on C. elegans physiology, transgenes of human β-amyloid peptide (Aβ1–42) were expressed with a signal sequence using different promoters in C. elegans. Pan-neuronal expression of human Aβ42 (Psnb-1::hAβ1–42) caused several neuronal deficits, including a chemotaxis defect toward the volatile chemoattractant benzaldehyde (Wu et al. 2006), an impaired associative avoidance response toward another volatile chemoattractant diacetyl (Dosanjh et al. 2010), impairment in serotonin-mediated behaviors, and mildly decreased lifespan compared with control animals (Dosanjh et al. 2010). These behavioral deficits suggest that human Aβ1–42 can affect synaptic function and has a detrimental effect on lifespan in C. elegans. In addition, the transgenic animals showed intraneuronal Aβ deposits, but no apparent neurodegeneration (Link 2005, 2006).
Expression of human Aβ in C. elegans muscles causes paralysis and intracellular Aβ accumulation
In humans, intracellular accumulation of Aβ in muscles leads to myopathy (inclusion body myositis; (Askanas and Engel 2006). Similarly, transgene expression of human Aβ1–42 in muscles led to the accumulation of intracellular Aβ and progressive paralysis during adulthood in C. elegans (Link 1995, 2001, 2006; Link et al. 2001). The paralysis phenotype was accompanied by oxidative stress and both were seen before Aβ fibril formation (Drake et al. 2003). Similarly, in transgenic mouse models, morphological and behavioral deficits preceded plaque formation (Jacobsen et al. 2006). Therefore, intracellular Aβ1–42 accumulation could play a role in AD pathogenesis, and the C. elegans human Aβ42 transgenic animals could be used to reveal the molecular mechanism of intracellular Aβ1–42 accumulation.
Surprisingly, despite the presence of a signal peptide in the C. elegans human Aβ1–42 model, only intracellular Aβ was detected (Link 1995, 2001; Link et al. 2001; Fonte et al. 2002). Furthermore, when the signal sequence is cleaved, two amino acids of Aβ1–42 are included in the cleavage, so that actually human Aβ3–42 is produced (McColl et al. 2009); we will refer to Aβ1–42 and Aβ3–42 produced in C. elegans collectively as Aβ42. Despite missing two amino acids, a C. elegans Aβ transgenic model may nevertheless provide insights into the intracellular Aβ pathogenesis for AD and human myopathy.
Reduced insulin/IGF-1 signaling protects against intracellular Aβ pathogenesis
Reducing insulin/IGF-1 signaling prolongs lifespan in C. elegans (Kenyon et al. 1993), flies (Tatar et al. 2001), and mice (Bluher et al. 2003; Holzenberger et al. 2003). Furthermore, polymorphisms in genes involved in the insulin signaling pathway have been associated with increased lifespan in humans (Suh et al. 2008; Willcox et al. 2008; Flachsbart et al. 2009). In C. elegans, reducing the activity of the insulin/IGF-1 receptor DAF-2 increases the activity of FOXO transcription factor DAF-16 (Lin et al. 1997; Ogg et al. 1997) and bZip transcription factor SKN-1 (Tullet et al. 2008), both of which regulate transcription of distinct, but overlapping sets of genes involved in detoxification, protein folding, stress resistance and longevity (Honda and Honda 1999; Barsyte et al. 2001; McElwee et al. 2003; An and Blackwell 2003; Murphy et al. 2003; Oliveira et al. 2009; Park et al. 2009). Reducing DAF-2 signaling protected against the progressive paralysis and increased the lifespan of a C. elegans transgenic strain expressing human Aβ42 in muscle (Cohen et al. 2006; Cohen et al. 2010). Interestingly, reduction in the insulin/IGF-1 signaling late in the worm lifespan was also sufficient to ameliorate the human Aβ42-induced progressive paralysis (Cohen et al. 2010). Although overall mRNA or protein levels of human Aβ42 were not altered (Cohen et al. 2006), those animals showed a shift from more soluble low molecular weight Aβ42 aggregates to high molecular weight human Aβ42 aggregates, which are presumed to be less toxic (Cohen et al. 2006, 2010). This active shift to high molecular weight Aβ42 aggregated species, the protection against paralysis, and the increase in lifespan required DAF-16 activity (Cohen et al. 2006, 2010). Although insulin/IGF-1 signaling directly regulates the transcription factor SKN-1 (Tullet et al. 2008), the requirement of skn-1 activity for the protection due to reduced daf-2 activity has not been investigated. However, activation of skn-1 by coffee extracts was required to delay progressive paralysis in the C. elegans transgenic strain expressing human Aβ42 in muscle (Dostal et al. 2010). By contrast, reducing skn-1 activity resulted in higher levels of human Aβ42 protein of both low and high molecular aggregates and progressive paralysis (Dostal et al. 2010). SKN-1 might be involved in general disaggregation of proteins and their degradation, since SKN-1 also protects against aggregation of artificial GFP::degron protein (Dostal et al. 2010). Moreover, pharmacological suppression of human Aβ42 aggregation and Aβ42-induced paralysis by amyloid-binding compounds required skn-1 but not daf-16 activity (Alavez et al. 2011), suggesting a general importance for SKN-1 in Aβ42 detoxification. Recently, polymorphisms in a skn-1 orthologue Nrf2 (NFE2L2) gene and in human IGF-1 have been associated with early-onset AD (von Otter et al. 2010) and an increased risk for AD (Vargas et al. 2011), respectively. Some AD patients develop brain-specific diabetes that alters their insulin signaling (Steen et al. 2005). In mice, reducing IGF-1 signaling increased lifespan (Holzenberger et al. 2003), decreased APP-induced mortality rate (Freude et al. 2009), and protected against neuronal cell loss and behavioral deficits (Cohen et al. 2009). Furthermore, reducing IGF-1 signaling in transgenic AD mice resulted in a shift to high molecular weight Aβ42 aggregates (Cohen et al. 2009), similar to what was seen in transgenic C. elegans Aβ42 animals (Cohen et al. 2006, 2010). Taken together, these findings suggest that reducing insulin/IGF-1 signaling might be a preventive strategy against AD pathogenesis.
Molecular interactions of human Aβ42 in C. elegans and AD patient brain tissue
Several approaches have been used in C. elegans to identify genes whose expression underlies intracellular human Aβ42 accumulation. One approach is to identify physical binding partners to human Aβ42. In transgenic animals that overexpress human Aβ42 in muscles, Aβ42 co-immunoprecipitated with six chaperone proteins: two proteins related to HSP70, which functions as an endoplasmic reticulum (ER) chaperone, a putative negative regulator of HSP70 R05F9.10, and three heat-shock proteins of the HSP-16 class with homology to αΒ-crystallin (Fonte et al. 2002). Interestingly, HSP-16/αΒ-crystallin also bound to Aβ42 in muscle fibers of human inclusion body myositis patients (Banwell and Engel 2000). A second approach is to take a more genomics approach and perform a microarray analysis to identify genes that are up- or downregulated in response to overexpression of human Aβ42 in C. elegans. Such microarray assays revealed strong and specific up-regulation of two HSP-16/αΒ-crystallin chaperone transcripts, supporting the immunoprecipitation results, and two transcripts with homology to the mammalian TNFAIP1 (tumor necrosis factor α–induced protein) family in C. elegans (Link et al. 2003). Moreover, over-expression of HSP-16/αΒ-crystallin or knockdown of R05F9.10, the negative regulator of HSP70, partially suppressed the paralysis induced by Aβ42 in transgenic animals that express human Aβ42 in muscles (Fonte et al. 2008). In general, HSP chaperone proteins mediate trafficking of non-native or unfolded proteins for degradation in the lysosomes (Kettern et al. 2010). RNAi knock-down of lysosomal components or associated genes enhanced the paralysis rate and caused higher Aβ42 levels of transgenic animals that overexpress human Aβ42 in muscles (Florez-McClure et al. 2007). Hence, HSP-16/αΒ-crystallin and HSP70 proteins become upregulated by the excessive Aβ42 levels and bind Aβ42 directly for clearance via lysosomes. To determine whether these results have relevance for AD, αΒ-crystallin and TNFAIPI transcripts in AD brain tissue were examined and found to be upregulated as well (Link et al. 2003). Thus, a common mechanism for the detoxification or clearance of intracellular human Aβ1–42 in neurons and muscle tissue is used by C. elegans and mammals.
Concluding remarks
A common theme in molecular processes has shown that once a pathway or mechanism has been established, it gets used over and over again in modified forms. The C. elegans APL-1 has multifunctional roles and acts non-cell autonomously. Either loss of APL-1 or overexpression of APL-1 results in lethality, showing the importance of maintaining APL-1 within homeostatic levels, particularly within neurons. Much remains to be understood about the mechanism of APL-1 function. Future genetic screens in C. elegans may reveal possible receptors to which sAPL-1 binds after its release. The greatest challenge in the field is to show the relevance of findings in C. elegans to mammalian biology or AD directly. Example of discoveries in C. elegans, such as the components of the γ-secretase complex, advanced the AD field tremendously. Furthermore, C. elegans has been a particularly useful model organism for outlining a pathway for clearance of intracellular Aβ deposits; mammals, including humans, use common elements in their clearance in both AD pathogenesis and human myopathy. Effects of intracellular Aβ accumulation are ameliorated by activation of general protein degradation pathways or longevity genes in C. elegans (Cohen et al. 2006; Florez-McClure et al. 2007; Fonte et al. 2008; Hassan et al. 2009). Drugs improving protein degradation might be a new strategy to fight Alzheimer’s disease.
References
Ahringer J (1997) Turn to the worm! Curr Opin Genet Dev 7(3):410–415. doi:S0959-437X(97)80157-8
Alavez S, Vantipalli MC, Zucker DJ, Klang IM, Lithgow GJ (2011) Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 472(7342):226–229. doi:10.1038/nature09873
Allinquant B, Hantraye P, Mailleux P, Moya K, Bouillot C, Prochiantz A (1995) Downregulation of amyloid precursor protein inhibits neurite outgrowth in vitro. J Cell Biol 128(5):919–927
An JH, Blackwell TK (2003) SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev 17(15):1882–1893. doi:10.1101/gad.11078031107803
Arimoto M, Koushika SP, Choudhary BC, Li C, Matsumoto K, Hisamoto N (2011) The Caenorhabditis elegans JIP3 protein UNC-16 functions as an adaptor to link kinesin-1 with cytoplasmic dynein. J Neurosci 31(6):2216–2224. doi:10.1523/JNEUROSCI.2653-10.2011
Askanas V, Engel WK (2006) Inclusion-body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition. Neurology 66(2 Suppl 1):S39–S48. doi:10.1212/01.wnl.0000192128.13875.1e
Banwell BL, Engel AG (2000) AlphaB-crystallin immunolocalization yields new insights into inclusion body myositis. Neurology 54(5):1033–1041
Bargmann CI, Horvitz HR (1991) Chemosensory neurons with overlapping functions direct chemotaxis to multiple chemicals in C. elegans. Neuron 7(5):729–742
Barsyte D, Lovejoy DA, Lithgow GJ (2001) Longevity and heavy metal resistance in daf-2 and age-1 long-lived mutants of Caenorhabditis elegans. Faseb J 15(3):627–634. doi:10.1096/fj.99-0966com15/3/627
Bird TD (2010) Early-onset Familial Alzheimer Disease. In: Pagon RA, Bird TD, Dolan CR, Stephens K (eds) GeneReviews. University of Washington, Seattle. http://www.ncbi.nlm.nih.gov/pubmed/20301414
Bluher M, Kahn BB, Kahn CR (2003) Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 299(5606):572–574
Borg JP, Ooi J, Levy E, Margolis B (1996) The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol Cell Biol 16(11):6229–6241
Botelho MG, Wang X, Arndt-Jovin DJ, Becker D, Jovin TM (2010) Induction of terminal differentiation in melanoma cells on downregulation of beta-amyloid precursor protein. J Invest Dermatol 130(5):1400–1410. doi:10.1038/jid.2009.296
Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77(1):71–94
Cabrejo L, Guyant-Marechal L, Laquerriere A, Vercelletto M, De la Fourniere F, Thomas-Anterion C, Verny C, Letournel F, Pasquier F, Vital A, Checler F, Frebourg T, Campion D, Hannequin D (2006) Phenotype associated with APP duplication in five families. Brain 129(Pt 11):2966–2976
Caille I, Allinquant B, Dupont E, Bouillot C, Langer A, Muller U, Prochiantz A (2004) Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development 131(9):2173–2181. doi:10.1242/dev.01103dev.01103
Carmine-Simmen K, Proctor T, Tschape J, Poeck B, Triphan T, Strauss R, Kretzschmar D (2009) Neurotoxic effects induced by the Drosophila amyloid-beta peptide suggest a conserved toxic function. Neurobiol Dis 33(2):274–281. doi:10.1016/j.nbd.2008.10.014
Chalfie M, Sulston JE, White JG, Southgate E, Thomson JN, Brenner S (1985) The neural circuit for touch sensitivity in Caenorhabditis elegans. J Neurosci 5(4):956–964
Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC (1994) Green fluorescent protein as a marker for gene expression. Science 263(5148):802–805
Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J et al (1991) Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature 353(6347):844–846
Chen F, Gu Y, Hasegawa H, Ruan X, Arawaka S, Fraser P, Westaway D, Mount H, St George-Hyslop P (2002) Presenilin 1 mutations activate gamma 42-secretase but reciprocally inhibit epsilon-secretase cleavage of amyloid precursor protein (APP) and S3-cleavage of notch. J Biol Chem 277(39):36521–36526. doi:10.1074/jbc.M205093200M205093200
Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, Inoue H, Shirotani K, Takahashi K, Gallyas F, Tabira T (1999) Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med 5(5):560–564. doi:10.1038/8438
Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A (2006) Opposing activities protect against age-onset proteotoxicity. Science 313(5793):1604–1610
Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW, Masliah E, Dillin A (2009) Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 139(6):1157–1169. doi:10.1016/j.cell.2009.11.014
Cohen E, Du D, Joyce D, Kapernick EA, Volovik Y, Kelly JW, Dillin A (2010) Temporal requirements of insulin/IGF-1 signaling for proteotoxicity protection. Aging Cell 9(2):126–134. doi:10.1111/j.1474-9726.2009.00541.x
Culetto E, Sattelle DB (2000) A role for Caenorhabditis elegans in understanding the function and interactions of human disease genes. Hum Mol Genet 9(6):869–877. doi:ddd102
Daigle I, Li C (1993) apl-1, a Caenorhabditis elegans gene encoding a protein related to the human beta-amyloid protein precursor. Proc Natl Acad Sci USA 90(24):12045–12049
Dosanjh LE, Brown MK, Rao G, Link CD, Luo Y (2010) Behavioral phenotyping of a transgenic Caenorhabditis elegans expressing neuronal amyloid-beta. J Alzheimers Dis 19(2):681–690. doi:10.3233/JAD-2010-1267
Dostal V, Roberts CM, Link CD (2010) Genetic mechanisms of coffee extract protection in a Caenorhabditis elegans model of beta-amyloid peptide toxicity. Genetics 186(3):857–866. doi:10.1534/genetics.110.120436
Drake J, Link CD, Butterfield DA (2003) Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid beta-peptide (1–42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging 24(3):415–420
Duerr JS, Han HP, Fields SD, Rand JB (2008) Identification of major classes of cholinergic neurons in the nematode Caenorhabditis elegans. J Comp Neurol 506(3):398–408. doi:10.1002/cne.21551
Ewald CY, Li C (2010) Understanding the molecular basis of Alzheimer’s disease using a Caenorhabditis elegans model system. Brain Struct Funct 214(2–3):263–283. doi:10.1007/s00429-009-0235-3
Flachsbart F, Caliebe A, Kleindorp R, Blanche H, von Eller-Eberstein H, Nikolaus S, Schreiber S, Nebel A (2009) Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc Natl Acad Sci USA 106(8):2700–2705. doi:10.1073/pnas.0809594106
Florez-McClure ML, Hohsfield LA, Fonte G, Bealor MT, Link CD (2007) Decreased insulin-receptor signaling promotes the autophagic degradation of beta-amyloid peptide in C. elegans. Autophagy 3(6):569–580
Fonte V, Kapulkin V, Taft A, Fluet A, Friedman D, Link CD (2002) Interaction of intracellular beta amyloid peptide with chaperone proteins. Proc Natl Acad Sci USA 99(14):9439–9444
Fonte V, Kipp DR, Yerg J III, Merin D, Forrestal M, Wagner E, Roberts CM, Link CD (2008) Suppression of in vivo beta-amyloid peptide toxicity by overexpression of the HSP-16.2 small chaperone protein. J Biol Chem 283(2):784–791
Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch R, Ruble C, Nye JS, Curtis D (2002) aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell 3(1):85–97
Freude S, Hettich MM, Schumann C, Stohr O, Koch L, Kohler C, Udelhoven M, Leeser U, Muller M, Kubota N, Kadowaki T, Krone W, Schroder H, Bruning JC, Schubert M (2009) Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer’s disease. Faseb J 23(10):3315–3324. doi:10.1096/fj.09-132043
Freude KK, Penjwini M, Davis JL, LaFerla FM, Blurton-Jones M (2011) Soluble amyloid precursor protein induces rapid neural differentiation of human embryonic stem cells. J Biol Chem 286(27):24264–24274. doi:10.1074/jbc.M111.227421
Glenner GG, Wong CW (1984) Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun 122(3):1131–1135
Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L et al (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349(6311):704–706
Goodman MB, Hall DH, Avery L, Lockery SR (1998) Active currents regulate sensitivity and dynamic range in C. elegans neurons. Neuron 20(4):763–772. doi:S0896-6273(00)81014-4
Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR (2000) Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156(1):15–20
Goutte C, Hepler W, Mickey KM, Priess JR (2000) aph-2 encodes a novel extracellular protein required for GLP-1-mediated signaling. Development 127(11):2481–2492
Haass C, Selkoe DJ (1993) Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell 75(6):1039–1042
Haass C, Koo EH, Mellon A, Hung AY, Selkoe DJ (1992) Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature 357(6378):500–503. doi:10.1038/357500a0
Haass C, Hung AY, Schlossmacher MG, Teplow DB, Selkoe DJ (1993) beta-Amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J Biol Chem 268(5):3021–3024
Hada K, Asahina M, Hasegawa H, Kanaho Y, Slack FJ, Niwa R (2010) The nuclear receptor gene nhr-25 plays multiple roles in the Caenorhabditis elegans heterochronic gene network to control the larva-to-adult transition. Dev Biol 344(2):1100–1109. doi:10.1016/j.ydbio.2010.05.508
Hassan WM, Merin DA, Fonte V, Link CD (2009) AIP-1 ameliorates beta-amyloid peptide toxicity in a Caenorhabditis elegans Alzheimer’s disease model. Hum Mol Genet 18(15):2739–2747. doi:10.1093/hmg/ddp209
Hayes GD, Frand AR, Ruvkun G (2006) The mir-84 and let-7 paralogous microRNA genes of Caenorhabditis elegans direct the cessation of molting via the conserved nuclear hormone receptors NHR-23 and NHR-25. Development 133(23):4631–4641. doi:10.1242/dev.02655
Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rulicke T, von Kretzschmar H, von Koch C, Sisodia S, Tremml P, Lipp HP, Wolfer DP, Muller U (2000) Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci 20(21):7951–7963
Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, Kretzschmar H, Sisodia S, Muller U (2004) Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J 23(20):4106–4115
Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y (2003) IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421(6919):182–187
Honda Y, Honda S (1999) The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J 13(11):1385–1393
Hoopes JT, Liu X, Xu X, Demeler B, Folta-Stogniew E, Li C, Ha Y (2010) Structural characterization of the E2 domain of APL-1, a Caenorhabditis elegans homolog of human amyloid precursor protein, and its heparin binding site. J Biol Chem 285(3):2165–2173. doi:10.1074/jbc.M109.018432
Hornsten A (2001) APL-1, a Caenorhabditis elegans protein related to the human amyloid precursor protein, is essential for viability. Dissertation, Boston University, Boston
Hornsten A, Lieberthal J, Fadia S, Malins R, Ha L, Xu X, Daigle I, Markowitz M, O’Connor G, Plasterk R, Li C (2007) APL-1, a Caenorhabditis elegans protein related to the human beta-amyloid precursor protein, is essential for viability. Proc Natl Acad Sci USA 104(6):1971–1976
Horvitz HR, Chalfie M, Trent C, Sulston JE, Evans PD (1982) Serotonin and octopamine in the nematode Caenorhabditis elegans. Science 216(4549):1012–1014
Hosono R, Sassa T, Kuno S (1987) Mutations affecting acetylcholine levels in the nematode Caenorhabditis elegans. J Neurochem 49(6):1820–1823
Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D (1995) Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron 15(5):1203–1218
Ikin AF, Annaert WG, Takei K, De Camilli P, Jahn R, Greengard P, Buxbaum JD (1996) Alzheimer amyloid protein precursor is localized in nerve terminal preparations to Rab5-containing vesicular organelles distinct from those implicated in the synaptic vesicle pathway. J Biol Chem 271(50):31783–31786
Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE (2006) Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 103(13):5161–5166
Johnson CD, Duckett JG, Culotti JG, Herman RK, Meneely PM, Russell RL (1981) An acetylcholinesterase-deficient mutant of the nematode Caenorhabditis elegans. Genetics 97(2):261–279
Kaether C, Skehel P, Dotti CG (2000) Axonal membrane proteins are transported in distinct carriers: a two-color video microscopy study in cultured hippocampal neurons. Mol Biol Cell 11(4):1213–1224
Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS (2000) Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 28(2):449–459. doi:S0896-6273(00)00124-0
Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B (1987) The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325(6106):733–736
Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R (1993) A C. elegans mutant that lives twice as long as wild type. Nature 366(6454):461–464
Kerr R, Lev-Ram V, Baird G, Vincent P, Tsien RY, Schafer WR (2000) Optical imaging of calcium transients in neurons and pharyngeal muscle of C. elegans. Neuron 26(3):583–594. doi:S0896-6273(00)81196-4
Kettern N, Dreiseidler M, Tawo R, Hohfeld J (2010) Chaperone-assisted degradation: multiple paths to destruction. Biol Chem 391(5):481–489. doi:10.1515/BC.2010.058
Kidd M (1964) Alzheimer’s disease—an electron microscopical study. Brain 87:307–320
Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ (2003) Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc Natl Acad Sci USA 100(11):6382–6387. doi:10.1073/pnas.10373921001037392100
Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL (1990) Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci USA 87(4):1561–1565
Koo EH, Squazzo SL, Selkoe DJ, Koo CH (1996) Trafficking of cell-surface amyloid beta-protein precursor. I. Secretion, endocytosis and recycling as detected by labeled monoclonal antibody. J Cell Sci 109(5):991–998
Korbel JO, Tirosh-Wagner T, Urban AE, Chen XN, Kasowski M, Dai L, Grubert F, Erdman C, Gao MC, Lange K, Sobel EM, Barlow GM, Aylsworth AS, Carpenter NJ, Clark RD, Cohen MY, Doran E, Falik-Zaccai T, Lewin SO, Lott IT, McGillivray BC, Moeschler JB, Pettenati MJ, Pueschel SM, Rao KW, Shaffer LG, Shohat M, Van Riper AJ, Warburton D, Weissman S, Gerstein MB, Snyder M, Korenberg JR (2009) The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci USA 106(29):12031–12036. doi:10.1073/pnas.0813248106
Kounnas MZ, Moir RD, Rebeck GW, Bush AI, Argraves WS, Tanzi RE, Hyman BT, Strickland DK (1995) LDL receptor-related protein, a multifunctional ApoE receptor, binds secreted beta-amyloid precursor protein and mediates its degradation. Cell 82(2):331–340. doi:0092-8674(95)90320-8
Kramer JM (2005) Basement membranes. WormBook 1–15. doi:10.1895/wormbook.1.16.1
Krigman MR, Feldman RG, Bensch K (1965) Alzheimer’s presenile dementia. A histochemical and electron microscopic study. Lab Invest 14:381–396
Kuester M, Kemmerzehl S, Dahms SO, Roeser D, Than ME (2011) The crystal structure of death receptor 6 (DR6): a potential receptor of the amyloid precursor protein (APP). J Mol Biol 409(2):189–201. doi:10.1016/j.jmb.2011.03.048
Kuo YM, Beach TG, Sue LI, Scott S, Layne KJ, Kokjohn TA, Kalback WM, Luehrs DC, Vishnivetskaya TA, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Weller RO, Roher AE (2001) The evolution of Abeta peptide burden in the APP23 transgenic mice: implications for Abeta deposition in Alzheimer disease. Mol Med 7(9):609–618
LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G (1995) The Alzheimer’s Abeta peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat Genet 9(1):21–30
LaFerla FM, Troncoso JC, Strickland DK, Kawas CH, Jay G (1997) Neuronal cell death in Alzheimer’s disease correlates with apoE uptake and intracellular Abeta stabilization. J Clin Invest 100(2):310–320. doi:10.1172/JCI119536
Lee RY, Sawin ER, Chalfie M, Horvitz HR, Avery L (1999) EAT-4, a homolog of a mammalian sodium-dependent inorganic phosphate cotransporter, is necessary for glutamatergic neurotransmission in Caenorhabditis elegans. J Neurosci 19(1):159–167
Lenkkeri U, Kestila M, Lamerdin J, McCready P, Adamson A, Olsen A, Tryggvason K (1998) Structure of the human amyloid-precursor-like protein gene APLP1 at 19q13.1. Hum Genet 102(2):192–196
Levitan D, Greenwald I (1995) Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature 377(6547):351–354
Levitan D, Doyle TG, Brousseau D, Lee MK, Thinakaran G, Slunt HH, Sisodia SS, Greenwald I (1996) Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc Natl Acad Sci USA 93(25):14940–14944
Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K et al (1995) Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269(5226):973–977
Li X, Greenwald I (1997) HOP-1, a Caenorhabditis elegans presenilin, appears to be functionally redundant with SEL-12 presenilin and to facilitate LIN-12 and GLP-1 signaling. Proc Natl Acad Sci USA 94(22):12204–12209
Li C, Kim K (2008) Neuropeptides in C. elegans. In: The C. elegans Research Community, WormBook. doi:10.1895/wormbook.1.142.1. http://www.wormbook.org
Li QX, Maynard C, Cappai R, McLean CA, Cherny RA, Lynch T, Culvenor JG, Trevaskis J, Tanner JE, Bailey KA, Czech C, Bush AI, Beyreuther K, Masters CL (1999) Intracellular accumulation of detergent-soluble amyloidogenic Abeta fragment of Alzheimer’s disease precursor protein in the hippocampus of aged transgenic mice. J Neurochem 72(6):2479–2487
Li H, Wang B, Wang Z, Guo Q, Tabuchi K, Hammer RE, Sudhof TC, Zheng H (2010a) Soluble amyloid precursor protein (APP) regulates transthyretin and Klotho gene expression without rescuing the essential function of APP. Proc Natl Acad Sci USA 107(40):17362–17367. doi:10.1073/pnas.1012568107
Li H, Wang Z, Wang B, Guo Q, Dolios G, Tabuchi K, Hammer RE, Sudhof TC, Wang R, Zheng H (2010b) Genetic dissection of the amyloid precursor protein in developmental function and amyloid pathogenesis. J Biol Chem 285(40):30598–30605. doi:10.1074/jbc.M110.137729
Lin K, Dorman JB, Rodan A, Kenyon C (1997) daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 278(5341):1319–1322
Link CD (1995) Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci USA 92(20):9368–9372
Link CD (2001) Transgenic invertebrate models of age-associated neurodegenerative diseases. Mech Ageing Dev 122(14):1639–1649
Link CD (2005) Invertebrate models of Alzheimer’s disease. Genes Brain Behav 4(3):147–156. doi:10.1111/j.1601-183X.2004.00105.x
Link CD (2006) C. elegans models of age-associated neurodegenerative diseases: lessons from transgenic worm models of Alzheimer’s disease. Exp Gerontol 41(10):1007–1013
Link CD, Johnson CJ, Fonte V, Paupard M, Hall DH, Styren S, Mathis CA, Klunk WE (2001) Visualization of fibrillar amyloid deposits in living, transgenic Caenorhabditis elegans animals using the sensitive amyloid dye, X-34. Neurobiol Aging 22(2):217–226
Link CD, Taft A, Kapulkin V, Duke K, Kim S, Fei Q, Wood DE, Sahagan BG (2003) Gene expression analysis in a transgenic Caenorhabditis elegans Alzheimer’s disease model. Neurobiol Aging 24(3):397–413
Lockery SR, Goodman MB (2009) The quest for action potentials in C. elegans neurons hits a plateau. Nat Neurosci 12(4):377–378. doi:10.1038/nn0409-377
Luo L, Tully T, White K (1992) Human amyloid precursor protein ameliorates behavioral deficit of flies deleted for Appl gene. Neuron 9(4):595–605
Luse SA, Smith KR Jr (1964) The ultrastructure of senile plaques. Am J Pathol 44(4):553–563
Mann DM, Esiri MM (1989) The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down’s syndrome. J Neurol Sci 89(2–3):169–179
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 82(12):4245–4249
McColl G, Roberts BR, Gunn AP, Perez KA, Tew DJ, Masters CL, Barnham KJ, Cherny RA, Bush AI (2009) The Caernorhabditis elegans Abeta1-42 model of Alzheimer’s disease predominantly expresses Abeta3-42. J Biol Chem. doi:10.1074/jbc.C109.028514
McElwee J, Bubb K, Thomas JH (2003) Transcriptional outputs of the Caenorhabditis elegans forkhead protein DAF-16. Aging Cell 2(2):111–121
McIntire SL, Jorgensen E, Kaplan J, Horvitz HR (1993) The GABAergic nervous system of Caenorhabditis elegans. Nature 364(6435):337–341. doi:10.1038/364337a0
Meléndez A, Tallóczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B (2003) Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301(5638):1387–1391
Mello C, Fire A (1995) DNA transformation. Methods Cell Biol 48:451–482
Mello CC, Kramer JM, Stinchcomb D, Ambros V (1991) Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10(12):3959–3970
Mucke L, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, Forss-Petter S, Pietropaolo M, Mallory M, Abraham CR (1994) Synaptotrophic effects of human amyloid beta protein precursors in the cortex of transgenic mice. Brain Res 666(2):151–167. doi:0006-8993(94)90767-6
Muller U, Cristina N, Li ZW, Wolfer DP, Lipp HP, Rulicke T, Brandner S, Aguzzi A, Weissmann C (1994) Behavioral and anatomical deficits in mice homozygous for a modified beta-amyloid precursor protein gene. Cell 79(5):755–765. doi:0092-8674(94)90066-3
Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C (2003) Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424(6946):277–283
Murrell J, Farlow M, Ghetti B, Benson MD (1991) A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science 254(5028):97–99
Nagel G, Brauner M, Liewald JF, Adeishvili N, Bamberg E, Gottschalk A (2005) Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr Biol 15(24):2279–2284. doi:10.1016/j.cub.2005.11.032
Nguyen M, Alfonso A, Johnson CD, Rand JB (1995) Caenorhabditis elegans mutants resistant to inhibitors of acetylcholinesterase. Genetics 140(2):527–535
Nikolaev A, Mclaughlin T, O’Leary DD, Tessier-Lavigne M (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457(7232):981–989
Niwa R, Hada K (2010) Identification of a spatio-temporal enhancer element for the Alzheimer’s amyloid precursor protein-like-1 gene in the nematode Caenorhabditis elegans. Biosci Biotechnol Biochem 74(12):2497–2500. doi:JST.JSTAGE/bbb/100450
Niwa R, Zhou F, Li C, Slack FJ (2008) The expression of the Alzheimer’s amyloid precursor protein-like gene is regulated by developmental timing microRNAs and their targets in Caenorhabditis elegans. Dev Biol 315(2):418–425
Nordstedt C, Caporaso GL, Thyberg J, Gandy SE, Greengard P (1993) Identification of the Alzheimer beta/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. J Biol Chem 268(1):608–612
Nunan J, Small DH (2000) Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett 483(1):6–10. doi:S0014-5793(00)02076-7
Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G (1997) The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389(6654):994–999
Oliveira RP, Porter Abate J, Dilks K, Landis J, Ashraf J, Murphy CT, Blackwell TK (2009) Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell 8(5):524–541. doi:10.1111/j.1474-9726.2009.00501.x
Paliga K, Peraus G, Kreger S, Durrwang U, Hesse L, Multhaup G, Masters CL, Beyreuther K, Weidemann A (1997) Human amyloid precursor-like protein 1-cDNA cloning, ectopic expression in COS-7 cells and identification of soluble forms in the cerebrospinal fluid. Eur J Biochem 250(2):354–363
Park SK, Tedesco PM, Johnson TE (2009) Oxidative stress and longevity in Caenorhabditis elegans as mediated by SKN-1. Aging Cell 8(3):258–269. doi:10.1111/j.1474-9726.2009.00473.x
Rand JB, Russell RL (1985) Molecular basis of drug-resistance mutations in C. elegans. Psychopharmacol Bull 21(3):623–630
Ranganathan S, Noyes NC, Migliorini M, Winkles JA, Battey FD, Hyman BT, Smith E, Yepes M, Mikhailenko I, Strickland DK (2011) LRAD3, A novel low-density lipoprotein receptor family member that modulates amyloid precursor protein trafficking. J Neurosci 31(30):10836–10846. doi:10.1523/JNEUROSCI.5065-10.2011
Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403(6772):901–906. doi:10.1038/35002607
Ring S, Weyer SW, Kilian SB, Waldron E, Pietrzik CU, Filippov MA, Herms J, Buchholz C, Eckman CB, Korte M, Wolfer DP, Muller UC (2007) The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J Neurosci 27(29):7817–7826
Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T et al (1995) Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376(6543):775–778
Rosen DR, Martin-Morris L, Luo LQ, White K (1989) A Drosophila gene encoding a protein resembling the human beta-amyloid protein precursor. Proc Natl Acad Sci USA 86(7):2478–2482
Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D (2006) APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 38(1):24–26
Sabo SL, Ikin AF, Buxbaum JD, Greengard P (2001) The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J Cell Biol 153(7):1403–1414
Salbaum JM, Ruddle FH (1994) Embryonic expression pattern of amyloid protein precursor suggests a role in differentiation of specific subsets of neurons. J Exp Zool 269(2):116–127. doi:10.1002/jez.1402690205
Schupf N, Kapell D, Nightingale B, Rodriguez A, Tycko B, Mayeux R (1998) Earlier onset of Alzheimer’s disease in men with Down syndrome. Neurology 50(4):991–995
Shaye DD, Greenwald I (2011) OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One 6(5):e20085. doi:10.1371/journal.pone.0020085PONE-D-11-05121
Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K et al (1995) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375(6534):754–760
Singh R, Sulston J (1978) Some observations on molting in C. elegans. Nematologica 24(1):63–71
Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, Del-Favero J, Cruts M, van Duijn CM, Van Broeckhoven C (2006) APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain 129(Pt 11):2977–2983
Slunt HH, Thinakaran G, Von Koch C, Lo AC, Tanzi RE, Sisodia SS (1994) Expression of a ubiquitous, cross-reactive homologue of the mouse beta-amyloid precursor protein (APP). J Biol Chem 269(4):2637–2644
Sprecher CA, Grant FJ, Grimm G, O’Hara PJ, Norris F, Norris K, Foster DC (1993) Molecular cloning of the cDNA for a human amyloid precursor protein homolog: evidence for a multigene family. Biochemistry 32(17):4481–4486
Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—is this type 3 diabetes? J Alzheimers Dis 7(1):63–80
Suh Y, Atzmon G, Cho MO, Hwang D, Liu B, Leahy DJ, Barzilai N, Cohen P (2008) Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci USA 105(9):3438–3442. doi:10.1073/pnas.0705467105
Sulston JE, Horvitz HR (1977) Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol 56(1):110–156
Sulston JE, White JG (1980) Regulation and cell autonomy during postembryonic development of Caenorhabditis elegans. Dev Biol 78(2):577–597
Sulston J, Dew M, Brenner S (1975) Dopaminergic neurons in the nematode Caenorhabditis elegans. J Comp Neurol 163(2):215–226. doi:10.1002/cne.901630207
Sulston JE, Schierenberg E, White JG, Thomson JN (1983) The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol 100(1):64–119. doi:0012-1606(83)90201-4
Sumakovic M, Hegermann J, Luo L, Husson SJ, Schwarze K, Olendrowitz C, Schoofs L, Richmond J, Eimer S (2009) UNC-108/RAB-2 and its effector RIC-19 are involved in dense core vesicle maturation in Caenorhabditis elegans. J Cell Biol 186(6):897–914. doi:10.1083/jcb.200902096
Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T (2003) The role of presenilin cofactors in the gamma-secretase complex. Nature 422(6930):438–441. doi:10.1038/nature01506nature01506
Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS (2001) A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 292(5514):107–110
Terry RD, Gonatas NK, Weiss M (1964) Ultrastructural studies in Alzheimer’s presenile dementia. Am J Pathol 44:269–297
Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, Liu S, Oliveira RP, Baumeister R, Blackwell TK (2008) Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 132(6):1025–1038. doi:10.1016/j.cell.2008.01.030
Vargas T, Martinez-Garcia A, Antequera D, Vilella E, Clarimon J, Mateo I, Sanchez-Juan P, Rodriguez–Rodriguez E, Frank A, Rosich-Estrago M, Lleo A, Molina-Porcel L, Blesa R, Gomez-Isla T, Combarros O, Bermejo-Pareja F, Valdivieso F, Bullido MJ, Carro E (2011) IGF-I gene variability is associated with an increased risk for AD. Neurobiol Aging 32(3):556 e553-511. doi:10.1016/j.neurobiolaging.2010.10.017
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M (1999) Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286(5440):735–741. doi:7936
von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, van der Ploeg LH, Price DL, Sisodia SS (1997) Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging 18(6):661–669
von Otter M, Landgren S, Nilsson S, Zetterberg M, Celojevic D, Bergstrom P, Minthon L, Bogdanovic N, Andreasen N, Gustafson DR, Skoog I, Wallin A, Tasa G, Blennow K, Nilsson M, Hammarsten O, Zetterberg H (2010) Nrf2-encoding NFE2L2 haplotypes influence disease progression but not risk in Alzheimer’s disease and age-related cataract. Mech Ageing Dev 131(2):105–110. doi:10.1016/j.mad.2009.12.007
Wang Y, Ha Y (2004) The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol Cell 15(3):343–353
Wasco W, Bupp K, Magendantz M, Gusella JF, Tanzi RE, Solomon F (1992) Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor. Proc Natl Acad Sci USA 89(22):10758–10762
Wasco W, Gurubhagavatula S, Paradis MD, Romano DM, Sisodia SS, Hyman BT, Neve RL, Tanzi RE (1993a) Isolation and characterization of APLP2 encoding a homologue of the Alzheimer’s associated amyloid beta protein precursor. Nat Genet 5(1):95–100. doi:10.1038/ng0993-95
Wasco W, Peppercorn J, Tanzi RE (1993b) Search for the genes responsible for familial Alzheimer’s disease. Ann N Y Acad Sci 695:203–208
Westlund B, Parry D, Clover R, Basson M, Johnson CD (1999) Reverse genetic analysis of Caenorhabditis elegans presenilins reveals redundant but unequal roles for sel-12 and hop-1 in Notch-pathway signaling. Proc Natl Acad Sci U S A 96(5):2497–2502
Wheelan SJ, Boguski MS, Duret L, Makalowski W (1999) Human and nematode orthologs–lessons from the analysis of 1800 human genes and the proteome of Caenorhabditis elegans. Gene 238(1):163–170. doi:S0378-1119(99)00298-X
White JG, Southgate E, Thomson JN, Brenner S (1986) The structure of the nervous system of the nematode C. elegans. Philos Trans Roy Soc Lond Ser B Biol Sci 314(1165):1–340
Wiese M, Antebi A, Zheng H (2010) Intracellular trafficking and synaptic function of APL-1 in Caenorhabditis elegans. PLoS One 5(9). doi:10.1371/journal.pone.0012790
Willcox BJ, Donlon TA, He Q, Chen R, Grove JS, Yano K, Masaki KH, Willcox DC, Rodriguez B, Curb JD (2008) FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci USA 105(37):13987–13992. doi:10.1073/pnas.0801030105
Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA (2001) Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett 306(1–2):116–120. doi:S0304-3940(01)01876-6
Wu Y, Wu Z, Butko P, Christen Y, Lambert MP, Klein WL, Link CD, Luo Y (2006) Amyloid-beta-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J Neurosci 26(50):13102–13113
Xu K, Tavernarakis N, Driscoll M (2001) Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca(2+) release from the endoplasmic reticulum. Neuron 31(6):957–971. doi:S0896-6273(01)00432-9
Yamada T, Sasaki H, Furuya H, Miyata T, Goto I, Sakaki Y (1987) Complementary DNA for the mouse homolog of the human amyloid beta protein precursor. Biochem Biophys Res Commun 149(2):665–671. doi:0006-291X(87)90419-0
Yochem J, Tuck S, Greenwald I, Han M (1999) A gp330/megalin-related protein is required in the major epidermis of Caenorhabditis elegans for completion of molting. Development 126(3):597–606
Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, Rogaev E, Smith M, Janus C, Zhang Y, Aebersold R, Farrer LS, Sorbi S, Bruni A, Fraser P, St George-Hyslop P (2000) Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 407(6800):48–54. doi:10.1038/35024009
Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR (1993) The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell 75(4):641–652. doi:0092-8674(93)90485-9
Zambrano N, Bimonte M, Arbucci S, Gianni D, Russo T, Bazzicalupo P (2002) feh-1 and apl-1, the Caenorhabditis elegans orthologues of mammalian Fe65 and beta-amyloid precursor protein genes, are involved in the same pathway that controls nematode pharyngeal pumping. J Cell Sci 115(Pt 7):1411–1422
Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, Stevens KA, Slunt HH, Sisoda SS, Chen HY, Van der Ploeg LH (1995) beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81(4):525–531
Acknowledgments
We wish to thank Alicia Melendez for her kind gift of the C. elegans strain carrying [RFP::BEC-1], Chris Link, Andrea Silva, and Greg O’Connor for permission to cite unpublished observations and members of our laboratory for many helpful discussions. This work was supported by grants from the Alzheimer’s Association, National Science Foundation, and National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ewald, C.Y., Li, C. Caenorhabditis elegans as a model organism to study APP function. Exp Brain Res 217, 397–411 (2012). https://doi.org/10.1007/s00221-011-2905-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00221-011-2905-7